
Abstract
Oxygen levels are an important variable during the in vitro culture of stem cells. There has been increasing interest in the use of low oxygen to maximize proliferation and, in some cases, effect differentiation of stem cell populations. It is generally assumed that the defined pO2 in the incubator reflects the pO2 to which the stem cells are being exposed. However, we demonstrate that the pO2 experienced by cells in static culture can change dramatically during the course of culture as cell numbers increase and as the oxygen utilization by cells exceeds the diffusion of oxygen through the media. Dynamic culture (whereby the cell culture plate is in constant motion) largely eliminates this effect, and a combination of low ambient oxygen and dynamic culture results in a fourfold increase in reconstituting capacity of human hematopoietic stem cells compared with those cultured in static culture at ambient oxygen tension. Cells cultured dynamically at 5% oxygen exhibited the best expansion: 30-fold increase by flow cytometry, 120-fold increase by colony assay, and 11% of human CD45 engraftment in the bone marrow of NOD/SCID mice. To our knowledge, this is the first study to compare individual and combined effects of oxygen and static or dynamic culture on hematopoietic ex vivo expansion. Understanding and controlling the effective oxygen tension experienced by cells may be important in clinical stem cell expansion systems, and these results may have relevance to the interpretation of low oxygen culture studies.
Introduction
N
These findings have implications for the in vitro expansion of stem cells for clinical applications. We [7] and others [8 –10] have demonstrated improved maintenance of self-renewal potential and in vitro expansion of hematopoietic stem/progenitor cells (HSPCs) when cultured at reduced oxygen. The cytokine cocktail used has hypoxia-mimicking effects and synergizes with low oxygen concentration in stem cell maintenance [11,12]. This is perhaps unsurprising given that the bone marrow, and in particular the HSC niche, is a low oxygen environment [8,13 –15]. There is increasing evidence for a critical role of HIF-dependent mechanisms in control of HSPC self-renewal [8,16 –18]. Hypoxia has also been shown to impact on human MSC expansion derived from bone marrow, adipose tissue, amniotic fluid, and umbilical cord blood (UCB) and appeared to depend on the source of the cells [19].
A critical question in interpretation of in vitro studies is whether the cells were actually exposed to the effective pO2 that was intended. More than 40 years ago, Balin et al. [20] showed the impact of stationary versus shaken cultures on the proliferative capacity of cells, while almost 20 years ago, Metzen et al. [21] demonstrated that the pericellular oxygen experienced by cells in monolayer culture may be substantially lower than the pO2 in the air to which the culture was exposed. This effect is due to the limited diffusion of oxygen through media, such that the utilization of oxygen by most confluent cell cultures exceeds the passive diffusion of oxygen through the medium [22], creating a gradient that can be directly measured [21] or visualized [23]. This effect has been modeled by Zhdanov et al. [24], who showed that the critical factors in determining the effective oxygen experienced by cells include atmospheric oxygen, sample geometry (eg, depth of media), cell density, and the respiration rate of cells. Given that stem cell expansion cultures can result in a many-fold increase in cell numbers over the course of the culture, we have measured the dissolved oxygen in HSPC cultures over 7 days. We have further investigated the functional impact of different atmospheric oxygen levels and of static and dynamic cultures on the repopulating capacity of HSPCs. These studies demonstrate the critical role of these variables on the outcome of HSPC cultures and the importance of directly measuring dissolved oxygen when performing studies on the effect of hypoxia on cell metabolism and function.
Materials and Methods
Ethics statement
Research with human samples and mice was performed in compliance with the local ethical guidelines and with the approval of the Institutional Review Boards. NSG (NOD.Cg-Prkdcscid Il2rgtm1Wjl /SzJ) mice, which were used for xenografting human UCB cells, were obtained from the Walter and Eliza Hall Institute and bred at the Monash Medical Centre Animal Facility under approval from the Office of the Gene Technology Regulator (IBC NLRD number: PC1-N30/09) and with animal ethics approval MMCA2013/28BC for breeding of the mice. NSG mice were bred and housed under specific pathogen-free conditions of an automated 12-h light–12-h dark cycle. Mice were fed ad libitum with standard mouse chow and autoclaved acidified water. After irradiation and transplantation, the mice were given Bactrim antibiotic in their water. Approval to undertake this project was obtained from Monash Medical Centre Animal Ethics Committee A (AEC number: MMCA 2012/83). UCB was collected with informed consent under Monash Health human research ethics committee approval (HREC number: 12387B). Collection was carried out in accordance with the National Statement on Ethical Conduct in Human Research (2007) produced by the National Health and Medical Research Council of Australia.
Cell culture
Isolation of CD34+ cells
CD34+ cells were isolated from freshly collected human UCB as described previously [25,26]. In short, diluted UCB was centrifuged using a Ficoll-Paque density gradient and mononuclear cells were obtained from the buffy coat. CD34+ cells were then isolated from mononuclear cells through magnetic-assisted cell sorting using MACS CD34 Microbead kits (Miltenyi Biotec). The viability and purity of the isolated cells were assessed by flow cytometry and cells were cryopreserved in liquid nitrogen until required.
HSPC expansion
CD34+ cells were cultured in serum-free medium, Stemspan Animal Component-Free (ACF) (Stem Cell Technologies) supplemented with 100 U/mL penicillin–100 μg/mL streptomycin (Life Technologies), 50 ng/mL stem cell factor (BD Biosciences), 50 ng/mL thrombopoietin, 80 ng/mL Fms-related tyrosine kinase ligand, and interleukin 6 (all three growth factors from Merck Millipore). Cells were seeded in triplicates onto the middle wells of 24-well tissue culture plates at 10,000 cells/well. Before seeding, the tissue culture wells were coated with 10 μg/mL RetroNectin® (Takara Bio, Inc.) for 1 h at room temperature.
The cells were cultured in either 20% O2 or 5% O2 under two conditions—static or dynamic. Cells cultured under static condition were placed on a shelf in the incubator and those cultured under dynamic conditions were placed on an orbital shaker at 20 rpm within the incubator. Two identical plates were set up for each experimental group whereby one plate was used to quantify CD34+ expansion, while the other was for measuring oxygen concentration daily. All groups were cultured in a humidified environment at 37°C and 5% CO2 for 7 days. Cells were harvested by gentle flushing, followed by two washes with phosphate-buffered saline (PBS). Viable cell counting was done with 1% trypan blue and the cells were further analyzed by flow cytometry and colony-forming assay.
Characterization of expanded HSPCs
After 7 days of culture, the HSPCs were harvested and counted using Trypan Blue exclusion. In addition, flow cytometric analysis for surface markers was performed with the following four antibody combinations: CD45 FITC + CD34 PE + 7AAD, CD34 FITC + CD38 PE + 7AAD, CD33 FITC + CD34 PE + 7AAD, and CD34 FITC + CD133 PE + 7AAD (all antibodies from BD Biosciences, except CD133 PE, which was from Miltenyi Biotec). PE- and FITC-labeled anti-mouse isotype immunoglobulin G stains were used as compensation controls. Antibody-stained cells were analyzed in an FACS Canto II (BD Biosciences) flow cytometer, using FACSDiva™ software. For determining the mean and standard error values, data from three experiments, with triplicate samples in each experiment, were used. Fold expansion of CD34+ and other lineage-specific cells was calculated by comparing the proportion of cells after 7 days of culture with the initial seeding of 1 × 104 cells.
Colony-forming unit (CFU) assay was carried out for all groups using Methocult media H4434 (Stem Cell Technologies) to assess the functionality of the expanded cells. The cells, resuspended in Methocult, were seeded in triplicates onto a 12-well plate at 1,000 cells/mL and cultured at 37°C, 5% CO2, and 20% O2 for 14 days, according to the manufacturer's instructions. The colonies were then manually counted to identify the burst-forming unit-erythroid, colony-forming unit-granulocyte, macrophage, and colony-forming unit-granulocyte, erythrocyte, macrophage, megakaryocyte.
Measurement of pO2
Dissolved oxygen concentrations were measured using OxyLite Pro (Oxford Optronix). A Bare Fiber fluorescent-containing oxygen sensor (Oxford Optronix) was placed as close as possible to the cells in a well of each experimental group (Fig. 1) to measure the dissolved oxygen concentration that the cells were exposed to at specific time points over a 7-day culture. The oxygen sensor was permanently attached to the tissue culture well for the duration of the culture to enable real-time pO2 to be measured.

Schematic diagram of the experimental setup showing placement of the oxygen sensor as close to the bottom of the culture well as possible to enable measurements of the oxygen concentration experienced by the cells.
The Oxylite Pro monitoring system was set on a 30 s update mode and eight measurements of pO2 were obtained over 5 min at each time interval. Dissolved oxygen was measured in mmHg and converted to percentage by dividing the pO2 measured by the atmospheric pressure and multiplying by 100.
NSG mouse repopulation assay
NSG mice were irradiated 6–8 h before transplantation with 325 cGy. The progeny of 5,000 CD34+ cells, post 7-day expansion, were injected into the tail vein of 8–12-week-old female and male mice (eight per group). The mice were killed 10–12 weeks after injection and the femurs and spleen were collected. Bones were flushed with 1–2 mL PBS/2% fetal bovine serum to remove bone marrow and the spleen was homogenized and filtered to obtain a single-cell suspension. Flow cytometric analysis was performed to identify mouse CD45 and human CD45, CD33, and CD19 (all antibodies from BD Biosciences)-positive cells after red blood cell lysis.
Statistical analysis
For flow cytometry analysis, 7,000–10,000 events were collected and analyzed. Selected cell populations were shown as a percentage among the total gated live cells based on the comparison with background staining shown when isotype controls of corresponding antibodies were used. Data of cell number, viability, cell surface markers, colony count, and mouse repopulation experiments were collected from three independent repeats (n = 3), and all measurements were performed in at least triplicate to avoid sampling error. Results were reported as mean ± standard error if not specified. Statistical relevance of the mean change in cell percentage and colony numbers during each experiment was determined through one-way analysis of variance (ANOVA), using GraphPad Prism 5 software. Two-way ANOVA with 95% confidence was performed to compare the effect of oxygen, movement (static vs. dynamic), and the combined effect of the two parameters on HSPC expansion. Significance level was set as (a P ≤ 0.05; b P ≤ 0.01; c P ≤ 0.001).
Results
pO2 in cultures under static and dynamic conditions
Dissolved oxygen was measured in static and dynamic HSPC cultures over 7 days at 5% ambient O2 (Fig. 2A) and 20% O2 (Fig. 2B). The pO2 equilibrated with the ambient oxygen levels between 4 and 19 h after initiating the culture, and levels remained stable until around 72 h. Static cultures in 5% atmospheric oxygen exhibited a gradual decrease in pO2 adjacent to the cells after 72 h and, by day 7, a pO2 of <1% was recorded. In the 5% dynamic culture, pO2 was maintained at ∼4% adjacent to the cells by day 7, even though the total cell number was 70% higher than in the 5% O2 static culture.

Change in percentage of diffused oxygen provided to the cells in static (S) and dynamic (D) conditions for a culture period of 7 days in hypoxic
Cultures grown in atmospheric O2 also showed a decrease in pO2 after the third day of culture, although the dynamic cultures showed a greater fall (to 14%) by day 7 compared with static cultures (to 16%). There was no statistically significant difference in cell numbers between dynamic and static cultures grown in 20% O2 (Fig. 4A).
Effect of pO2 and dynamic culture on HSPC expansion
In vitro analysis
The expansion of HSPCs was assessed by phenotypic analysis (Figs. 3 and 4) and clonogenic assay (Fig. 5) after a 7-day expansion culture. Fluorescence-activated cell sorting (FACS) staining was performed in several combinations to mark different lineage-specific cells. CD34PE+ CD45FITC+ cells mark multipotent and committed progenitor cells and CD34PE+ CD38 FITC− mark primitive HSPCs. CD133PE+ CD34FITC+ include HSPCs, multipotential progenitors, and some early committed progenitor cells, whereas CD33PE+ CD34FITC+ represent committed myeloid lineage stem cells.

Dot-plot analysis of HSPCs with lineage-specific surface markers (comparison of representative pre- and postculture cells).

Fold expansion of the HSPC subpopulation based on phenotypic analysis: the effect of oxygen and movement on the fold expansion of target populations was determined by manual counts and detection of cell surface markers by flow cytometry. All analyses were gated for viable cells only 7-aminoactinomycin D (7-AAD). Fold expansion of the viable cells were analyzed for total cells
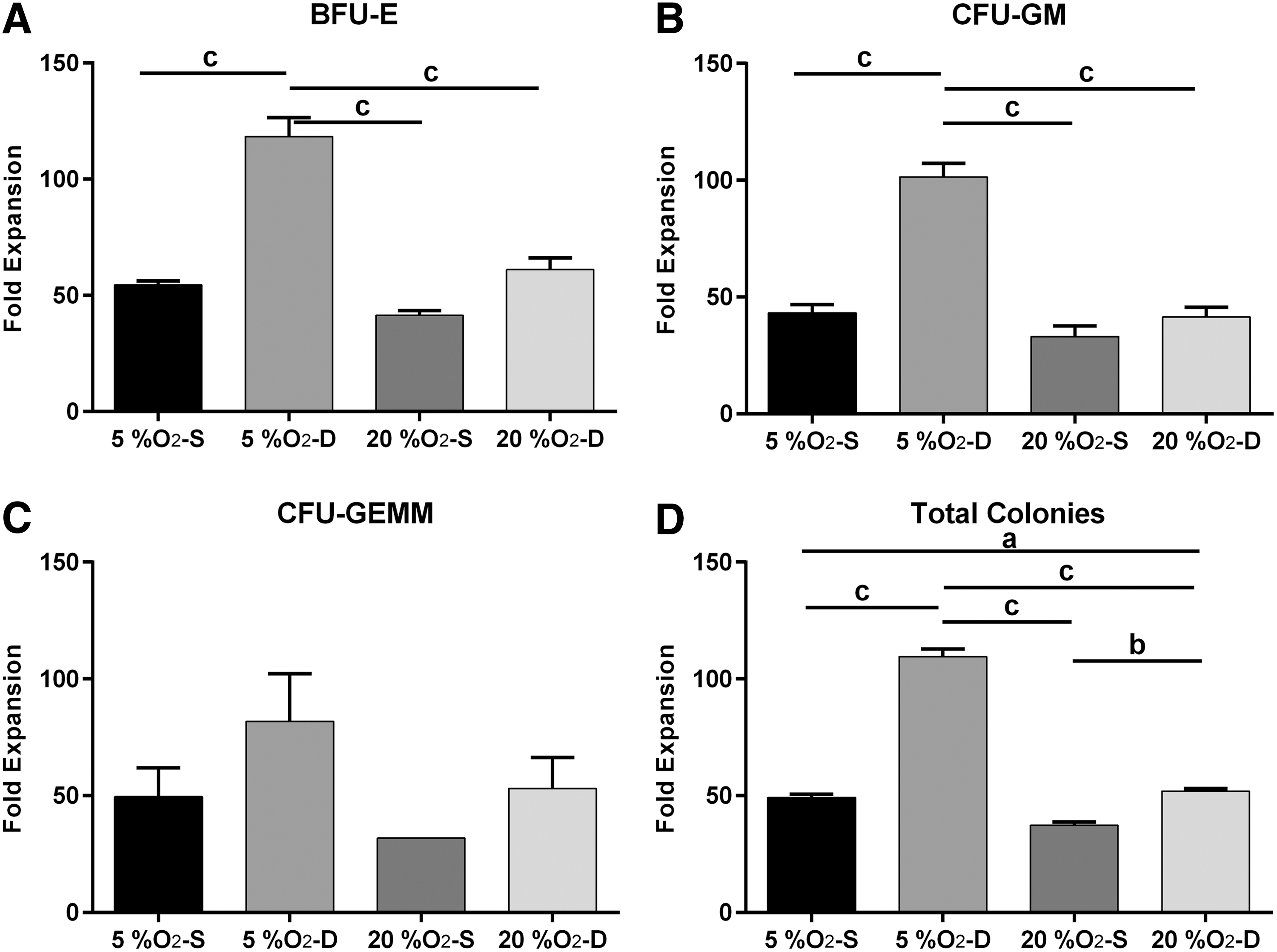
Fold expansion of the HSPC subpopulation based on CFU assay: the effect of oxygen and movement on the fold expansion of target populations was determined by manual counts and colony counts. Functional assessment of multipotent cells was performed with CFU assays for
As shown in Fig. 4, cells cultured at 5% O2 in a dynamic environment demonstrated the highest CD34 expansion, with almost 30-fold increase by FACS analysis and 120-fold by CFU assay. In general, cells cultured in 5% O2 showed significantly greater expansion than those cultured in 20% O2 (P < 0.01). Within each of the oxygen concentration group, cells cultured dynamically expanded more than those cultured statically (P < 0.001). Furthermore, an interaction between oxygen concentration and static versus dynamic culture was observed in that cells cultured dynamically in 20% O2 exhibited comparable or better expansion than the 5% O2 static group (P < 0.05). Similar results were obtained with the clonogenic assay whereby there were more colonies when cultured at 5% O2 (P < 0.001) in a dynamic environment (P < 0.001) (Fig. 5). Additionally, there was a significant interaction between oxygen and movement on HSPC expansion (P < 0.001).
In vivo analysis
To examine the long-term repopulating ability of UCB-HSPCs, progeny of 5,000 expanded cells were transplanted into NSG mice. After 10–12 weeks, there were significantly more human CD45+ cells detected in mice that were transplanted with HSPCs expanded at 5% O2 dynamically than statically, with ∼11% human CD45+ cells found in the bone marrow in the dynamic culture group (P < 0.001) (Fig. 6). The engraftment in the bone marrow for this group was fourfold or greater compared with 20% O2 static culture. Engraftment of human CD45+ cells in the spleen demonstrated similar results. Mice transplanted with HSPCs expanded at 20% O2 dynamically exhibited comparable levels of engraftment in the bone marrow with those expanded at 5% O2 statistically. The expression of lineage markers on the reconstituted human CD45+ cells was also analyzed (Fig. 6). All the recipients transplanted with the expanded cells possessed CD33+ myeloid cells and CD19+ lymphoid cells, confirming complete repopulation. While engraftment was significantly affected by both oxygen concentration and static versus dynamic culture, no significant effect was observed between the interactions of these two parameters.
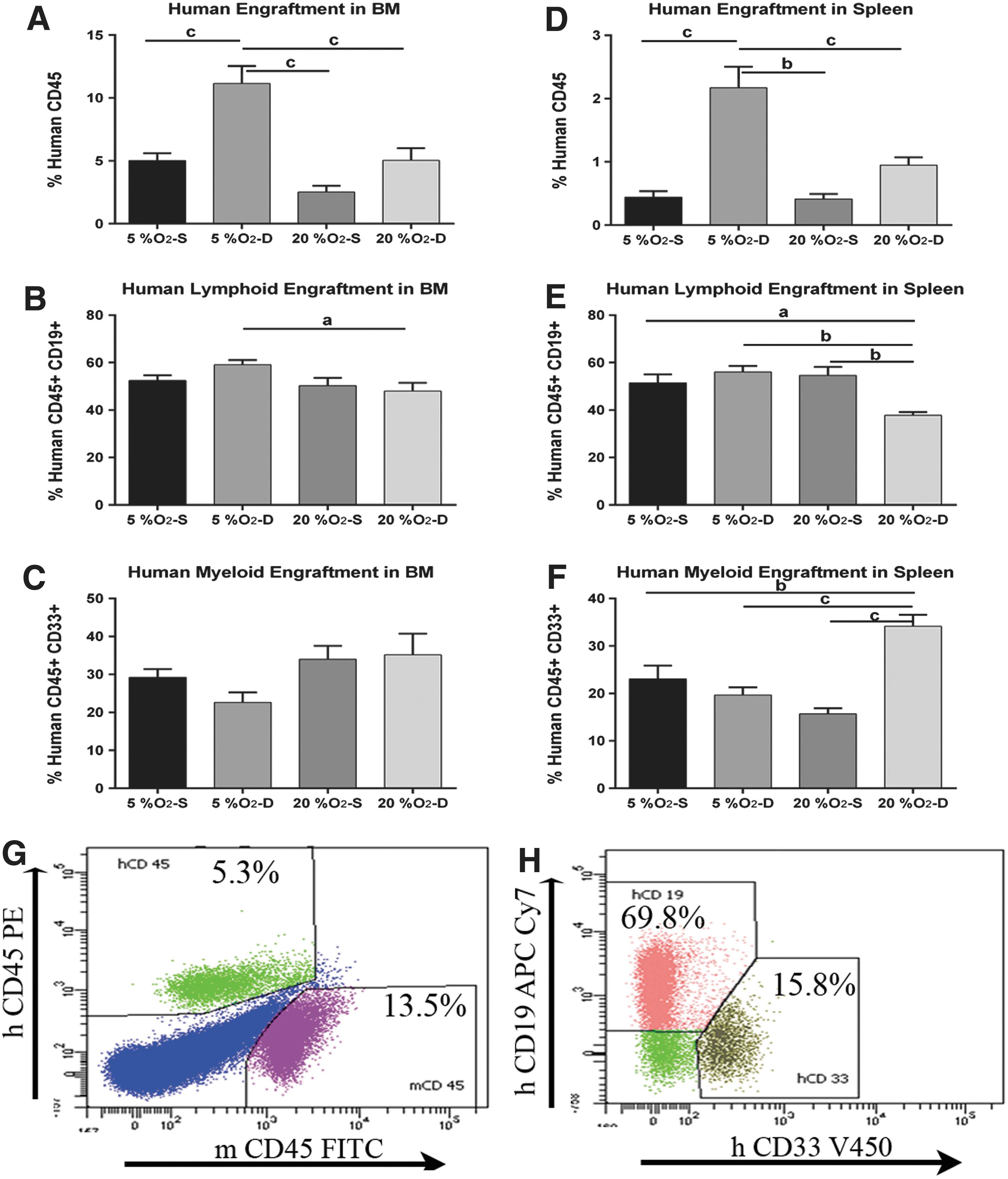
NOD/SCID engraftment assay. The effect of oxygen and movement on the engraftment of human CD34+ cells into NOD-SCID mice was determined by detection of human cell surface markers on mouse bone marrow and spleen-derived cells by flow cytometry. Percentage of human cell engraftment in mouse bone marrow
Discussion
The small number of HSPCs present in a UCB unit limits the use of this source of stem cells for transplantation, particularly in adults. The strong correlation between cell dose and outcome after cord blood transplantation has led to a progressively increased demand for large cord blood units from public banks [27 –30]. Expansion is seen as critical to overcome this problem by enabling the use of the hundreds of thousands of smaller units currently stored in public cord blood banks around the world for making cord blood transplantation safer and less costly.
The importance of oxygen levels in the proliferation and differentiation of stem cell populations, particularly HSPCs, is well established [31 –33]. We have previously demonstrated the importance of oxygen in determining the characteristics of HSPC-supportive stromal cells [34], and a number of groups have identified the importance of oxygen in the efficiency of expansion of HSPCs [9,35]. Low oxygen has been reported to promote quiescence and regulate their differentiation, thereby maintaining stem cell phenotype. Even extremely low oxygen concentrations are permissive for slow self-renewing divisions of hematopoietic stem cells, while higher (atmospheric) oxygen promotes terminal differentiation [36 –38].
As part of the validation of a clinical-grade dynamic HSPC expansion system, we have sought to understand the in vitro environment and, in particular, oxygenation of cells grown in our system compared with that in standard (static) culture conditions. In particular, we have examined the relationship between atmospheric oxygen, cell number during expansion, static/dynamic culture conditions, and the effective pO2 experienced by cells in a culture system.
Oxygen is relatively poorly soluble in water, with a slow diffusion constant. Balin et al. [20] in the mid 1970s showed a relationship between oxygen levels, dynamic (shaken) cultures, and proliferation rate. Metzen et al. [21] demonstrated the impact of cell number on effective oxygenation in monolayer cultures and derived an equation for the relationship between pO2 in the incubator [pO2(gas)], the tissue-specific rate of oxygen utilization (UO2, nmol O2 × min−1 × mg cellular protein−1), the protein content of the cell layer (tProt), the diffusion constant of O2 (D = 3.3 × 10−5 cm2 × s−1), the oxygen solubility constant (α, 0.94 μmol O2 × mL−1 × atm−1), the height of media in the culture (h, cm), and the area of the culture vessel (A, cm2):
Metzen's equation assumes that the tissue-specific rate of O2 consumption, U O2, is constant, whereas Balin's study demonstrated that consumption is inversely related to available oxygen. Nevertheless, these studies form the basis for understanding the relationship between cell number (as represented by tProt), oxygen consumption, diffusion, and available oxygen. Metzen showed that the measured pericellular oxygen was highly dependent on the cell type in culture, confirming that the tissue-specific rate of oxygen consumption can vary considerably. The application of these calculations to standard tissue culture is also complicated by the fact that the number of cells (and hence tProt) varies through the course of the culture. Pettersen et al. [39] measured pericellular oxygen in subconfluent, confluent, and superconfluent cultures of the breast cancer cell line, T47D, and showed that the effective oxygen being experienced by the cells differed substantially from the gaseous oxygen as the cultures progressed.
We have measured the pericellular oxygen concentration in both static and dynamic HSPC cultures over 7 days under conditions that result in 30–60-fold expansion of cell numbers. The probe for the measurements was placed immediately above the plastic surface (1–2 mm) and so represented the conditions experienced by cells adhering to the tissue culture plastic. HSPCs are semiadherent cells and will settle to the bottom of the plate under static conditions.
Our results demonstrate that the doubling time for HSPCs varied from 32.5 h at 5% O2 under dynamic conditions to 39.5 h at 20% O2 under static conditions. This difference resulted in a twofold difference in total cell numbers and an almost fourfold difference in CD34+ cells over the 7 days of culture used in these experiments. As shown in Supplementary Fig. S1, no significant change in cell viability was observed between the different conditions. This result suggests that the combination of dynamic culture and lower O2 levels may promote maintenance of a more primitive phenotype, which was supported by a similar fourfold difference in human CD45+ engraftment in NSG mice. While total engraftment was significantly affected by oxygen concentration and movement, the ratio of myeloid versus lymphoid engraftment did not show any significant changes. The percentage of hCD45+CD19+ cells was 47%–60% compared with 22%–35% of hCD45+CD33+ cells in the mouse bone marrow and confirms the findings of Noort et al. [40], where they demonstrated that after transfer of CD34+ cells from UCB, the main cell lineage recovering in the NOD/SCID mice is of B cells.
This difference in expansion may be directly related to changes in effective O2 experienced by the HSPCs during the culture. As shown in Fig. 2, cells grown in 5% atmospheric oxygen under static conditions experienced extremely low (<1%) oxygen by day 7 of the culture. Such hypoxic conditions may promote relative quiescence of primitive HSCs and so may inhibit expansion. Our results demonstrate that cells exposed to static cultures at 5% oxygen exhibited a pO2 under 1% by day 7, supporting the concept those actual oxygen measurements in clinical cultures should be part of routine validation studies under GMP.
Jing et al. [41] have reported that in dynamic culture, an agitation speed of 30 rpm was better than that of 45, 60, and 80 rpm for the expansion of HSPCs. However, we found even lesser cell death at 20 rpm compared with 30 rpm. According to Cabrita et al. [42], cells in static culture accumulate at the bottom of culture plates, generating oxygen and cytokine concentration gradients, which can negatively affect both cell proliferation and phenotype. However, in a dynamic culture system, the fluid shear stress can directly act as mechanical signal acting on cells. Under agitation, an optimum degree of hypoxia may be maintained as the culture grows. This may operate alongside removal of negative regulators of growth from the cells and their surroundings, as well as mechanical stimulation. Yang et al. [43] demonstrated that static cultures favored the expansion of HSPCs and stirred cultures were more effective in preserving functional HSCs so that static culture and stirred culture may be combined to guarantee both the quantity and quality of HSCs, providing helpful clues for further developing novel culture systems. Hosseinizand et al. [44], who used a different model for dynamic cultures, have shown that the agitation increases expansion of cord blood hematopoietic cells and promotes their differentiation into myeloid lineage.
In summary, although some studies have compared the effect of static versus dynamic cultures for HSPC expansion, while others have investigated the effect of O2 levels on HSC expansion, the current study compared the combined effect of oxygen and static versus dynamic culture on UCB-HSPC ex vivo expansion. In this UCB expansion system, we have shown that there is a marked discrepancy between the oxygen concentration in the atmosphere of the incubator and the concentration to which the cells are directly exposed. Our results also indicate that HSPC expansion ex vivo and engraftment in vivo was significantly enhanced at 5% oxygen and dynamic culture system, suggesting that this culture condition can further improve transplantation outcomes.
Footnotes
Acknowledgments
This study was supported by funding from Cytomatrix Ltd. to G.J. and M.A.K. The authors are grateful to Miller-Jenkin group members for their assistance in mouse experiments. This work is supported by the Victorian Government's Operational Infrastructure Support Program.
Author Disclosure Statement
M.A.K. is Chief Scientific Officer of Cytomatrix Ltd., a for-profit biotechnology company. Other authors declare no conflicts of interest.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.