
Abstract
Success in the differentiating human embryonic stem cells (hESCs) into insulin-secreting β cells raises new hopes for diabetes treatment. In this work, we demonstrated the feasibility of developing islet organoids from hESCs within biomimetic 3D scaffolds. We showed that such a 3D microenvironment is critical to the generation of pancreatic endoderm and endocrine from hESCs. The organoids formed consisted of pancreatic α, β, δ, and pancreatic polypeptide (PP) cells. A high-level co-expression of PDX1, NKX6.1, and NGN3 in these cells suggests the characteristics of pancreatic β cells. More importantly, most insulin-secreting cells generated did not express glucagon, somatostatin, or PP. The expression of mature β cell marker genes such as Pdx1, Ngn3, Insulin, MafA, and Glut2 was detected in these 3D-induced cell clusters. A high-level expression of C-peptide confirmed the de novo endogenous insulin production in these 3D induced cells. Insulin-secretory granules, an indication of β cell maturity, were detected in these cells as well. Glucose challenging experiments suggested that these cells are sensitive to glucose levels due to their elevated maturity. Exposing the cells to a high concentration of glucose induced a sharp increase in insulin secretion.
Introduction
D
Stepwise pancreatic differentiation approaches that mimic in vivo critical events of pancreatogenesis have been developed and used to differentiate hESCs into insulin-secretion cells [5 –9]. A recent groundbreaking work reported by Melton's group demonstrated the generation of mature β cells from hPSCs [3]. The transplantation of these insulin-secreting cells in immune-competent mice led to long-term glycemic control, suggesting potential use of these cells for diabetes therapy [10]. The generation of patient-specific β cells from type 1 diabetic patients has also been demonstrated recently [11]. On the other hand, a number of groups have been working to mature hPSC-derived pancreatic progenitors in vivo by transplanting immature hPSC-derived pancreatic progenitors into mice [3,12]. However, the difficulty of controlling the fate of these cells after they are grafted in the body poses a challenge to diabetes treatment.
The generation of islet organoids containing mature β cells has yet to be demonstrated. The generation of islet organoids will allow for drug screening and pathological studies of diabetes development and progression. Previously, we reported success in generating islet-like cell clusters from mouse embryonic stem cells (mESCs) within 3D collagen scaffolds [13]. Here, we report the development of islet organoids from hESCs within a biomimetic 3D scaffold.
A line of evidence reveals that the extracellular matrix (ECM) plays a critical role in cell proliferation and development. Cell-matrix interactions have been shown to improve β cell proliferation [14,15], insulin secretion [16,17], and islet development [16,18,19]. Blocking islet-matrix interaction by using β1 integrin antibodies results in decreased insulin gene expression and islet-cell apoptosis [20]. Collagen has been used widely for 3D stem cell cultures [21]. A collagen scaffold constitutes of a soft and flexible fibrous network that supports the maintenance of cell morphology and allows cells to freely reach out, migrate, and form 3D structures. Nevertheless, collagen alone is insufficient in providing the multiple cues and the sophisticated geometry and composition that exist in a native ECM. We hypothesized that the augmentation of a collagen scaffold with Matrigel, a mixture of basement membrane proteins such as laminin, collagen IV, fibronectin, heparin sulfate proteoglycans, and entactin can improve islet development from hESCs. Oberg-Welsh have shown that Matrigel significantly enhances the insulin secretion of fetal porcine islet-like cell clusters in vitro [22]. The combination of collagen with Matrigel has been used to reconstruct cardiac muscle uterus and kidney in vitro [23,24]. We demonstrated that the combination of collagen with Matrigel (C-M) creates better 3D niches islet organoid development from hESCs.
Materials and Methods
hESC culture
hESCs H9 acquired from WiCell Research Institute (Madison, MI) were cultured on Matrigel (BD Biosciences, Bedford, MA)-coated cell culture plates in a mTeSR1 medium (Stem Cell Technologies, Inc., Vancouver, Canada). A solution of 1 mg/mL dispase in the DMEM/F12 medium was used to passage cells every 3–4 days. The cultures were incubated at 37°C in an atmosphere that was supplemented with 5% CO2, with the cell culture medium exchanged daily.
3D scaffold preparation
Collagen type I scaffolds were prepared at a 1.5 mg/mL concentration under sterile conditions by diluting a rat tail tendon-derived collagen I solution (BD Biosciences, Bedford, MA) with 10× phosphate-buffered saline (PBS; Mediatech, Inc., Manassas, VA) and cell culture grade distilled water (Thermo Fisher Scientific, Inc., Waltham, MA). The pH of the mixture was neutralized immediately to 7.4 by using 1 N NaOH. The C-M scaffolds were prepared by mixing growth factor-reduced Matrigel with the collagen I solution at 10%, 35%, or 50% (v/v). The solutions were then neutralized to pH 7.4, aliquoted into 48-well plates, and incubated at 37°C for 1 h to induce gelation. No cross-linking agent was used.
3D hESC pancreatic differentiation
H9 cells were treated with the ROCK inhibitor, Y-27632, in the cell culture medium for 2 h before dissociation. After incubation with Accutase (Stem Cell Technologies, Inc.) for 7 min, cell suspension was prepared post pelleting and resuspension in the mTeSR1 medium. For 3D cultures, a 10% volume of cell suspension (1.2 × 106 cells) was mixed with 0.5 mL mixture of collagen I and Matrigel solution. The solution composed was then added to a 24-well plate to form a scaffold that has an initial radius of 8 mm and a thickness of 2.5 mm. The 3D constructs were placed in a CO2 incubator for 1 h at 37°C for gelation. Once the scaffolds were cast, 0.5 mL of the cell culture medium was added to the top of the scaffolds. The cell-laden scaffolds were then placed in a CO2 incubator and used for subsequent differentiation experiments. Two-dimensional cultures, where H9 cells were grown on Matrigel-coated culture plates, were also prepared for comparison. The cells were allowed to grow within the scaffolds or on the Matrigel-coated plates in an mTeSR1 medium that was supplemented with 10 μM Y-27632 for 24 h for attachment and proliferation. A stepwise protocol (Fig. 1), modified from Zhang et al.'s protocol [7], was used for hESC pancreatic differentiation.
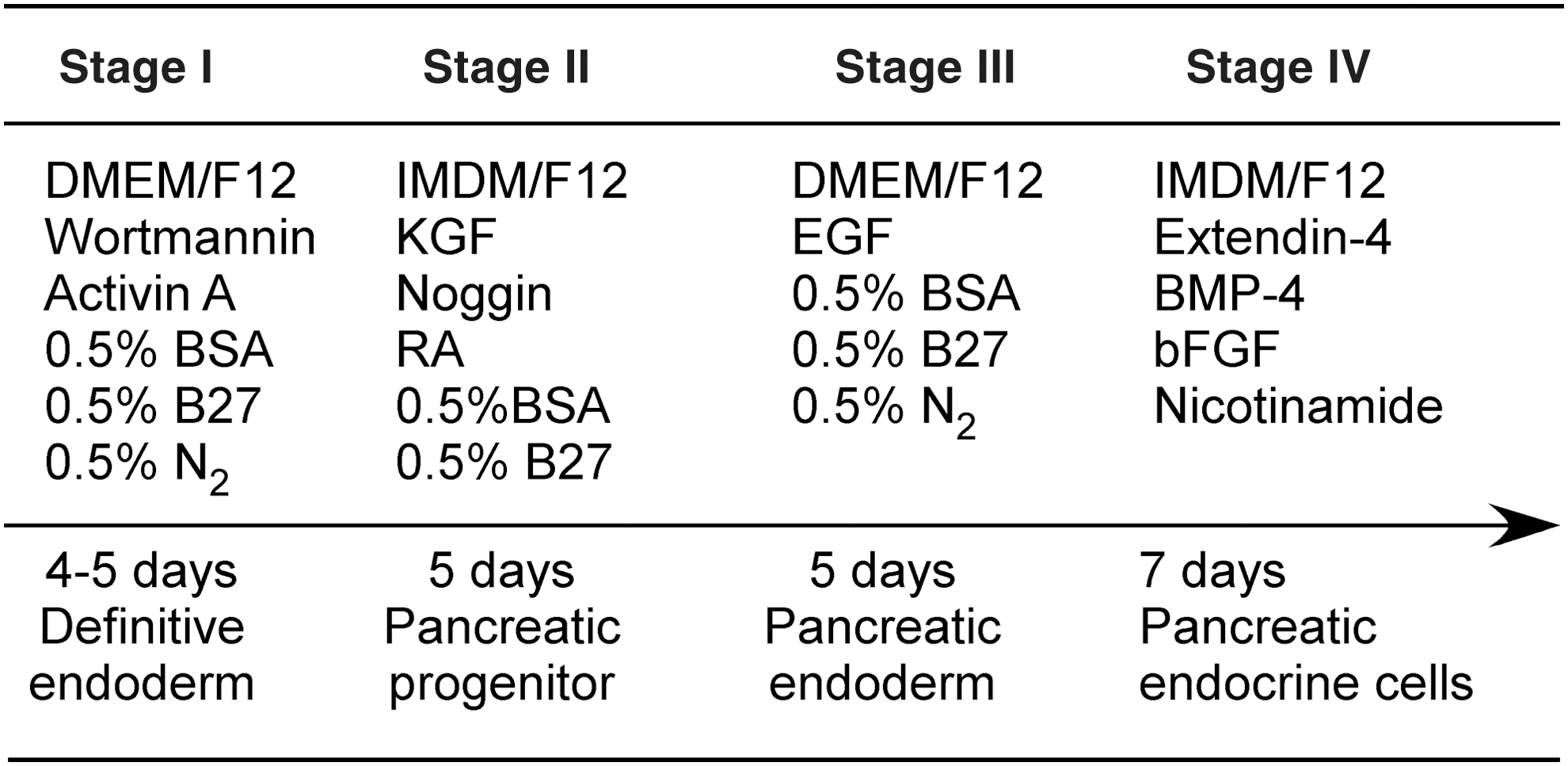
Stepwise differentiation of hESCs into islet organoids. Stage I: hESCs were induced into definitive endoderm (DE) in the presence of activin A and wortmannin. Stage II: the DE cells were treated with RA, KGF, Noggin, and ILV to induce pancreatic progenitor formation. Stage III: the cells were exposed to EGF to endorse pancreatic endoderm lineage-specific differentiation. Stage IV: a cocktail of factors, including nicotinamide, extendin-4, BMP4, and bFGF, was used to promote the formation of islet organoids and the maturation of β cells. KGF, keratinocyte growth factor; RA, retinoic acid; EGF, epidermal growth factor; BMP4, bone morphogenetic protein 4; BSA, bovine serum albumin; N2, a chemically defined serum-free supplement; bFGF, basic fibroblast growth factor; hESC, human embryonic stem cell.
Scanning electron microscopy
In preparation for scanning electron microscopy (SEM), cell-laden scaffolds were washed with PBS and fixed in 2.5% (v/v) glutaraldehyde (Electron Microscopy Sciences, Hatfield, PA) overnight at room temperature. After fixation, the scaffolds were washed three times with distilled water. The samples were then snap frozen in liquid nitrogen (−196°C) and broken with cold forceps. A fragment of the frozen scaffolds was transferred into a freeze dryer chamber for low-temperature (−100°C) high-vacuum dehydration. Dried samples were mounted on SEM sample stages by using carbon tape and were sputter coated with gold-plutonium (palladium) alloy (Pd/Au) under vacuum by using a Palaron SC7620 sputter coater (Watford, United Kingdom). Samples were then analyzed by using the Jeol Field Emission SEM (JSM-6335F; JEOL, Peabody, MA) with a low-vacuum mode (0.75 Torr), at 15 kV accelerating voltage.
Quantitative real-time polymerase chain reaction
Cell-scaffold constructs were mechanically homogenized by using a Tissue-Tearor (BioSpec Products, Bartlesville, OK). Total RNA was extracted by using an RNeasy Plus Mini kit (Qiagen, Valencia, CA). RNA samples were digested with DNase I to remove any contaminated genomic DNA. A portion of 1 μg of total RNA was used for reverse transcription by using a high-capacity cDNA reverse transcription kit from Invitrogen. Quantitative real-time polymerase chain reaction (qRT-PCR) was carried out by using a power SYBR green PCR master mix (Applied Biosystems) and a Realplex Real-Time PCR system (Eppendorf, Realplex4). Initial enzyme activation was performed at 95°C for 15 min, followed by 40 cycles of denaturation at 95°C for 15 s and primer annealing/extension at 60°C for 1 min. Melting curve analysis was performed at 95°C for 1 min, 60°C for 30 s, and 95°C for 30 s. Primers used are listed in Table 1. Human adult pancreatic RNA from Invitrogen was used as a control. The expression levels of target genes were normalized against endogenous control β-actin. All samples were analyzed in triplicate.
Immunofluorescence staining
Cell-laden scaffolds were fixed in 4% freshly made paraformaldehyde for 15 min. They were then washed three times with PBS. Permeabilization was carried out with 0.1 Trixton X-100 for 5 min. Cryosectioning of the scaffolds was also performed, as described elsewhere [25]. Scaffold-cell constructs were fixed in 10% neutral-buffered formalin solution for 3 h. After overnight infiltration with a series of 20% sucrose and sucrose/OCT solutions, the scaffolds were placed in a peel-away mold (VWR, South Plainfield, NJ) within a tissue freezing medium (Tissue-Tek OCT Compound, Sakura-Fintek). All of the molds were then placed into the MICROM cryostat (Richard-Allan Scientific) chamber, and the samples were allowed to freeze at −20°C for at least 30 min. Routine frozen sectioning was performed by collecting 8-μm sections onto positively charged slides (Electron Microscopy Sciences, Hatfield, PA). The scaffold sections were either collected or air dried for a minimum of 30 min. Before staining, the sections were hydrated in PBS for 10 min, then permeabilized with 0.1 Trixton X-100 for 5 min, and finally incubated in a blocking buffer (PBS containing 5% normal goat serum, 0.3% Triton X-100) for 1 h. Nonspecific antibody binding was blocked by 10% normal serum from the species in which the secondary antibodies were raised. Cells were incubated with primary antibodies overnight at 4°C, followed by secondary antibody incubation for 1 h at room temperature. To stain cells cultured in tissue culture plates (2D), cells seeded on Matrigel-coated plates were fixed with 4% paraformaldehyde (Sigma). Cells were then treated with the blocking buffer to inhibit nonspecific labeling (45 min at room temperature). Cells were then labeled with primary and secondary antibodies, as described earlier. DAPI (Research Organics) were used to counterstain the cell nucleus. Images were captured under an inverted fluorescence microscope (Olympus, IX71, Japan). The primary antibody and secondary antibodies used are listed in Table 2.
Glucose-stimulated insulin secretion assay
To determine whether hESC-derived pancreatic cells are capable of secreting insulin on glucose challenging, cells generated from hESCs in either 2D or 3D conditions were washed three times with a Krebs-Ringer buffer containing 0.1% bovine serum albumin and 10 mM HEPES. Cells were first incubated with a Krebs-Ringer buffer free of glucose at 37°C for 60 min and were then exposed to the Krebs-Ringer buffer containing 5.5, 15, or 25 mM glucose at 37°C for 90 min. Supernatants were collected and analyzed for insulin secretion from cells by using an insulin ELISA kit (Alpco Diagnostics). The amounts of insulin secretion were normalized by measuring total intracellular protein using a BCA protein assay kit (PIERCE).
Transmission electron microscopy
Both 2D cultured cells and cell-laden scaffolds were fixed in 3% glutaraldehyde in 0.1 M cacodylate buffer (pH 7.4) at room temperature for 2 h. After rinsing with 0.05 M cacodylate buffer, the samples were post-fixed in 1% osmium tetroxide for 2 h, followed by rinsing with distilled water. After pre-staining with 0.5% uranyl acetate at 4°C overnight, the samples were dehydrated once through a graded ethanol series of 30%, 50%, 70%, 80%, 95%, and 3 changes of 100%, then rinsed in propylene oxide, and finally embedded in Spurr's epoxy. Eighty-nanometer-thick sections were cut on a MT2-B ultramicrotome and stained with 2% uranyl acetate and lead citrate. The samples were examined by using a Philips EM 410LS transmission electron microscope (TEM) operated at 80 kV.
Statistical analysis
All experiments were carried out in at least triplicate. Unless otherwise indicated, all analytical measurements were repeated three times. All data were expressed as means ± standard deviations. Statistical analysis was performed with Student's t-test, with P < 0.05 considered statistically significant.
Results
Matrigel enhances hESC definitive endoderm differentiation toward pancreatic lineages within C-M scaffolds
We have developed a tissue-engineered collagen scaffold for differentiating mESCs into glucose-responsive, insulin-secreting cells [13]. When working on hESCs, we found that collagen scaffolds became weaker and partially collapsed after 10–15 days of differentiation (data not shown). To overcome this issue, we mixed collagen with Matrigel during scaffolding. We expected that the incorporation of Matrigel into collagen scaffolds would better support hESC pancreatic differentiation. We investigated the effect of different concentrations of Matrigel on hESC pancreatic differentiation. The SEM suggested that Matrigel has a profound effect on the microstructures of the scaffolds. As shown in Fig. 2A–F, the scaffolds containing 10% Matrigel appeared to be less interconnected and less uniform. The scaffolds became more homogenous and formed tightly packed fibrils and smaller pores with an increase in the concentration of Matrigel from 10% to 35%, and then to 50%. It was evident that the increases of Matrigel concentration gave rise to a substantial decrease in both void and size of the interconnected pores. To investigate whether these changes in the microstructures of the scaffolds affect hESC pancreatic differentiation, we performed hESC definitive endoderm (DE) differentiation within these scaffolds. DE differentiation is the first and also the most critical step in hESC pancreatic differentiation. After 4 days of differentiation, cells were harvested and analyzed for DE marker expression. As illustrated in Fig. 2G, the incorporation of Matrigel in collagen scaffolds improved hESC DE differentiation. We observed an increase of 2- to 4.5-fold in Sox17, Foxa2, and CXCR4 expression in scaffolds containing 35% Matrigel, as compared with those in collagen-only scaffolds.
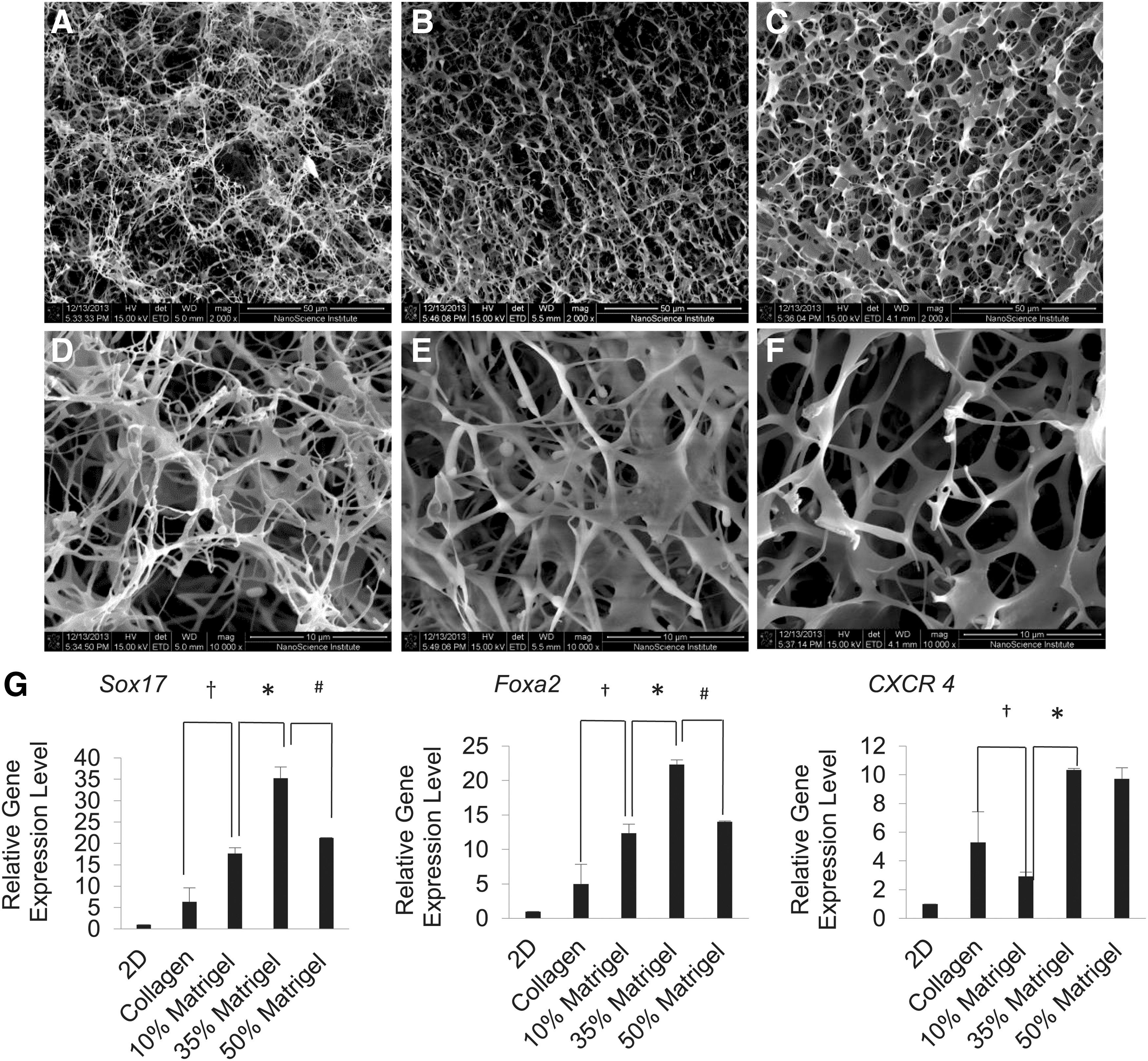
Effect of Matrigel on hESC DE differentiation within C-M scaffolds.
(-)-indolactam V treatment promoted the generation of pancreatic progenitor cells
Next, we interrogated the effect of (-)-indolactam V (ILV) on the commitment of hESCs to PDX1+ pancreatic progenitors. ILV was previously found to specifically induce pancreatic progenitors from DE [26]. We intended to ascertain whether treating cells with ILV at Stage II would enhance the commitment of hESC toward pancreatic lineages within the scaffolds.
First, we performed 2D hESC pancreatic differentiation in the presence and absence of ILV. An examination of the morphology of cell clusters formed under these conditions revealed the critical effect of ILV treatment on cell cluster formation during differentiation (Fig. 3A–F). We found that pancreatic progenitor cells generated in the presence of ILV tended to form cell clusters and co-expressed a high level of PDX1 and HNF6, as compared with those differentiated in the absence of ILV. PDX1 and HNF6 play a key role in regulating lineage-specific endoderm differentiation during pancreatogenesis. Although both are initially expressed throughout the pancreatic epithelium within cell types [27], the expression of PDX1 is restricted more primarily to β cells and is maintained at low levels in acinar cells when endocrine cells develop and form mature islets. The expression of HNF6 turns into silence in mature endocrine cells but maintains a high expression level in surrounding exocrine and duct cells. To this end, controlling the divergent expressions of PDX1 and HNF6 is critical to pancreatic islet development during pancreatogenesis.
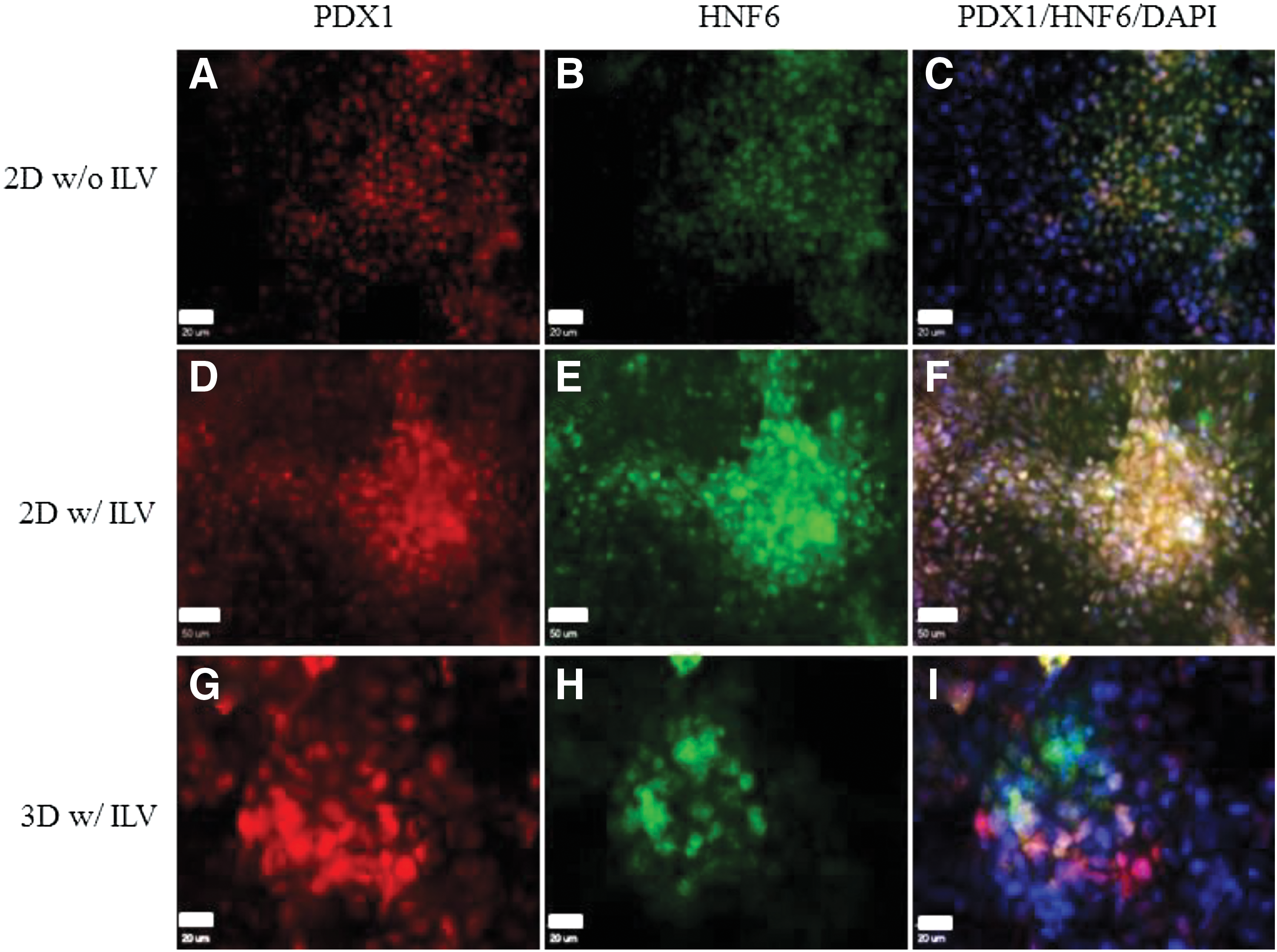
The effect of (-)-indolactam V (ILV) on hESC pancreatic commitment from DE.
Next, we investigated whether the differentiation of hESCs within scaffolds in the presence of ILV improves the divergent expression of PDX1 and HNF6 in these cells. As shown in Fig. 3G–I, a majority of cells differentiated under these conditions expressed a high level of PDX1, not HNF6, indicating their commitment toward pancreatic endocrine progenitors. A high-level expression of HNF6, not PDX1, was observed in small groups of cells in the clusters, suggesting their commitment toward pancreatic exocrine progenitors. Very few cells in the cell clusters expressed both PDX1 and HNF6, implying that these cells were still at the early stages of pancreatic development. These experiments suggested that the combinatory effect of 3D environments and signaling molecule ILV improved the branching of hESC pancreatic differentiation toward the pancreatic endoderm.
The formation of islet organoids within C-M scaffolds
Finally, we explored the feasibility of generating islet organoids from hESCs. We used a four-stage differentiation strategy to differentiate hESCs into pancreatic endoderm and to further mature them into islet organoids within the C-M scaffolds. We analyzed pancreatic marker gene expression throughout the entire differentiation process by using qRT-PCR analysis. We observed an elevated expression of DE marker gene Sox17 at the end of Stage I (Fig. 4A). Its expression level dropped sharply with progression of the differentiation toward pancreatic progenitor cells. The expression of both Pdx1 and Ngn3 in these cells was detected at the end of Stage II, indicating the branching of differentiation toward pancreatic endoderm cells (Fig. 4B, C). Their expression levels reached a peak at the end of Stage III. The detection of insulin gene expression revealed a 5-fold increase (P < 0.05) in cell clusters formed at the end of Stage IV in 3D conditions, as compared with those formed in 2D conditions (Fig. 4D). Glut2 is a glucose transporter found in the cell membrane of mature β cells. It is not only an important indicator for β cell maturity but also required for glucose-induced insulin release. Its expression emerged at the end of Stage II, elevated at Stage III, and reached a peak at the end of Stage IV. We observed a 4.3-fold increase in Glut2 gene expression in 3D-formed cell clusters (Fig. 4E), as compared with those formed in 2D conditions. MafA is another hallmark of mature pancreatic endocrine cells. The expression of MafA was observed at the end of Stage III. It was elevated to a higher level at the end of Stage IV, indicating the maturation of these hESC-derived β cells (Fig. 4F). We observed a fivefold increase in MafA gene expression in cell clusters formed in 3D conditions, as compared with those formed in 2D conditions, suggesting the significant elevation of hESC-derived β cell maturity within the scaffolds.

To determine whether these cell clusters exhibited a characteristic islet-like structure, we performed dual-color immunofluorescence staining. The co-expression of PDX1 with NKX6.1 (Fig. 5A–C) and NGN3 (Fig. 5D–F) was observed in these cell clusters collected at the end of Stage IV, suggesting the pancreatic β cell characteristics of these cells. The detection of a high-level C-peptide in insulin-secreting cells further confirmed the de novo endogenous insulin production in these cells (Fig. 5G–I). Furthermore, the expression of endocrine hormones such as insulin, glucagon, somatostatin, and pancreatic polypeptide (PP) in these cell clusters was analyzed. As shown in Fig. 5J–R, glucagon, somatostatin, and PP were expressed only in small subsets of cells that were mixed with insulin-secreting cells. This is consistent with the hormone expression pattern of mature islets where small subsets of cells, such as α, δ, and PP cells, secrete glucagon, somatostatin, and PP, respectively. These results suggest the formation of islet-like organoids during the maturation of pancreatic endoderm at Stage IV. More importantly, most insulin secreting cells do not express somatostatin, glucagon, or PP, suggesting a high-degree maturity of these cells. It is well known that immature pancreatic β-like cells express insulin along with somatostatin, glucagon, or PP. The detection of insulin-expressing cells suggests the generation of pancreatic β-like cells.

Dual-color immunofluorescence microscopy of islet organoids differentiated from hESCs within CM scaffolds. Cell clusters formed at Stage IV within CM scaffolds were dual-color stained by using primary antibodies against PDX1
To further determine the maturity of β cells formed, we detected the insulin secretory granules in these cells through TEM (Fig. 6). Insulin secretory granules are pivotal organelles of mature β cells and represent a key participant of glucose homeostasis. The TEM unveiled many insulin secretory granules in cells generated within scaffolds (Fig. 6A). These granules are characterized by a round electron-dense crystalline core surrounded with a distinctive large, clear halo. In contrast, only very few insulin secretory granules were detected in cells differentiated in 2D conditions (Fig. 6B). The inner crystalline core of insulin granules is less electron opaque in mature β cells than in those found in glucagon-containing and somatostatin-containing α and δ cells, which further confirmed the generation of mature insulin-producing β cells. The number of insulin secretory granules detected in cells generated in 3D conditions was ∼12 times (counted from 5 individual cells from each sample) more than that in cells generated in 2D conditions.

Glucose-responsive, insulin secretion of islet organoids generated from hESCs within C-M scaffolds.
Determination of insulin secretion from islet organoids on glucose challenge
Finally, we detected glucose-responsive insulin secretion from islet organoids developed within C-M scaffolds. Cell clusters collected at Stage IV were exposed to 5.5, 15, and 25 mM of glucose, respectively. We observed a sharp increase in insulin secretion from the cell clusters when exposed to a high concentration of glucose (Fig. 6C). We detected the secretion of 44 ± 4.9 μIU (international unit) insulin/mg cellular proteins from 3D-induced cell clusters when exposed to 25 mM glucose. This represents a seven-fold increase in insulin secretion, as compared with what was detected in cells exposed to a low concentration of glucose (5.5 mM). In contrast, 2D-formed insulin-secreting cells are less sensitive to glucose challenge. Only 10.8 ± 3.2 μIU insulin/mg cellular protein secretion was detected when 2D-induced cells were exposed to 25 mM glucose, representing a 3.7-fold increase in insulin secretion, as compared with that detected in cells exposed to a low concentration of glucose (5.5 mM). These results demonstrated that 3D-induced insulin-secreting cells are more sensitive to a glucose challenge due to their elevated maturity.
Discussion
The study of hESC pancreatic differentiation has been focused mainly on mimicking the sequence of signaling events that underlie cell commitment and tissue formation during pancreatogenesis. Apart from soluble growth factors that are the keys to cell lineage specification, the importance of ECM in hESC pancreatic differentiation and islet development has not been fully elucidated. In this study, we demonstrated the feasibility of generating islet organoids from hESCs by using a biomimetic ECM scaffold. We investigated the combinatory effects of physiological cues and ILV offered by collagen and Matrigel scaffolds on the generation of islet organoids from hESCs. As previously shown [13,28], collagen I scaffolds offer better niches for promoting in vitro differentiation and maturation of mESC-derived pancreatic endocrine β cells. Nevertheless, a collagen scaffold is not strong enough to support hESC pancreatic differentiation. These scaffolds collapse during differentiation. The addition of Matrigel to collagen scaffolds seems to improve the mechanical strength of the scaffolds. Although we did not measure the Young's modulus of these scaffolds, it has been discovered that the Young's moduli of collagen-Matrigel scaffolds is proportional to the concentration of Matrigel incorporated [29]. Thus, we speculated that the addition of Matrigel to a collagen scaffold improves its mechanical strength, which better supports the development of islet organoids from hESCs. The addition of Matrigel not only improves the mechanic strength of the scaffolds but also affects the fibrillary structure of the scaffolds. The SEM analysis suggested that the scaffolds become more uniform with a fine fiber structure at a low concentration of Matrigel (10% v/v). Larger fibrils were formed with an increase in the concentration of Matrigel. Our experimental results showed that 35% Matrigel gave rise to the highest expression of DE marker genes such as Sox17, Foxa2, and CXCR4. The efficient generation of DE cells would facilitate subsequent hESC pancreatic specification. These experimental results suggested that the differentiation of hESCs into pancreatic endocrine cells requires an optimal fibril structure and mechanical strength of scaffolds. These effects on islet organoid development need to be further characterized.
It has been long known that the ECM of mature human islets entails rich laminin and collagen IV [30], which are major ECM proteins of Matrigel [31]. It is presumed that the inclusion of Matrigel in the collagen scaffolds offers preferential environments that allow hESCs to differentiate and mature into islet organoids. This observation was supported by many recent studies. A line of evidence indicates that ECM provides a spatially and temporally controlled environment for stem cell differentiation and tissue maturation [32].
Activin A has long been used to induce DE cells. Nevertheless, a recent study reported by McLean et al. showed that the specification of hESCs into DE lineages occurs only when phosphatidylinositol 3-kinase (PI3K) signaling is suppressed [33]. Thus, we used wortmannin, a P13K inhibitor, to suppress P13K signal when inducing hESC DE differentiation. We showed that more than 95% of cells were Sox17+ when hESC DE differentiation was carried out within C-M (35% v/v) scaffolds. Furthermore, we added Noggin (an inhibitor of bone morphogenetic protein signaling), retinoic acid, and fibroblast growth factor 7 (FGF7 or KGF) [34 –37] sequentially to the cell culture media to differentiate DE cells into pancreatic progenitors. (-)-indolactam (ILV) has been discovered to efficiently induce PDX1+ pancreatic progenitor cells [26]. We showed that the effect of ILV on hESC pancreatic lineage specification becomes more profound when it is used in combination with a 3D scaffold. A higher level of expression of PDX1, not HNF6, was observed in hESC-derived pancreatic progenitors differentiated within a C-M scaffold in the presence of ILV, suggesting the divergent expression of PDX1 and HNF6 during pancreatic lineage specification under these conditions. It has been known that both PDX1 and HNF6 are initially expressed throughout the pancreatic epithelium within cell types. The expression of PDX1 will be more restricted to β cells and will be maintained at a low expression level in acinar tissues when endocrine cell clusters form mature islets [27]. In contrast, HNF6 remains at a high expression level in surrounding exocrine tissues and ducts whereas it becomes silent in mature endocrine cells. The observation of the co-expression of both PDX1 and HNF6 in hESC-derived pancreatic cells differentiated under 2D conditions suggested their immaturity. These observations were further confirmed by examining the tissue structures of both 2D- and 3D-formed cell clusters through various assays. As revealed by immunohistochemical microscopy, the cell clusters formed under 3D conditions consist of four types of pancreatic endocrine cells, including insulin-secreting β cells, glucagon-secreting α cells, somatostatin-secreting δ cells, and polypeptide-secreting PP cells, similar to those observed in mature islets. These tissue organizations were not observed in cell clusters formed under 2D conditions. Moreover, the co-expression of insulin, C-peptide, PDX1, and NKX6.1 was observed in 3D-induced insulin-producing cells. This is considered a characteristic of mature β cells [38]. The expression of pancreatic marker genes revealed a significant elevation of β cell-specific transcription factors, including Pdx1, insulin, and Glut2 in 3D-induced cell clusters. More cells expressed insulin in 3D-induced cell clusters, suggesting a high efficiency of hESC pancreatic differentiation under these conditions. Moreover, we demonstrated the glucose-responsive insulin secretion of these cell clusters. The detection of insulin-secretory granules further suggests that these insulin-secreting cells are close to adult mature islet β cells. It is worthy of mention that more characterizations such as transplanting these islet organoids in animal models need to be performed to further confirm the biological functions of these organoids. In addition, more hESC cell lines, including hiPSCs, need to be tested to determine the robustness of the approach developed through this work.
It should be pointed out that the generation of mature β cells from hPSCs both in vitro and in vivo has been well demonstrated recently by Melton's group [3,11]. They showed that mature β cells can be generated from hPSCs in a suspension culture that can easily be scaled up for the large-scale production of therapeutic β cells from hPSCs. The animal studies demonstrated the potential application of these cells for long-term glycemic control in diabetic mice [10]. Here, we demonstrated the feasibility of generating islet organoids from hESCs by using Matrigel-collagen scaffolds. Although neither Matrigel nor rat tail type I collagen is a Food and Drug Administration–approved material for clinical applications, the study shown here demonstrates the feasibility of generating islet organoids from hESCs. The cell clusters can be purified by enzymatically digesting scaffolds that remove scaffold materials from cell clusters. Another alternative is to use porcine decellularized ECMs to construct scaffolds. We are currently testing these ECMs for generating islet organoids from hiPSCs.
Footnotes
Acknowledgments
This work was partially supported by NSF CBET-0756455 and 1445387, and the Juvenile Diabetes Research Foundation (JDRF) 5-2009-381.
Author Disclosure Statement
No competing financial interests exist.