
Abstract
Endothelial progenitor cells/endothelial cells (EPCs/ECs) have great potential to treat pathological conditions such as cardiac infarction, muscle ischemia, and bone fractures, but isolation of EPC/ECs from existing cell sources is challenging due to their low EC frequency. We have isolated endothelial progenitor (EP)-like cells from rat oral mucosa and characterized their yield, immunophenotype, growth, and in vivo angiogenic potential. The frequency of EP-like cells derived from oral mucosa is thousands of folds higher than EPCs derived from donor-match bone marrow samples. EP-like cells from oral mucosa were positive for EC markers CD31, VE-Cadherin, and VEGFR2. Oral mucosa-derived EP-like cells displayed robust uptake of acetylated low-density lipoprotein and formed stable capillary networks in Matrigel. Subcutaneously implanted oral mucosa-derived EP-like cells anastomosed with host blood vessels, implicating their ability to elicit angiogenesis. Similar to endothelial colony-forming cells, EP-like cells from oral mucosa have a significantly higher proliferative rate than human umbilical vein endothelial cells. These findings identify a putative EPC source that is easily accessible in the oral cavity, potentially from discarded tissue specimens, and yet with robust yield and potency for angiogenesis in tissue and organ regeneration.
Introduction
C
Asahara et al. [1] isolated circulating endothelial progenitor cells (EPCs) from adult peripheral blood (PB), a discovery which questioned whether vasculogenesis was restricted only to the embryonic development period [1]. Since then, their utility as a predictive biomarker of cardiovascular disease and as cell source for vascular regeneration has been explored [2 –5]. A subfraction, endothelial colony-forming cells (ECFCs or late outgrowth ECs) have been isolated from human peripheral as well as umbilical cord blood. In contrast to other subfractions, ECFCs display strong proliferative potential and also new blood vessel formation in vivo in immunodeficient mice [6 –8].
Recent research has shown significant interest with respect to the therapeutic potential of EPCs in re-/neovascularization and regeneration in multiple principles, including cardiac infarct repair [2,3,9] and bone regeneration [10 –15]. Accordingly, it is imperative to have a reliable cell source for the isolation of EPCs for potential treatments that promote vascularization repair and/or regeneration. Although bone marrow (BM) has long been regarded as the primary source of circulating EPCs [16 –19], a recent study using the CD34+CD133−CD146+ surface marker combination to enrich endothelial precursors from PB (normal or mobilized PB), cord blood, and BM has led to a different conclusion [8]. The authors in this study have demonstrated that endothelial precursors resided within the CD34+CD133−CD146+ mononuclear cell fraction, which both BM and mobilized PB only possessed at a negligible level [8]. Regardless of where the circulating EPCs originated, frequency of EPCs in PB or BM is known to be extremely low [6,8,17,20 –23]. Other studies using rat lung tissues or human renal arteries have shown success in isolating functional ECFCs, but it will be difficult to translate the same approaches to clinical settings [24,25].
The orofacial complex is among the most vascularized tissues in the body [26,27]. However, whether endothelial stem/progenitor cells reside within orofacial tissues has not been investigated. In contrast to the extensive study on stem/progenitor cells from the mesenchymal compartment of dental pulp and periodontal ligament [28], little is known about the presence or functions of endothelial stem/progenitor cell populations in oral mucosa. Recent reports have demonstrated that the gingival mucosa comprised a population of multipotent stem cells that express characteristic mesenchymal stromal cell (MSC) markers and can differentiate into osteogenic, chondroblastic, and adipogenic lineages [29 –34]. We hypothesize that oral mucosa carries EPCs at a higher level than lung, PB, or BM and possesses angiogenic potential in vascular regeneration. The overall goal of this study is to identify a clinically relevant cell source for the isolation of postnatal ECs. Oral mucosa samples are easily obtainable from individuals who undergo orthodontic or gum replacement treatment, and the procedure is considered significantly less invasive than lung or BM biopsy, which will make oral mucosa a better clinical cell source for the isolation of EPCs/ECs.
Materials and Methods
Sample collections and cell culture
Approximately 5 × 6 × 2 mm pieces of oral mucosa tissue from the buccal area, including both epithelial and lamina propria layers, were collected from 2- to 3-month-old female Sprague-Dawley rats (Harlan Laboratories, South Easton, MA) or GFP transgenic rats (Dr. DongMing Sun, Rutgers University, New Brunswick, NJ). All samples were rinsed extensively with cold phosphate-buffered saline (PBS) supplemented with 2 × penicillin/streptomycin (pen/strep, Cat. No. 15140-122; Invitrogen, Grand Island, NY). After rinsing, samples were cut into small pieces and placed in 5 mL StemPro Accutase Cell Dissociation buffer (Cat. No. A11105-01; Invitrogen) at 37°C with shaking for an hour, 2 h, or at 4°C overnight. Supernatant from each sample was collected and centrifuged for 7 min at 1,600 rpm. Cell pellets were resuspended in 10 mL endothelial growth medium (Cat. No. CC-3162; Lonza, Allendale, NJ), which comprised basal medium, human epidermal growth factor (hEGF), gentamicin/amphotericin-B, VEGF, hFGF-B, R3-IGF-1, ascorbic acid, heparin, and 20% fetal bovine serum (FBS, Cat. No. 10439-024; Invitrogen). Cells were seeded on 10 cm tissue culture dishes precoated with 0.1% collagen-I (Cat. No. 354249; BD Biosciences, San Jose, CA) and allowed to culture in the same medium for 2–3 days before replacing with fresh medium. Attached cells were continued to culture in endothelial growth medium until cobblestone-appearing cell colonies become prominent for isolation. Cloning cylinders lined with vacuum grease were placed at the premarked areas. Fifty microliters of trypsin/EDTA was added into each cylinder tube, and the cell culture dish was incubated at 37°C for 5 min before the dissociated cells in the trypsin/EDTA solution were collected and plated onto fresh 6- or 12-well culturing plates. Culture medium was changed every other day until ready for further analysis. Any samples that were not processed immediately were stored at 4°C in PBS.
Rat primary aortic endothelial cells (RAECs, Cat. No. R2196; Cell Biologics, Chicago, IL) were cultured in tissue plates precoated with gelatin-based coating solution (Cat. No. 6950) in EC medium (Cat. No. M1266) as instructed by the supplier. The culture medium included basal medium, EGF,
Human umbilical vein endothelial cells (HUVECs, Cat. No. C2519A; Lonza) were cultured in the same endothelial growth medium as the oral mucosa-derived EPC-like cells (Cat. No. CC-3162; Lonza).
Method used to isolate donor-match BM stem cells isolated from rat tibiae was as previously described [35] but without fractionation. Human dental pulp (hDP) tissues were obtained from the mandibular first premolar of a 34-year-old male patient, and isolation of MSCs from these tissues was using conditions established from previous studies [36,37]. The hDP-derived MSCs (hDP-MSCs) were cultured and expanded in low-glucose Dulbecco's modified Eagle's medium (Cat. No. 11885-092; Invitrogen) supplemented with 10% FBS. Isolation and short-term culturing conditions of rat preameloblasts were described by Jiang et al. [38].
All animal care are under the guidelines of the Columbia University Institutional Animal Care and Use Committee (IACUC) and the use of human dental pulp tissues was approved by the Columbia University IRB committee.
Immunofluorescence staining
Cobblestone-appearing cells from individual colonies were fixed with 1% formaldehyde for 10 min, rinsed with PBS, and fixed with methanol at −20°C for 5 min. Cells were rinsed in PBS before permeabilization with 0.1% Triton X-100 for 3 min and incubated with 1° antibodies in incubation buffer [5% bovine serum albumin (BSA), 5% FBS, 0.1% saponin, and 0.02% NaN3 in PBS] at 4°C overnight. Cells were rinsed with PBS and incubated in 2° antibodies in incubation buffer for an hour in the dark before mounting for microscopic analysis.
VE-Cadherin (Cat. No. ab166715; Abcam, Cambridge, MA), CD31 (PECAM-1, Cat. No. sc-1506; Santa Cruz Biotechnology, Inc., Dallas, TX), and keratin-14 (CK14, Cat. No. sc-17104; Santa Cruz Biotechnology) antibodies were used at 5, 10, and 12.5 μg/mL, respectively. Goat anti-rabbit IgG conjugated with Alexa 488 (1:1,000; Invitrogen), goat anti-mouse IgG conjugated with Alexa 555 (1:500; Invitrogen), and donkey anti-goat IgG conjugated with Alexa 488 (1:1,000; Invitrogen) were used as secondary antibodies.
DiI-AcLDL uptake assay
Cobblestone-appearing cells from individual colonies were incubated in the presence of DiI-labeled acetylated low-density lipoprotein (AcLDL, Cat. No. L3484; Invitrogen) for 5 h. Cells were rinsed with PBS and recultured in fresh culture medium before microscopic analysis.
Flow cytometry
Cobblestone-appearing cells from individual colonies were collected for flow cytometric analysis at passage 2, 3, or 4. Cell pellets were resuspended in PBS supplemented with 0.5% BSA and incubated with mouse anti-rat Fc blocker, CD32 (Cat. No. 550271; BD Biosciences, San Jose, CA), for 10 min. Preconjugated antibodies that targeted CD31 (Cat. No. FAB3628G; R&D Systems, Minneapolis, MN or Cat. No. NB100-63704; Novus Biologicals, Littleton, CO), CD34 (Cat. No. sc-7324 PE; Santa Cruz Biotechnology), or VEGFR2 (Cat. No. NB100-2382G; Novus Biologicals) were added to separate cell preparations for 30 min on ice. Cells were washed with 0.5% BSA/PBS twice and fixed with 1% fresh paraformaldehyde before flow cytometric analysis (BD Calibur). Twenty thousand events were measured during the readout of each sample. All data were analyzed by FlowJo_V10 software. Significance and P values were determined by Student's t-test analysis.
In vitro tube formation assay
To prepare tube formation assay, 0.5 mL of Matrigel (Cat. No. 354230; BD Biosciences) was added to each well in a 24-well plate and incubated at 37°C for 30 min to allow the gel to solidify. 2 × 105 of rat oral mucosa (rOM)-derived EPC-like cells, GFP+ rat oral mucosa (GrOM)-derived EPC-like cells, HUVECs, RAECs, or hDP-derived MSCs were overlaid on Matrigel with 200 μL Lonza ECM-2 supplemented with 2% serum (L-EDM). Tube formation efficiency of RAECs was also tested with Cell Biologics ECM supplemented with 2% serum (C-EGM).
In vivo subcutaneous implantation experiments
1 × 106 GrOM-derived EPC-like cells (GFP expressing) were resuspended in 50 μL of culture medium and infused directly into a medical graded collagen sponge (Cat. No. 1690ZZ; Integra Lifesciences Corporation, Plainsboro Township, NJ) before subcutaneous implantation. Eight-week-old athymic mice (Harlan) were used as a host. Before cell seeding, each collagen sponge was cut roughly into two equal-sized pieces and presoaked in fresh endothelial culture medium for a few minutes. Excess medium in the collagen sponge was removed by dry padding with a sterile cotton gauze before cell seeding (Cat. No. 22-037-986; Fisher Scientific). The same number of HUVEC-seeded collagen sponges was implanted on the contralateral side of the same animal as control. Scaffolds were collected at 4- or 8-week postimplantation.
All animal care and procedures were performed in compliance with Columbia University Medical Center Institute of Comparative Medicine (ICM) and Institutional Animal Care and Use Committee (IACUC) guidelines.
Growth curve kinetics
2 × 104 HUVECs, oral mucosa-derived endothelial progenitor (EP)-like cells, or RAECs were seeded in 12-, 6-well, 60-mm, and 10-cm culture plates at day 0 in the respective culture medium. Medium was replaced every other day, and overconfluent cell preparations were not used for counting. Cells from triplicate wells/dishes of HUVECs, oral mucosa-derived EP-like cells, or RAECs were collected each day and counted by a hemocytometer. Results are calculated as the mean data ± standard error of the mean from three separate experiments.
Microscopy
Photomicrographs were taken by digital cameras (Leica DFC 365 FX or Leica DFC 310 FX, Buffalo Grove, IL) connected to an inverted fluorescence microscope (Leica DMI4000 B) or upright fluorescence microscope (Leica DM5000 B). Images were processed by LAS V4.2 software. Confocal microscopy was performed using the Zeiss LSM 710 confocal scanning microscopy, and digital images were analyzed using Zen 2012 lite software (Carl Zeiss, Inc., Thornwood, NY).
Results
Robust number of EPC-like colonies can be isolated from rat buccal mucosa tissues
Figure 1A depicts the protocol conditions for isolation of EPC-like colonies from oral/buccal mucosa (rOM). rOM samples were collected from 2- to 3-month-old Sprague-Dawley rats (n > 10) comprised with both epithelial and lamina propria layers to identify optimal conditions for isolation of progenitor cells. We collected dissociated cells after enzymatic digestion at different time points and seeded the cells on collagen I (ColI) precoated 10-cm culture dishes for prolonged culturing. Small clusters of cells (∼5–20 cells) could be seen as early as 5 days after plating, but majority of the EPC-like colonies would appear around 8–10 days of culturing. High homogenous EPC-like colonies were selected for expansion and analysis. Among the three conditions we have tested (1 h, 2 h, and overnight), we found the 2-h enzyme incubation time produced the most robust number of EPC-like colonies (Fig. 1A, A2).
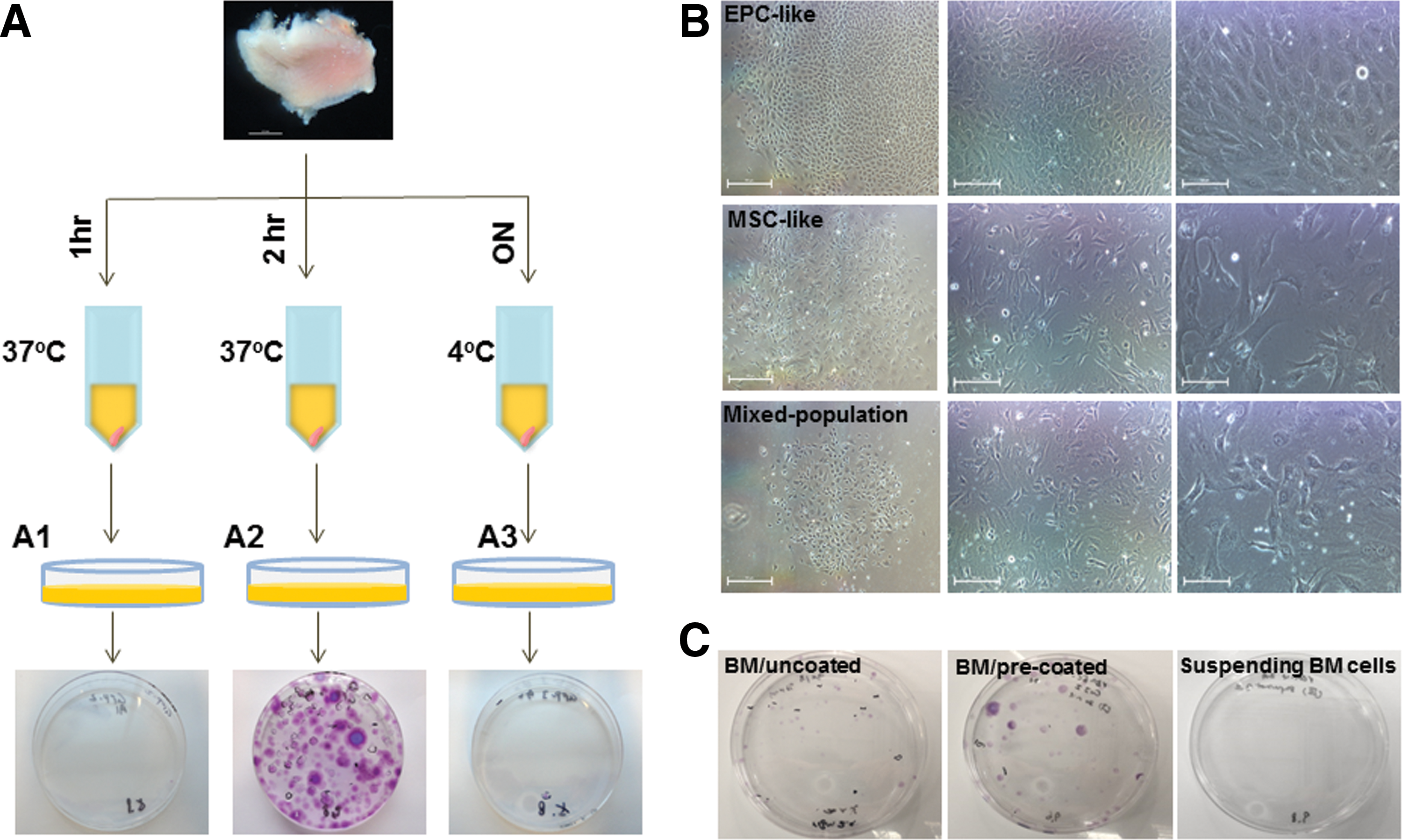
Protocol diagram depicts conditions used in rOM-derived EPC-like colony isolation.
After culturing for 12–14 days, cells isolated from rOM samples (n = 3) and rat bone marrow (rBM) samples (n = 3) were fixed and stained with crystal violet. Numbers of colony-forming unit (CFU) and numbers of EPC-like colony from each sample are recorded and summarized in Table 1. Representative photomicrographs of rOM-derived EPC-like, MSC-like, and mixed-population colonies are demonstrated in Fig. 1B. Only highly homogenous cobblestone-appearing cell colonies were scored as EPC-like colonies (Fig. 1B, top panel). MSC-like colonies (Fig. 1B, middle panel) or mixed-population colonies (Fig. 1B, bottom panel) were only scored as CFU. Examples of crystal violet-stained, rBM cell-seeded culture plates are shown in Fig. 1C, and live cell image of rBM-derived colony is shown in Supplementary Fig. S1 (Supplementary Data are available online at
CFU, colony-forming unit; ECFC, endothelial colony-forming cell; rBM, rat bone marrow; rOM, rat oral mucosa.
rOM-derived cobblestone-appearing cells possessed AcLDL uptake functionality. After establishing the condition for cell isolation, we proceeded to characterize the functionality and immunophenotype of the rOM-derived cobblestone-appearing colonies. HUVECs and RAECs were included in the study for comparison. Compact, circular cell colonies (one of the characteristics of ECFC colonies [6,7]) were picked around day 8–10 and cultured in endothelial growth medium containing hEGF, gentamicin/amphotericin-B, VEGF, hFGF-B, R3-IGF-1, ascorbic acid, heparin, and 20% fetal bovine serum (L-EGM) (Fig. 2A, left panel). L-EGM was also used to culture HUVECs (Fig. 2B, middle panel). RAECs were kept in either L-EGM (Fig. 2A, right panel) or C-EGM (basal medium with EGF,

Cell colonies isolated from rOM resemble HUVECs and expressed endothelial markers.
We then set up the low-density lipoprotein ingestion assay to assess the functionality of the rOM-derived cobblestone-appearing cells. After culturing in L-EGM supplemented with DiI-AcLDL for 5 h, uptake of the fluorescence-labeled lipid by the cells was documented by fluorescence microscopy (Fig. 2A, bottom panel). Our assay showed that all rOM-derived EP-like cells were capable of ingesting a high level of DiI-AcLDL intracellularly (Fig. 2A, bottom panel).
rOM-derived cobblestone-appearing cells expressed EC markers. To characterize the immunophenotype of the rOM-derived cobblestone-appearing cells, we performed immunofluorescence staining and flow cytometric analysis. Figure 2B shows that a high level of the EC marker, VE-Cadherin (CD144), was expressed in rOM-derived EP-like cells (Fig. 2B, top panel) as well as two previously established EC lines, RAECs (Fig. 2B, second panel) and HUVECs (Fig. 2B, third panel). Staining on rOM-derived EP-like cells in the absence of the primary antibody confirmed the specificity of the VE-Cadherin antibodies used in this study (Fig. 2B, bottom panel). Since rOM tissues have been known to contain a high frequency of epithelial cells, we also used an epithelial marker, keratin 14, to confirm that no epithelial cells were present in the EPC-like colonies (Fig. 2C).
Our flow cytometric analysis showed that a large population of the rOM-derived EP-like cells was VEGFR2+ (>80%), whereas ∼35% of the cells were CD31+ (Fig. 3A, B). Interestingly, majority of the RAECs expressed CD31 (>91%), and ∼20% of the cell population expressed VEGFR2 (Fig. 3A, B). RAECS also carry a small subpopulation that expressed CD34 (∼17%).
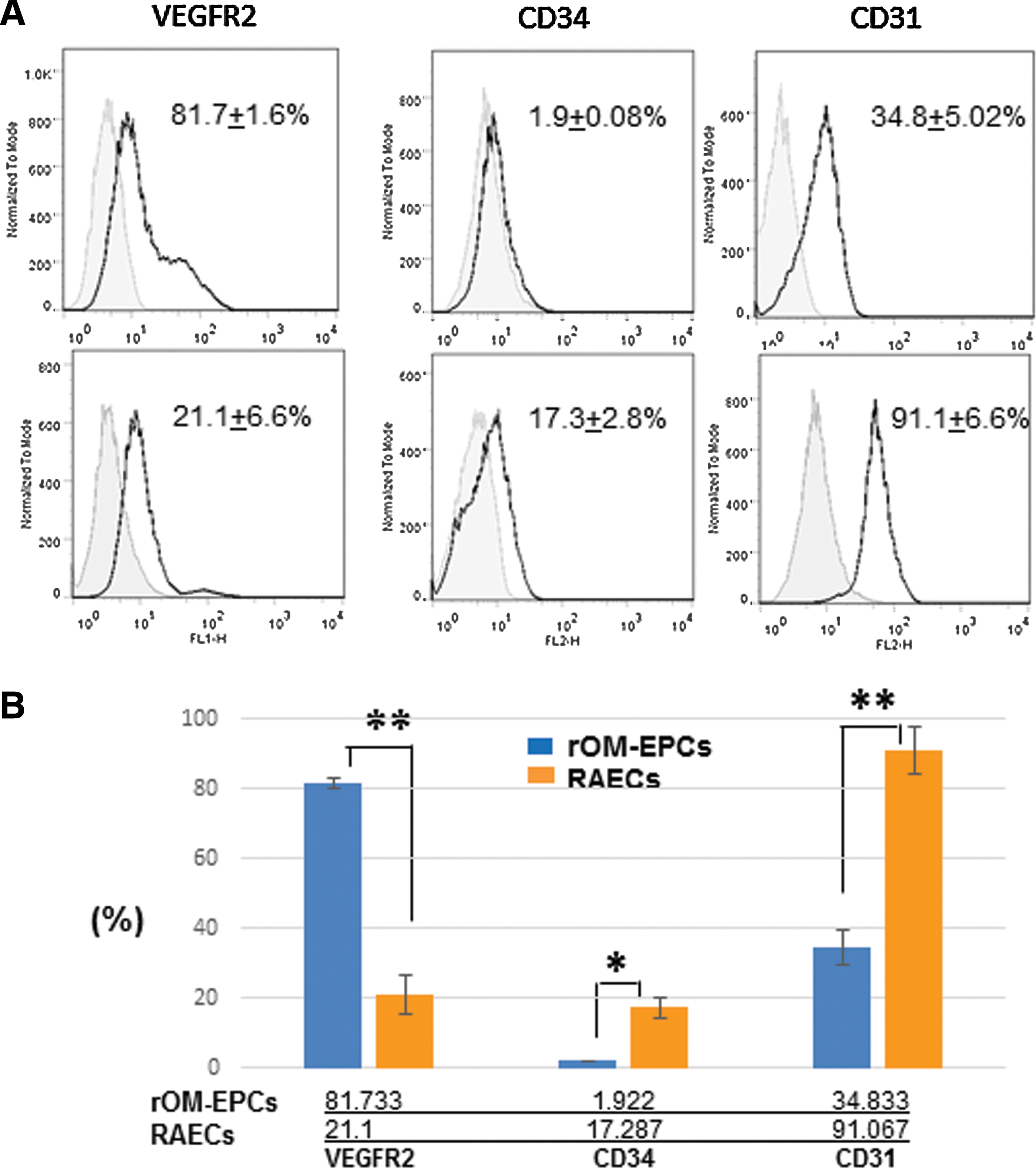
Flow cytometric analysis of rOM-derived EP-like cells and RAECs.
rOM-derived EP-like cells formed extensive and stable capillary networks on Matrigel. To further assess the endothelial functionality of the rOM-derived EP-like cells, we have investigated their tube formation potential on Matrigel. In this assay, we also included rOM-derived EP-like cells isolated from GFP transgenic rats [39] (GrOM-EPCs) (Fig. 4A), HUVECs, RAECs, and hDP-MSCs (Fig. 4A, B). Except for the RAEC preparation cultured in L-EDM, all cell types were successful in producing lattice structures within hours of being placed on Matrigel (Fig. 4). However, hDP-MSC-derived capillary network quickly diminished and retracted into a spheroid-like structure after 24 h (Fig. 4B, bottom panel), whereas networks derived from other cell types appeared stable at the same time point. On day 2, tube formation of HUVECs also disappeared and was replaced by spheroid-like structures (Fig. 4A, bottom panel). Although RAECs cultured in C-EGM also started to lose most of the lattice structure on day 2, small sections of tube formation remained intact (Fig. 4B, top panel). After 72 h, only rOM-EP-like cells and GrOM-EPC-like cells have maintained extensive network formation, and the networks remained stable until the end of the study (day 7) (Fig. 4A, bottom panel and Supplementary Fig. S2).
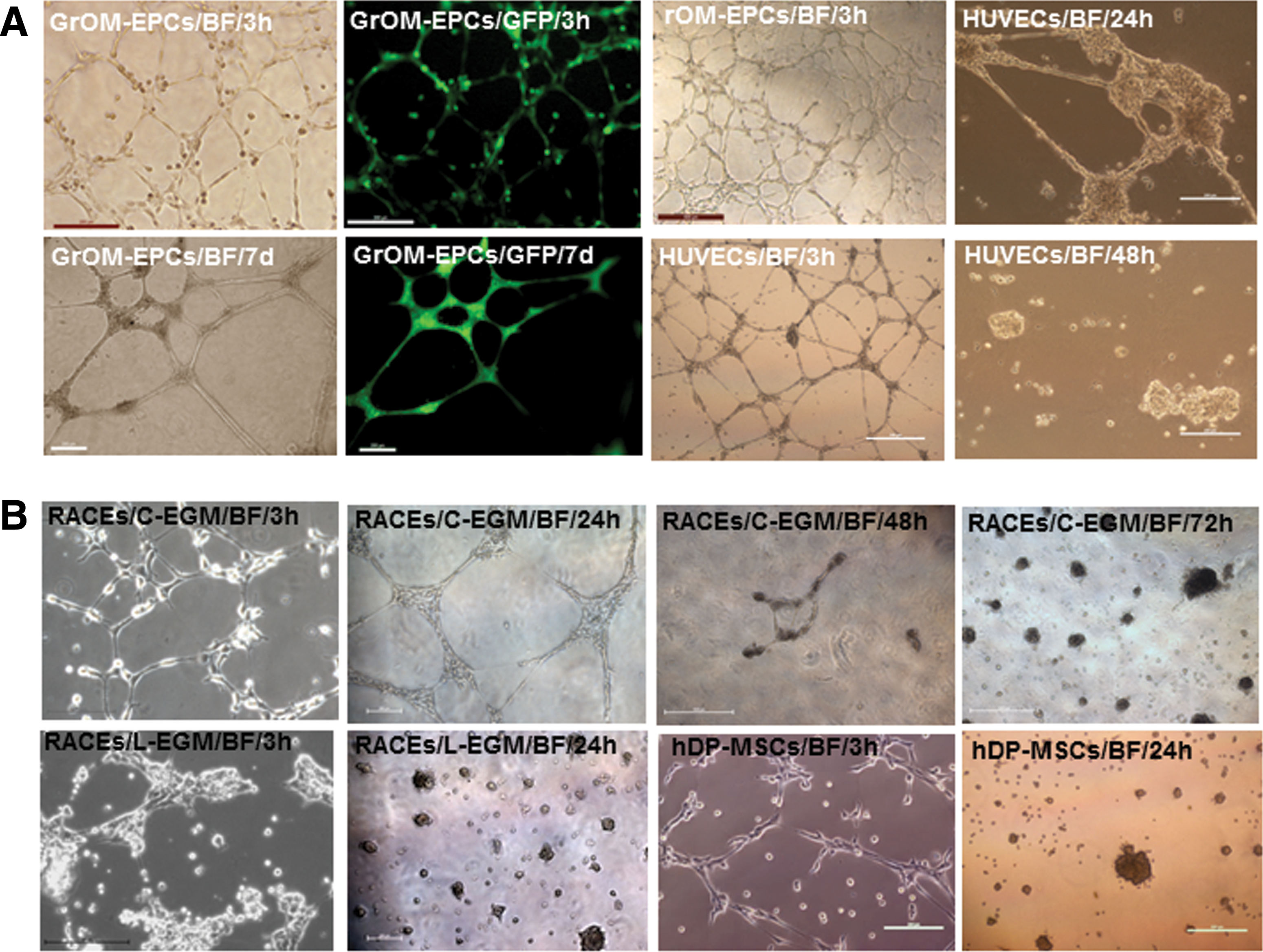
Matrigel lattice formation from rOM-derived EP-like cells, HUVECs, RAECs, and hDP-MSCs.
In vivo angiogenic potential of GrOM-derived EP-like cells. To determine whether rOM-derived EP-like cells were indeed true EPCs, we have implanted 1 million of GrOM-derived EP-like cells into an athymic host subcutaneously, using collagen sponges as carriers (Fig. 5A). Collagen sponge infused with the same number of HUVECs was placed at the contralateral side of the host animal as control (Fig. 5A). All scaffolds were collected at either 4- or 8-week postimplantation (Supplementary Fig. S3). Vascular networks were apparent within all samples. Macroscopic images showed that the GFP fluorescence signal was detected from scaffolds infused with GrOM-derived EP-like cells but not with HUVECs, thus eliminating the possibility of this being an artifact created by nonspecific autofluorescence (Fig. 5A, bottom panel). In GrOM-derived, EP-like cell-infused scaffolds, blood vessels containing GFP-positive cells were readily detected from both 4- and 8-week samples, indicating that not only were these cells angiogenic but the blood vessels they formed were also highly stable after engraftment (Fig. 5B, top and second panels). Immunofluorescence staining of whole mount (Fig. 5B, third panel) and sections (Fig. 5B, bottom panel) from GrOM-derived, EP-like cell-infused scaffolds confirmed strong expression of CD31 in the GFP-positive cells.
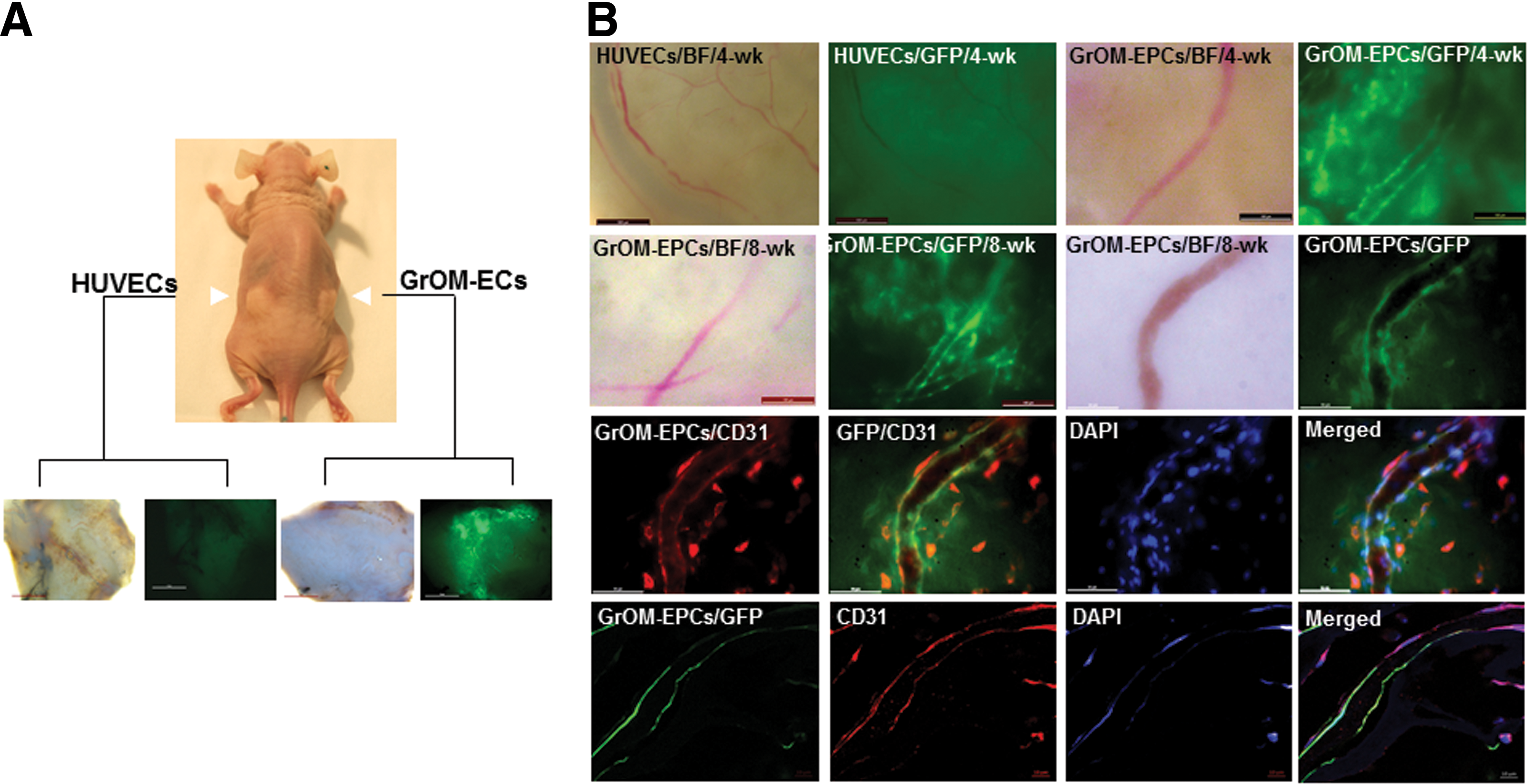
GrOM-derived EP-like cells incorporated into blood vessels in vivo.
rOM-derived EP-like cells are highly proliferative. One of the characteristics of ECFCs/endothelial late outgrowth cells (EOCs) is their high proliferative rate [7,8,40]. During cell expansion analysis, we did observe extraordinary high proliferation from the rOM-derived EP-like cells. To quantify their proliferation efficiency, we carried out a 5-day growth curve analysis on the cells, using RAECS and HUVECs for comparison (Fig. 6). Our results showed that the number of rOM-derived EP-like cells increased ∼200-fold (8.04 × 104 ± 2.05 × 104, 5.13 × 105 ± 5.18 × 104, 1.07 × 106 ± 1.99 × 105, 2.04 × 106 ± 2.95 × 105, and 3.84 × 106 ± 1.03 × 106), the number of RAECs increased ∼100-fold (6.75 × 104 ± 2.25 × 104, 2.76 × 105 ± 4.34 × 104, 7.96 × 105 ± 1.42 × 105, 1.3 × 106 ± 2.76 × 105, and 1.8 × 106 ± 2.98 × 105), and the number of HUVECs increased ∼30-fold (2.41 × 104 ± 7.5 × 103, 6.58 × 104 ± 2.9 × 103, 1.93 × 105 ± 5.1 × 104, 2.77 × 105 ± 5.95 × 104, and 6.37 × 105 ± 1.89 × 105) by day 5 (Fig. 6).
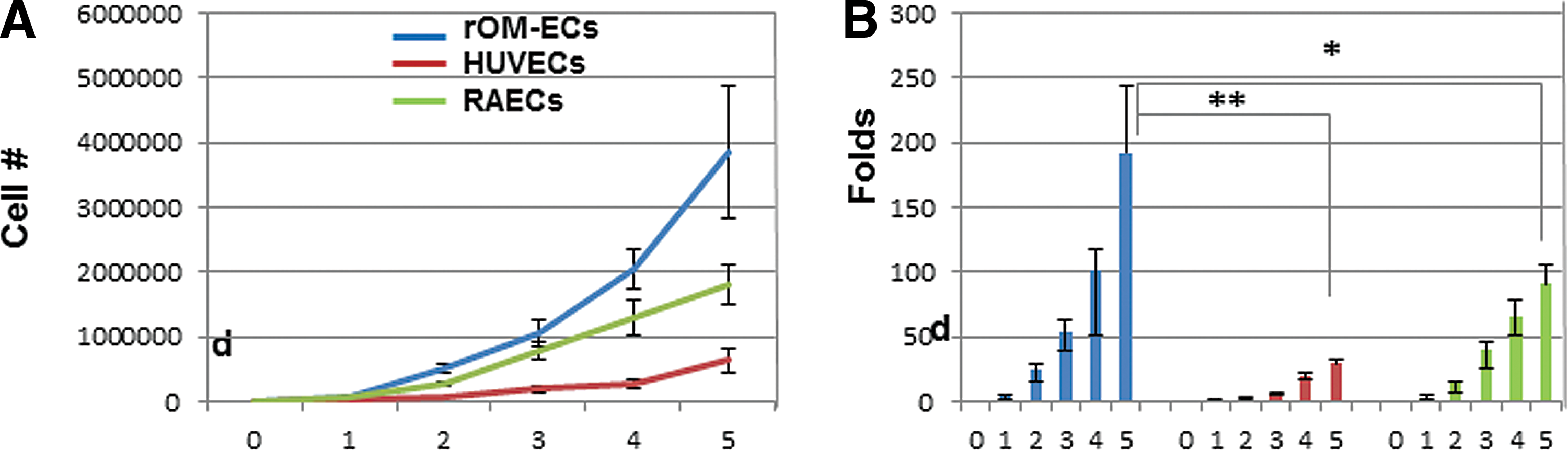
Analysis of cell growth kinetics. rOM-derived EP-like cells, HUVECs and RAECs in 5-day growth curve analysis.
Discussion
Amid the difficulty to isolate postnatal ECs from existing cell sources, we decided to explore oral mucosa as an alternative tissue choice for isolation of postnatal ECs or EPCs.
Our study showed that a robust number of cobblestone-appearing cell colonies could be readily isolated from the oral/buccal mucosa (rOM) (Fig. 1). We have repeated the experiments using donor-match BM samples to show that the EPC frequency was thousands of folds higher in oral mucosa than in BM aspirates (Table 1) [8,17], which makes oral mucosa the most prolific tissue for isolation of postnatal ECs that have been identified to date.
Characterization of the rOM-derived cobblestone-appearing cells was done by assessment of EC function through AcLDL ingestion (Fig. 2A), examination of EC marker expression (Figs. 2 and 3), and their ability to form lattice networks on Matrigel (Fig. 4). Notably, the choice of EC culture medium was critical for the survival of the rOM-derived EP-like cells and the EC functionality of RAECs (Fig. 4B). We have tested two different EC growth media, L-EGM (basal medium with hEGF, gentamicin/amphotericin-B, VEGF, hFGF-B, R3-IGF-1, ascorbic acid, heparin, and 20% FBS) and C-EGM (basal medium with EGF, L-glutamine, antibiotics/antimycin solution, and 2% FBS), on both rOM-derived EP-like cells and RAECs. rOM-derived EP-like cells grew well in L-EGM (Figs. 1 and 2) but could not survive in C-EGM (not shown), whereas RAECs could grow and proliferate well in both media (Fig. 2A, right panel).
In the immunofluorescence staining experiments, rOM-derived EP-like cells, RAECs, and HUVECs have all shown strong expression level of VE-Cadherin protein (Fig. 2B). However, our flow cytometry study showed that the EP-like cells from oral mucosa carried a rather different EC marker expression profile than RAECs. In rOM-derived EP-like cells, the majority of the cell population expressed VEGFR2 (>80%), but only ∼35% of cell population was positive for CD31 (Fig. 3). In contrast, only ∼20% of the RAEC population was positive for VEGFR2 but >90% were positive for CD31 (Fig. 3). The different expression patterns of the VEGFR2 and CD31 markers in rOM-derived EP-like cells and RAECs suggest that they probably comprise EPCs at different maturation stages, given that VEGFR2 (KDR/Flk1) is an earlier maker for EPCs, whereas CD31 is a late marker for ECs [17,41]. The VEGFR2hi/CD31lo expression profile of our rOM-EP-like cells is very similar to the VEGFR2hi/CD31lo expression profile of the human embryonic stem cell-derived EC-KDR+ cells from the previous study [42], whereas the VEGFR2lo/CD31hi expression profile of RAECs is almost identical to the VEGFR2lo/CD31hi expression profile of HUVECs and adult late outgrowth cells/ECFCs [6,8,42,43].
We also observe a small cell population from either rOM-EPCs or RAECs expressed CD34 (Fig. 3). Since CD34 is often used as an endothelial marker for human progenitor cells but not for other species such as murine EPCs [1,2,6,44,45], we suspected that its limited expression in the rOM-EPCs may be the result of species-specific targeting. Taken together, our flow cytometric data have placed the rOM-EP-like cells closer to the earlier endothelial precursors than the more mature well-differentiated ECs in the hierarchical order of the progression EPs to ECs.
We have used both C-EGM and L-EDM (same as L-EGM but with 2% serum) to culture RAECs for the flow cytometric analysis and tube formation assay. Although there was no apparent difference in the EC surface marker expression profile (flow cytometric analysis) with either medium, we found that L-EDM has a negative impact on RAECs in tube formation (Fig. 4B). When cultured in C-EGM, RAECs could readily form extensive networks and the network integrity lasted ∼3 days on Matrigel (Fig. 4B, top panel). However, tube formation was not successful when RAECs were cultured in L-EDM (Fig. 4B, bottom panel). The tube formation from RAECs in C-EGM was more stable than HUVECs and hDP-MSCs (Fig. 4). However, among our four test groups, only capillary networks from rOM-derived EP-like cells could remain intact at the end of our 7-day study (Fig. 4A, bottom panel and Supplementary Fig. S2). On the contrary, lattice structures from the DP-MSCs were very transient and would dissemble into spheroid-like structures overnight (Fig. 4B, bottom panel). Our study suggests that rOM-derived EP-like cells have the best and most stable capillary network formation potential.
Through in vivo subcutaneous implantation experiments, we have established the status of rOM-derived EP-like cells as true EPCs. GrOM-EPCs were infused into medical-grade collagen sponge before the implantation procedure (Fig. 5A), and our data showed that GrOM-derived EP-like cells could anastomose with host blood vessels and expressed CD31 (Fig. 5B). In addition, GFP+ vasculature was readily detected at the end of an 8-week study (Fig. 5B, second panel), indicating that the vasculature from GrOM-derived EP-like cells remained stable long after the engraftment. This observation was consistent with the data from our tube formation assay (Fig. 4A, bottom panel).
One of the distinct features of ECFCs/EOCs is their robust proliferative potential [6,7]. In our study, we did observe an extremely high proliferation rate from rOM-derived EP-like cells and our growth curve analysis showed that their proliferative rate was approximately eight times higher than HUVECs and two times higher than RAECs (Fig. 6). Faced with constant challenges from the environment and the physiological aging process, the body needs to continuously replace its cells. This occurs particularly for tissues that are most exposed to the environment, such as the mucosal epithelia, skin, and immune system. Wounds in the oral mucosa heal by regeneration, and the healing rate is faster than in other tissues [46,47]. Taken together, we conclude that the rOM-derived EP-like cells are indeed true EPCs and can be used to repair or regenerate vascular network in tissue engineering.
The importance of blood vessels to bone formation was noted and documented for more than two centuries [48], but it was not until the mid-1900s that the interest in bone vasculature was revived [49]. In that study, the author suggested that a “vascular stimulating factor (VSF)” was present and operating at sites of bone damage [49]. These early predictions have proven to be remarkably accurate. Inadequate or inappropriate bone vascularity is associated with decreased bone formation and bone mass [50,51] and has also shown to be the major cause for delayed union or nonunion during fracture healing [52 –54]. EPCs have been successfully used in regenerative medicine to promote neovascularization in patients after myocardial infarction and limb ischemia [2,3,9,55 –57].
Investigators, including our group, have recently started to examine the potential of ECs (or EPCs) to treat critical-sized bone defects, either on their own [10,58,59] or in combination with MSCs [60 –63]. Our preliminary study shows that there is a synergistic effect between ECs (HUVECs) and MSCs in promoting bone regeneration when implanted at the fracture gap of the large bone defects created by osteotomy at the tibial epiphysis of host recipients (orthotopic implantation) (preliminary data). These results are consistent with previous studies [60,62]. However, since HUVECs are not postnatal cells, their clinical relevance is unclear. In order for us to move forward with our study, we have to resolve the challenges of isolating postnatal ECs or EPCs from existing cell sources. In this study, we have successfully identified oral mucosa as a novel cell source that can be used to isolate a high level of putative postnatal EPCs, which display robust vascular capillary formation potential under both in vitro and in vivo environments. With this new information, we can design our experiments to identify a potential bone regeneration treatment that can easily translate to clinical applications.
Footnotes
Acknowledgments
The work of the authors is supported by NIH R01AR065023, R01EB009663, and Gatorade Trust Start-Up Fund 00123001. The authors thank Dr. J. Kitajewski for helpful discussion and Dr. E. Williamson for critical review of the article.
Author Disclosure Statement
No competing financial interests exist.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.