
Abstract
Compact bones (CB) are major reservoirs of mouse mesenchymal stem cells (mMSC). Here, we established a protocol to isolate MSC from CB and tested their immunosuppressive potential. Collagenase type II digestion of BM-flushed CB from C57B/6 mice was performed to liberate mMSC precursors from bone surfaces to establish nondepleted mMSC. CB cells were also immunodepleted based on the expression of CD45 (leukocytes) and TER119 (erythroid cells) to eliminate hematopoietic cells. CD45−TER119− CB cells were subsequently used to generate depleted mMSC. CB nondepleted and depleted mMSC progenitors were cultured under hypoxic conditions to establish primary mMSC cultures. CB depleted mMSC compared to nondepleted mMSC showed greater cell numbers at subculturing and had increased functional ability to differentiate into adipocytes and osteoblasts. CB depleted mMSC had high purity and expressed key mMSC markers (>85% Sca-1, CD29, CD90) with no mature hematopoietic contaminating cells (<5% CD45, CD11b) when subcultured to passage 5 (P5). Nondepleted mMSC cultures, however, were less pure and heterogenous with <72% Sca-1+, CD29+, and CD90+ cells at early passages (P1 or P2), along with high percentages of contaminating CD11b+ (35.6%) and CD45+ (39.2%) cells that persisted in culture long term. Depleted and nondepleted mMSC nevertheless exhibited similar potency to suppress total (CD3+), CD4+ and CD8+ T cell proliferation, in a dendritic cell allostimulatory one-way mixed lymphocyte reaction. CB depleted mMSC, pretreated with proinflammatory cytokines IFN-γ, TNF-α, and IL-17A, showed superior suppression of CD8+ T cell, but not CD4+ T cell proliferation, relative to untreated-mMSC. In conclusion, CB depleted mMSC established under hypoxic conditions and treated with selective cytokines represent a novel source of potent immunosuppressive MSC. As these cells have enhanced immune modulatory function, they may represent a superior product for use in clinical allotransplantation.
Introduction
T
The exact mechanisms by which human BM-derived MSC function in vivo; the route, dose, timing of MSC patient infusion, and safety concerns with regards to MSC; off target effects; tumorgenicity [4 –7]; and differentiation into other cell types [8 –10] remain unresolved. Together these issues limit the wide application of human BM MSC therapy into the clinic. Therefore, preclinical mouse models are widely utilized to address these issues associated with MSC therapy.
Human BM-derived MSC compared to their mouse MSC (mMSC) counterparts have been isolated and expanded ex vivo with high purity [11]. The rarity of mMSC from the BM, however, represents a major obstacle for research aiming to dissect mechanisms of MSC function in vivo, optimize therapeutic strategies, and address safety concerns of BM MSC therapy. Mouse MSC have an extremely low incidence in BM compared to human MSC, and require modified growth conditions and media supplements for ex vivo expansion [12,13]. The major challenge in BM mMSC isolation protocols is to obtain a homogenous and highly purified population of mMSC, with less contaminating hematopoietic cells and other non-MSC cells or progenitor populations that coexist in the BM niche [12 –14].
Previous attempts to eliminate these contaminating cells in mMSC cultures included immunodepletion and (or) unique culture systems such as frequent media change, low cell density cultures, alteration to the standard mMSC culture media components or cell culture surfaces to promote mMSC adherence [15 –21]. Despite these attempts, isolation of mMSC from whole BM remained problematic due to poor yields or purity [21 –23]. Absence of standardization on protocols in BM mMSC isolation protocols may be a contributing factor to the conflicting results on the in vivo therapeutic efficacy of mMSC in preclinical mouse models of diseases including allotransplantation. Most in vivo studies on the immunosuppressive function of MSC in preclinical murine models of allotransplantation use the traditionally isolated plastic-adherent selected BM mMSC [24 –28], which contain numerous accessory cell populations.
In this study, we established a protocol to enrich a highly purified and homogenous population of compact bone (CB) mMSC with potent in vitro immunosuppressive capacity that would be beneficial in an allotransplant setting. We employed collagenase type II digestion of BM depleted CB combined with immunodepletion of CD45 (lymphocyte common antigen) and TER119 (erythroid lineages) expressing cells as well as hypoxia preconditioning to establish primary mMSC cell lines. CB depleted mMSC were easily isolated from CD45− TER119− CB progenitor cells with high initial cell yields at passage 1 (P1). Based on this isolation protocol, CB mMSC were also able to potently suppress in vitro total T cells and subset (CD4+ and CD8+) T cell proliferation in dendritic cells (DC) allostimulatory one-way mixed lymphocyte reaction (MLR), which mimic an in vivo allotransplant rejection response.
Materials and Methods
Animals
All experimental procedures were approved by the Animal Ethics Committee of the University of Adelaide and conform to the guidelines established by the Australian Code of Practice for the Care and Use of Animals for Scientific Purposes (ethics approval M-2013-033).
Mouse MSC
Dissection of hind limbs
mMSC were isolated from BM or CB (tibia and femur) of C57BL/6 mice (4–8 mice per mMSC isolation, 6–7 weeks old). Animals were humanely killed by carbon dioxide or isoflurane overdose followed by cervical dislocation. Tibia and femur were dissected from the hind limbs in a laminar flow hood using sterile instruments and washed thoroughly with cold α-MEM (50 U/mL penicillin/50 μg/mL streptomycin). Limbs were bisected by cutting through the knee joint. Muscle and connective tissues from bones were removed by scrapping the diaphysis of the bone, then pulling the tissue toward the end of the bone. The ends of the marrow cavity were then bisected and bones were stored in cold α-MEM on ice until further processing.
Harvesting BM cells from the tibia and femur
BM cells were harvested using a 21-gauge needle, inserted into the distal end of the femur or tibia. Five–10 mL 5% FBS/HBSS was drawn through the marrow cavity, and then rapidly flushed out into the tube. This process was repeated until the bone became completely white indicating that all marrow cells have been harvested. The resulting cell suspension was passed through a 70 μm nylon cell strainer to remove bone spicules or muscle and cell clumps. The viability and yields were determined by trypan blue exclusion assay. Approximately 70 × 106 BM cells can be obtained from each mouse donor.
Mouse MSC isolation from CB
CB was crushed into small pieces (1–2 mm) using a sterile blade, allowing for subsequent enzymatic digestion to release maximum number of cells. CB cells were then incubated with collagenase (at varying concentrations) and DNase I (50 U/mL) for 1–2 h at 37°C on a shaking platform at 240 rpm. The resultant cell suspension (nondepleted mMSC precursors) was strained using a 70 μm nylon cell strainer and bone chips were discarded. To isolate CB depleted mMSC, mouse CB cells were further purified using the EasySep™ Mouse Mesenchymal Progenitor Enrichment Kit (StemCell Technologies; catalogue #19771) according to the manufacturer's instructions. CB cells were depleted of cells of hematopoietic origin using antibodies against CD45 and TER119.
Nondepleted (5 × 105 cells/cm2) and depleted (1 × 104 cells/cm2) CB cells were seeded in tissue culture flasks to establish the primary CB mMSC cell lines in primary MSC media containing α-MEM, 20% (v/v) heat-inactivated fetal bovine serum, 2 mM
Mouse MSC growth culture conditions were optimized using different batches of FBS. The effect of different FBS on nondepleted mMSC progenitor cell enrichment and in mMSC morphology and growth potential following mMSC subculturing were tested. The three FBS batches examined included (1) SAFC Batch #10C126 (SAFC)–previously used in mMSC isolation protocols, (2) Invitrogen Batch #1153562 (Invitrogen)–new non-MSC qualified FBS, and (3) Invitrogen Batch #981394 (Invitrogen MSC)–new human MSC qualified FBS.
BM cell harvested from eight mice were seeded into a T-75 flask and primary BM mMSC cell lines were established at 37°C in normoxic conditions.
Mouse MSC characterization
MSC precursors form fibroblast colony-forming unit (CFU-F) when cultured in vitro [30,31]. For CFU-F assays, BM or depleted CB cells were plated in a six-well plate with primary MSC media in triplicate wells at 1 × 105 cells/well or 9 × 105 cells/well for 5 days. Cells were incubated at 37°C in a humidified tissue culture incubator under normoxic (21% O2) or hypoxic conditions (5% O2). Nuclei of cells were stained with Toluidine blue stain and colonies were counted under an inverted light microscope (Olympus CKX41). A colony was defined as a cluster of ≥50 cells. Cobblestone-shaped colonies were excluded for CFU-F counts. Immunophenotype of culture expanded CB depleted and nondepleted CB mMSC were characterized by flow cytometry [32] (Table 1). Samples were acquired on BD FACSCantoII (BD Biosciences) flow cytometer and analyzed by FCS express V4. CB mMSC were induced to differentiate into adipocytes and osteocytes as published [11]. Mouse MSC at passages 3–6 were used in experiments.
Cytokine treatment of CB mMSC
CB depleted mMSC (5 × 105 cells) were seeded into a T-75 tissue culture flask. Cells were allowed to adhere overnight and replaced with fresh MSC media with either no cytokines or with recombinant mouse cytokines IL-17A (50 ng/mL; eBioscience), IFN-γ (500 U/mL; Peprotech), and (or) TNF-α (20 ng/mL; Peprotech) [33].
Dendritic cells
DC were isolated and propagated from BALB/c mice BM precursors for 5 days in RPMI 1640 medium supplemented with 10% FBS, β-mercaptoethanol (2 μL/500 mL), 1,000 U/mL recombinant mouse granulocyte macrophage colony-stimulating factor (GM-CSF; R&D Systems), and 500 U/mL recombinant mouse IL-4 (R&D Systems) as previously described [34 –36]. On day 5, 200 ng/mL lipopolysaccharide (LPS; Sigma) was added to the BM-DC culture for 24 h to induce DC maturation. CD11c+ mature DC (mDC) were positively selected using the EasySep™ mouse CD11c positive selection kit II (StemCell Technologies; catalogue #18780A) according to the manufacturer's instructions. Purified CD11c+ mDC expressed mDC markers (>90% CD11b, CD11c, CD40, CD80, CD83, and CD86 and MHC class II).
Mouse T cells
Splenic CD3+ T cells were isolated from C57BL/6 mice using the Mouse T cell Enrichment Column (R&D Systems; MTCC-5/10) according to the manufacturer's instructions (>90% purity). Purified CD3+ T cells were labeled with carboxyfluorescein succinimidyl ester (CFSE; Invitrogen) for mouse T cell proliferation assays as per manufacturer's instructions. T cells (10 × 106 cells/mL) were stained with 0.7 μL CFSE per 1 mL cell suspension.
Mixed lymphocyte reaction
Irradiated depleted or nondepleted mMSC (30 Grays) at 1 × 102 (0.1%), 1 × 103 (1%), or 1 × 104 cells (10%) were seeded overnight into 96-well round bottom plates in a total volume of 0.2 mL complete RPMI 1640 medium. CFSE-labeled responder CD3+ T cells (1 × 105cells) and 30 Grays irradiated stimulator mDC (1 × 104cells) at a DC to T cell ratio of 1:10. Cells were cultured for 3 or 5 days and T cells were harvested for analysis of CFSE-labeled T cell proliferation. Harvested CFSE-labeled T cells were also cell-surface stained with anti-CD4 and anti-CD8 antibodies to detect proliferation levels of CD4+ and CD8+ T cells, respectively (Table 1).
Statistics
Statistical significances (P < 0.05) were assessed by one- or two- analyses of variance (ANOVA) with post-Sidak multiple comparison tests or Kruskal–Wallis with post-Dunn multiple comparison tests. Statistical analyses were performed using the GraphPad Prism software (GraphPad Software,
Results
Optimization of compact bone mMSC isolation and culture
We aimed to establish a protocol to enrich and isolate a highly purified and homogenous population of CB mMSC. Collagenase digestion is an essential step to isolate mMSC progenitors from CB. CB-derived Sca-1+ cells (stem cell antigen-1) are previously known to be precursor cells of mMSC [37 –39], providing an indication whether the bone digestion step was sufficient to release mMSC precursors from the CB. Collagenase II (3 mg/mL, 1 h) resulted in high yield of CB cells and the highest of Sca-1+ cells (34.66%) immediately after CB digestion compared to all the other CB digestion conditions (Table 2). The colony forming efficacy of the differently digested bone fragments confirmed that the digestion protocol was sufficient to release mMSC precursor cells from CB, where collagenase II (3 mg/mL, 1 h) showed the highest CFU-F efficacy with low toxicity (>90% viability).
CB cell yield immediately after CB digestions and the % Sca-1+ cells were determined. Yield of cells was counted as the number of CB cells released immediately after CB digestion.
CB, compact bones; CFU-F, colony-forming unit fibroblasts.
FBS batches vary in quality and composition. This may affect adherence, mitotic expansion of primary MSC cultures, and the retention of MSC in undifferentiated states [40]. All FBS batches showed no difference in the enrichment of mMSC progenitor cells evident by the comparable number of CFU-F in primary mMSC cultures and no aberrant changes in mMSC morphology (Fig. 1A, B). Invitrogen FBS was optimal for mMSC cultures as it favored mMSC growth, particularly from P2 to P3 when compared to Invitrogen MSC FBS (Fig. 1C) and was subsequently used in all mMSC isolations and cultures.
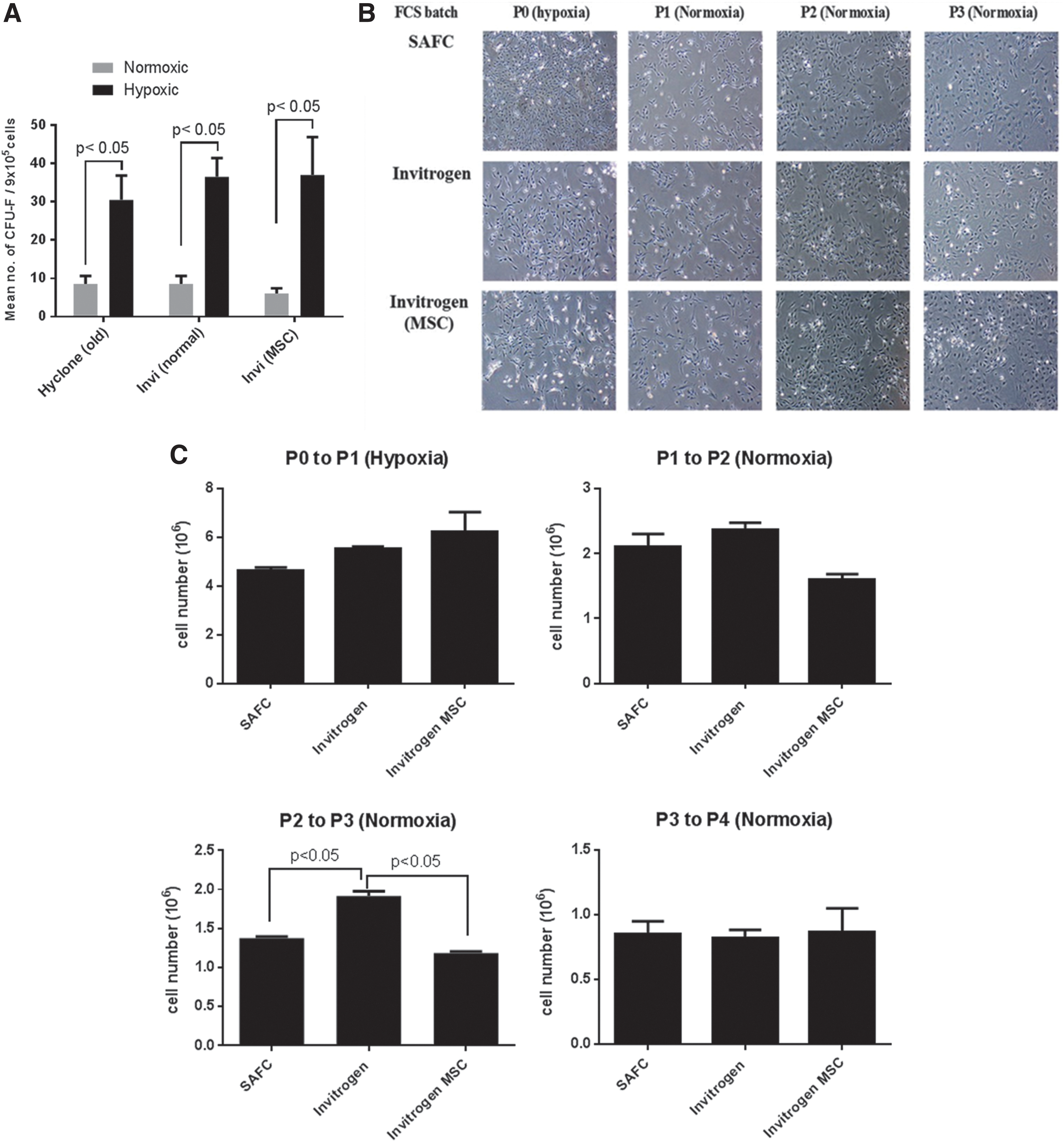
Optimization of CB mMSC isolation and culture conditions.
Hypoxic culture conditions favored the enrichment and growth of mMSC progenitor cells evident by the high numbers of CFU-F derived from these CB cells (Fig. 1A and Table 3). Precursors of mMSC were highly prevalent in the CB rather than in the BM fraction as CB-derived mMSC had higher colony forming efficiency (Table 3).
Based on the optimization of culture conditions of CB nondepleted mMSC isolation, we have established a protocol to maximize the enrichment and expansion of mMSC. Collagenase II (3 mg/mL, 1 h) digestion of the CB followed by hypoxia culture isolated, selected, and enhanced the clonogenicity of CB-derived mMSC precursors. This protocol was used as a standard operating procedure for the culture and establishment of nondepleted and depleted mMSC cell lines from CB precursors. Depleted mMSC cell lines were established using these culture conditions in addition to lineage depletion of CD45 (lymphocyte common antigen) and TER119 cells (erythroid lineages) of the digested CB fragments with the aim to isolate highly pure CB mMSC precursors compared to nondepleted mMSC.
CD45 and TER119 depletion of compact bone cells enhances mMSC precursor enrichment
CB depleted mMSC precursors obtained following CD45 and TER119 immunodepletion represented only a very small fraction (0.02%–1%) of the digested CB cells. Following 5 days of culture under hypoxia with media change every 2 days, there was a 5.9-fold increase in the numbers of CB depleted mMSC when subcultured from P0 to P1 (Fig. 2C). Nondepleted mMSC precursors were initially seeded at a 50x higher seeding density to obtain comparative density and growth rates as observed with depleted mMSC at day 5 under hypoxia. There was, however, a decrease in cell numbers at P0 to P1 due to the low frequency of mMSC precursors in the CB nondepleted fraction (Fig. 2C). There was also presence of cells with a cobblestone appearance, which contaminated the nondepleted mMSC cultures, whereas depleted mMSC cultures showed homogeneity of cell morphology, exhibiting a fibroblastic MSC-like appearance as early as P0 and P1 (Fig. 2B). CB depleted mMSC also showed highest enrichment of Sca-1+ cells (90.6% ± 1.141%) and were homogenous in Sca-1 expression, compared to CB nondepleted mMSC (72.4% ± 3.635%) at P1 (Fig. 2A). The level of Sca-1 expression was also highest in CB depleted mMSC (Fig. 2A).
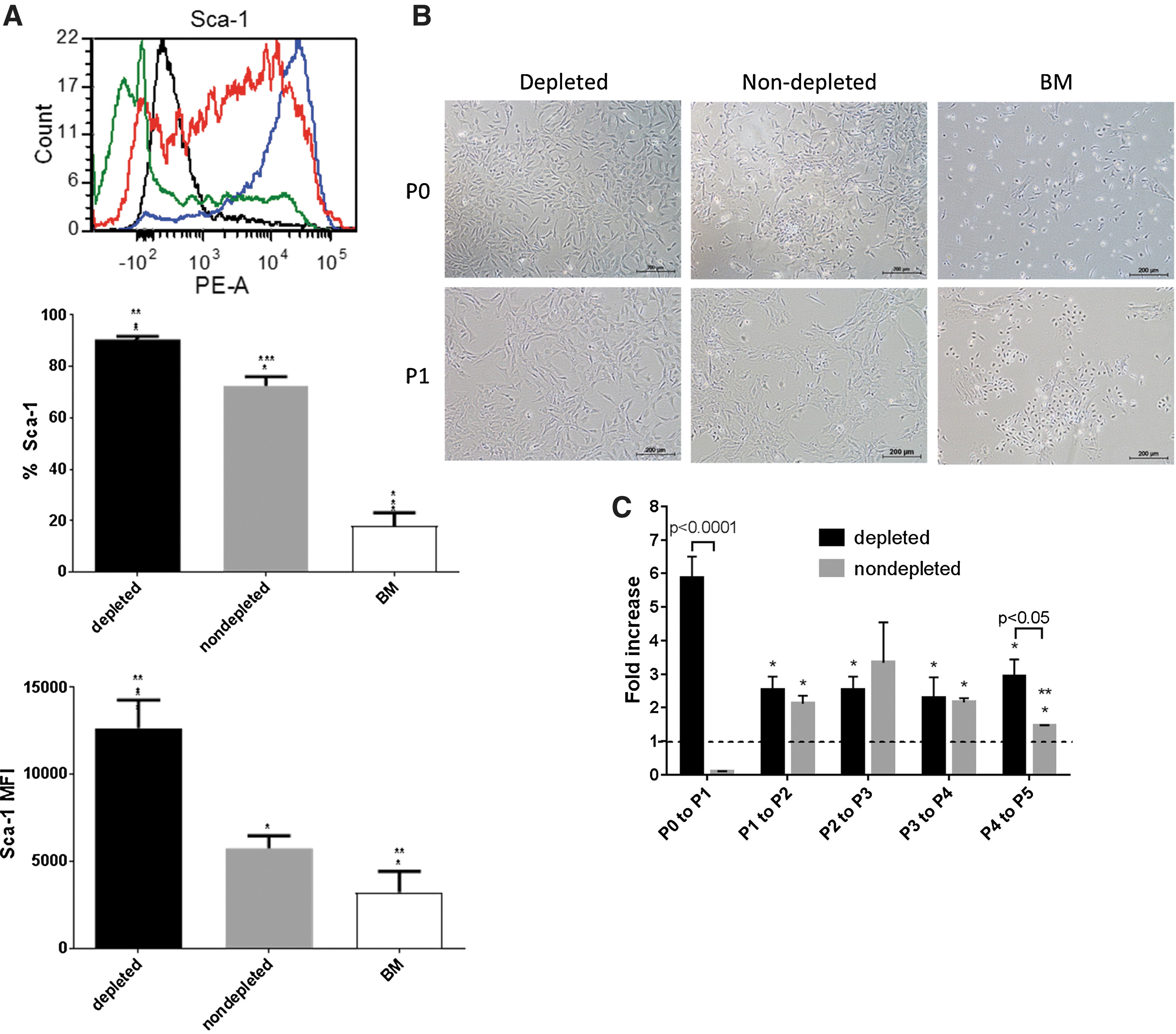
Enrichment of mMSC progenitors by CD45 and TER119 depletion of compact bones. Bone marrow and compact bones (depleted and nondepleted) mMSC cell lines isolated from C57BL/6 mice were established under normoxia or hypoxia, respectively. CB nondepleted mMSC progenitors were obtained following collagenase digestion. CB depleted mMSC precursors were isolated by depleting CD45 and TER119 cells from the collagenase digested CB cell fraction.
Additionally, both depleted and nondepleted mMSC showed similar increase in fold proliferation from P1 to P4 (Fig. 2C). Nondepleted mMSC, however, had reduced proliferation potential when subcultured to higher passages (P4 to P5; 1.5-fold) and when compared to depleted mMSC (3.0-fold).
Interestingly, BM mMSC initially established under normoxia at the highest seeding density with frequent media change exhibited lowest Sca-1 percentages (18.0% ± 5.083%) and expression levels (Fig. 2A). BM mMSC cultures were heterogenous with cobblestone colonies, round adherent cells and cells were less elongated and fibroblastic-like (Fig. 2B). Culture of BM mMSC was unsuccessful as they failed to proliferate beyond P1 and was therefore not used in this study.
CB depleted mMSC have high purity and greater functional differentiation potential
In subsequent experiments, we further compared and characterized the immunophenotype and differentiation potential of CB nondepleted and depleted mMSC. At a low passage (P2), depleted mMSC cultures had higher percentages of cells expressing key MSC markers Sca-1 (94.3%), CD29 (85.0%), and CD90 (86.7%) compared to nondepleted mMSC with 71.4%, 54.2%, and 63.2% positive cells, respectively (Fig. 3A). Sca-1 was also highly expressed by depleted mMSC relative to nondepleted mMSC at P2. Nondepleted mMSC at a higher passage (P5) nevertheless showed increased percentages of Sca-1+ and CD90+ when compared to P2 and were similar to depleted mMSC. Other standard positive MSC markers CD44 and CD105 were expressed similarly by both mMSC groups regardless of the passage.
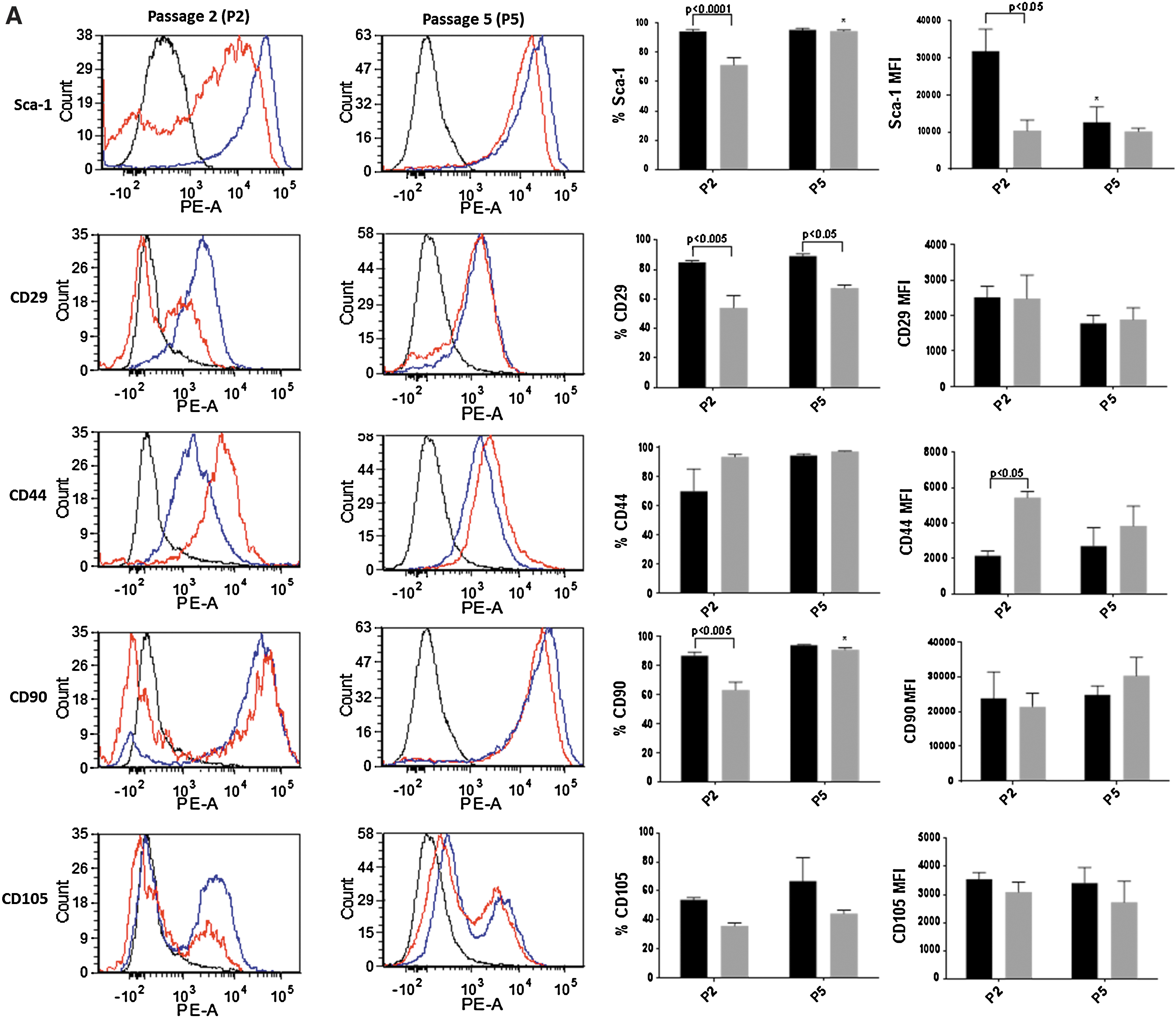
Immunophenotype comparison of compact bone depleted and nondepleted mMSC. The expression of standard positive

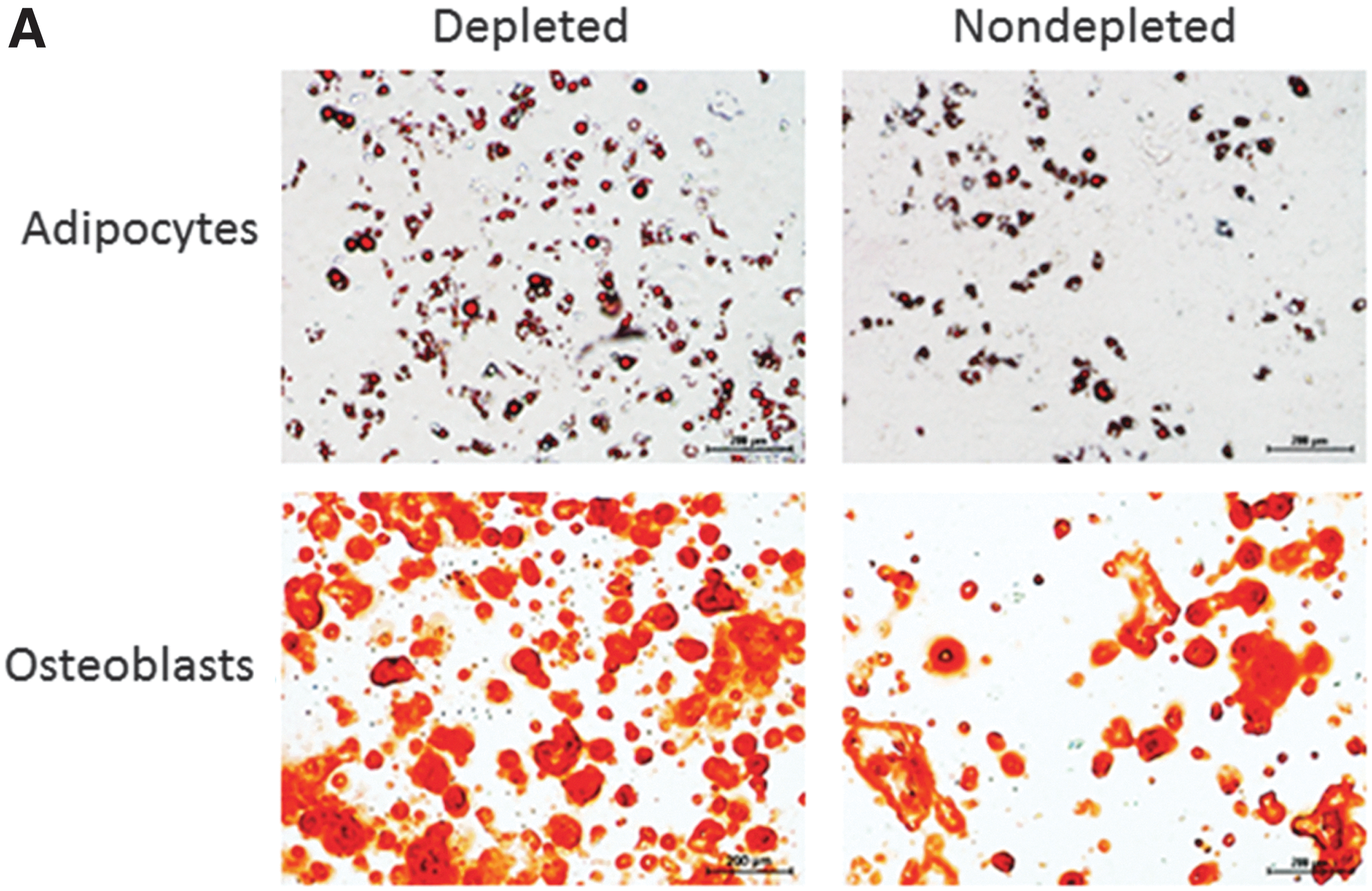
Early nondepleted mMSC cultures (P2) were contaminated with CD11b+ (35.6%) and CD45+ (39.2%) cells expressing high levels of these markers (Fig. 3B). The percentages and the expression levels of CD11b (20.8%) and CD45 (10.3%) decreased in nondepleted cultures upon subculturing. Low and high passage depleted mMSC had <5% contaminating CD11b+ and CD45+ cells. Depleted mMSC also exhibited higher functional capacity and could differentiate more efficiently into adipocytes and osteoblasts (Fig. 4A). The high intensity of Alizarin Red staining indicated increased number of osteocytes containing calcium deposit in depleted mMSC. Similarly, greater lipid formation detected by Oil Red O staining suggested the enhanced differentiation capacity of depleted mMSC into adipocytes (Fig. 4A).
Depleted and nondepleted mMSC inhibit allogeneic T cell proliferation in a dose-dependent manner.
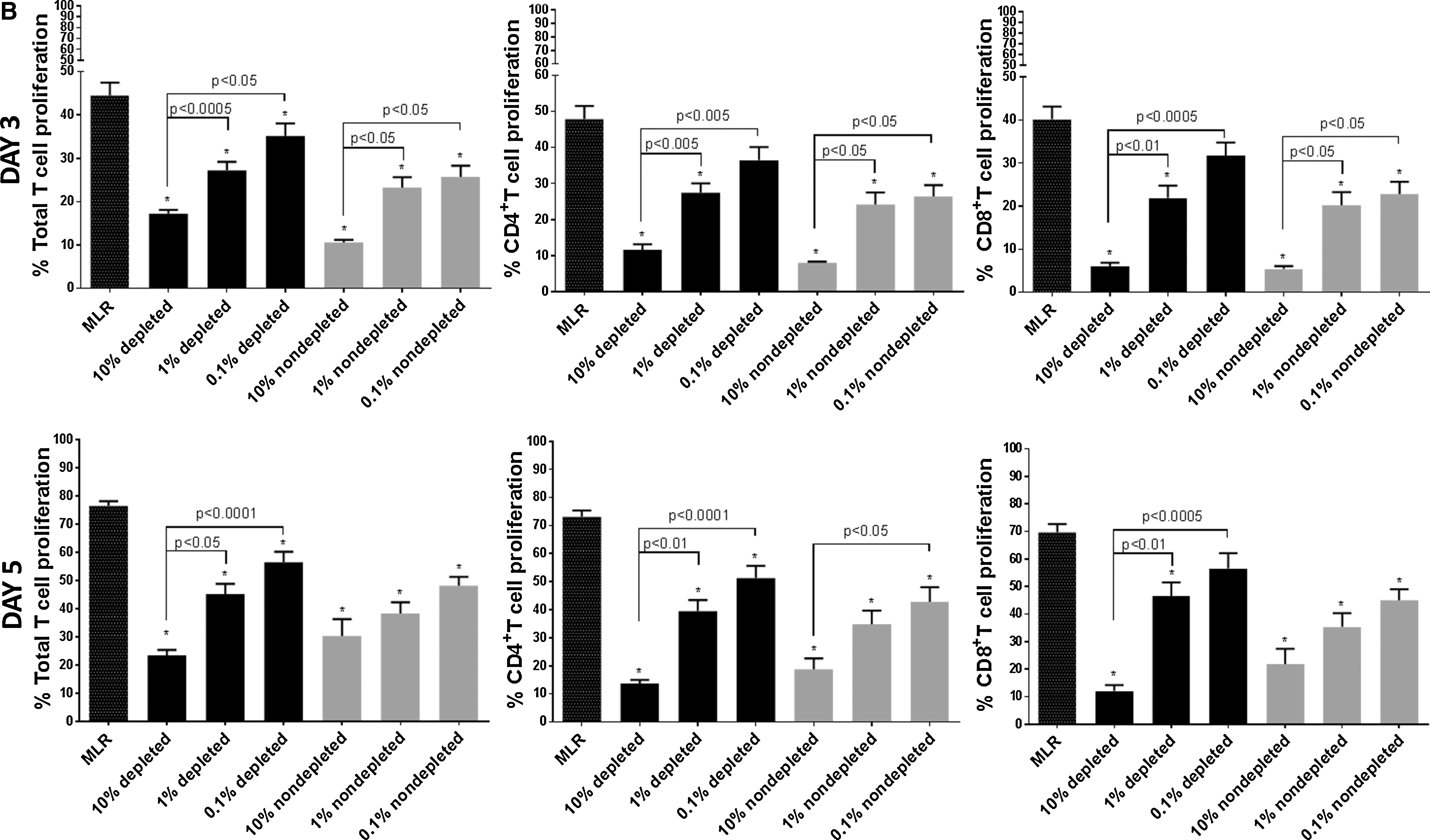
Depleted and nondepleted mMSC exhibit comparable inhibition of T cell proliferation
Nondepleted and depleted mMSC inhibited allogeneic DC stimulated T cell proliferation in a dose-dependent manner at days 3 and 5 of the MLR assay with similar efficacy (Fig. 4B). Maximal immunosuppression of T cells was observed at 10% mMSC dose whereby CD4+ T cell proliferation was suppressed by up to 83.4% and 81.2% at days 3 and 5 of MLR, respectively, while CD8+ T cell proliferation was inhibited by up to 86.5% and 82.6% (Fig. 4B cumulative data; Table 4). Both nondepleted and depleted mMSC retained their immunosuppressive capacity on total, CD4+ and CD8+ T cells when used at lower doses (1% or 0.1%).
Depleted (dep) and nondepleted (nondep) UT-mMSC at 0.1%, 1%, or 10% doses were cocultured with CD11c+ mDC stimulated CD3+ T cells for 3 or 5 days. CD4+ and CD8+ T cell proliferation was measured in a one-way MLR. Data are presented as percentages of CD4+ and CD8+ T cell proliferation. Bold fonts indicate P < 0.05 versus MLR determined by Kruskal–Wallis with post-Dunn multiple comparison test. Error bars depict means of ± SEM.
CB depleted mMSC-17 are not superior suppressors of T cells
We have previously established that human IL-17A pretreated MSC (MSC-17) conformed to untreated MSC (UT-MSC) immunophenotype and are superior suppressors of T cells [33]. Since CD45 and TER119 depletion of CB cells yields a highly purified and homogenous population of mMSC compared to nondepleted mMSC, subsequent experiments were conducted using only depleted mMSC. Here, we aimed to determine whether murine CB depleted mMSC treated with IL-17A (mMSC-17) have similar characteristics to human MSC-17 and retained UT-MSC phenotype. Immunophenotype of mMSC-17 was similar to untreated mMSC (UT-mMSC) following 5 days of IL-17A treatment (Supplementary Fig. S1A; Supplementary Data are available online at
TNF-α enhances the immunosuppressive effect of CB depleted mMSC
MSC pretreated with other inflammatory cytokines including IFN-γ and TNF-α and in a cytokine combination cocktail have previously been shown to enhance MSC immunosuppression [33,41,42]. Hence, we further explored the capacity of 5 days IFN-γ (MSC-γ), and TNF-α (MSC-TNF) treated mMSC on T cell suppression. MSC-γ showed no improvement in MSC inhibition of T cells, comparable to UT-MSC (Fig. 5). Interestingly, CB depleted MSC-TNF exhibited most potent suppression on allogeneically activated CD8+ T cells at day 5 MLR by 92.6% (Fig. 5).
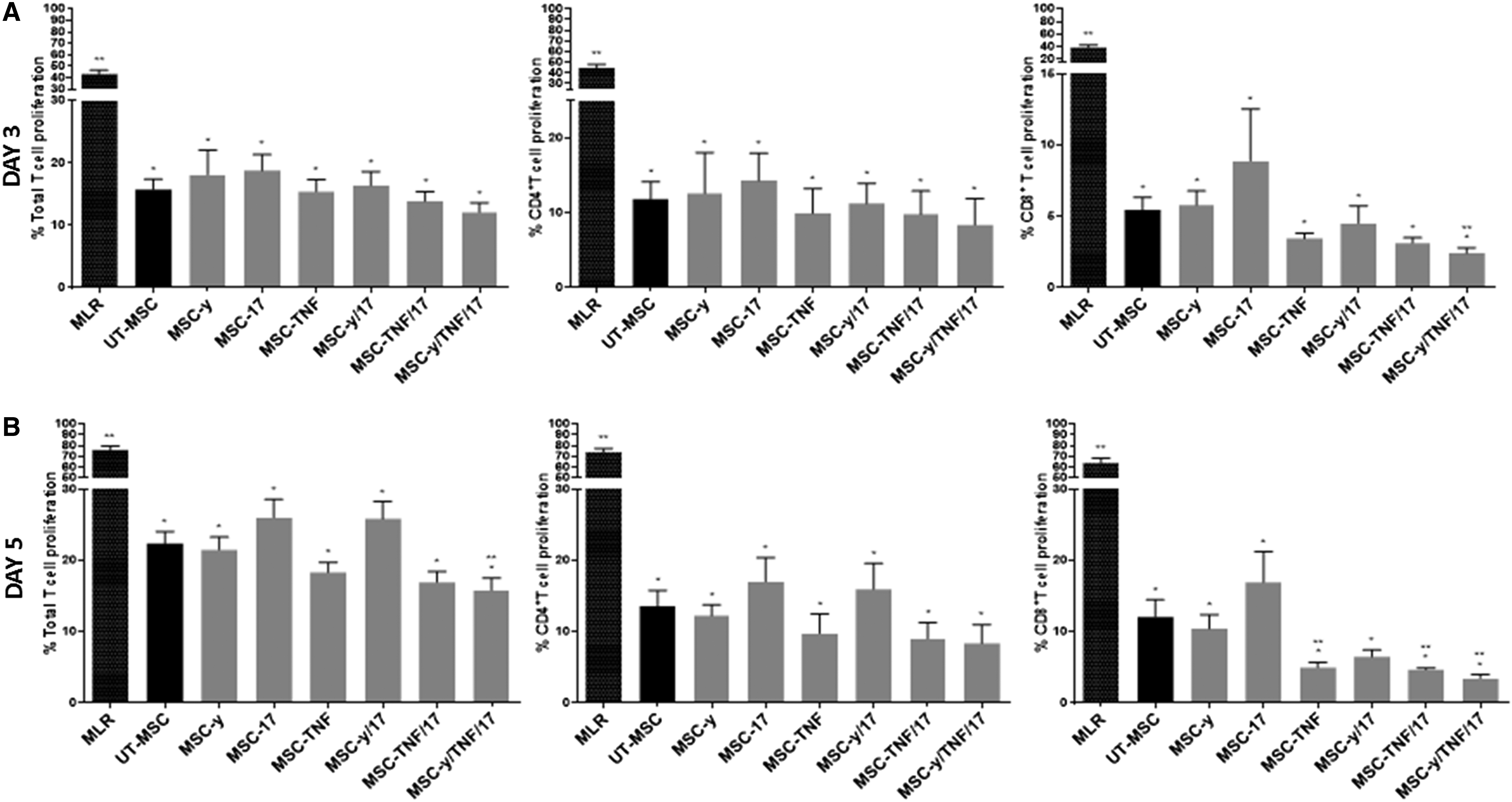
TNF-α enhanced mMSC suppression of T cell proliferation. CB depleted mMSC were either untreated (
We also investigated whether IL-17A could act synergistically with IFN-γ (MSC-γ/17), TNF-α (MSC-TNF/17), or in combination with both IFN-γ and TNF-α (MSC-γ/TNF/17) to enhance mMSC immunosuppression. Although MSC-TNF/17 augmented mMSC inhibition on CD8+ T cells relative to UT-MSC by 3.2-fold, no additional T cell suppression was observed with respect to mMSC treated with TNF-α alone (MSC-TNF) (Fig. 5B). MSC-γ/17/TNF were most potent among all other cytokine pretreatment groups at suppressing CD8+ T cell proliferation at day 3 and 5 MLR, by 2.3- and 4.2-fold, respectively (Fig. 5A, B). MSC-γ/TNF/17 were also superior to UT-MSC at inhibiting total T cell proliferation at day 5 MLR (Fig. 5B). The enhancement of T cell immunosuppression by MSC-TNF, MSC-TNF/17, and MSC-γ/TNF/17 was only evident with high mMSC doses (10%) (Fig. 5B and Supplementary Fig. S3). It should be noted that TNF-α in mMSC cultures drastically altered MSC morphology, changing their fibroblastic-like appearance to a hypertrophic flattened irregular shape. TNF-α also significantly inhibited mMSC growth potential (data not shown).
Discussion
In this study, we have shown that compact bones (CB) have emerged to be one of the major reservoirs of clonogenic MSC or progenitor cells in the murine system [22,23,37,43]. The stem cell antigen-1 (Sca-1) has recently emerged as a potential marker to isolate mMSC precursors with higher clonogenicity potential, cell yields, and the capacity to maintain CB mMSC in an undifferentiated state [37 –39]. Precursors of mMSC were mostly prevalent in the CB rather than in the BM fraction as CB-derived mMSC had higher colony forming efficiency and percentages of highly expressing Sca-1+ cells, indicating greater enrichment of mMSC. Mouse MSC are traditionally isolated from the BM but mMSC are present at low frequencies where BM mMSC preparations are highly heterogeneous with many contaminating hematopoietic cells, monocytes, granulocytes, and pre-B cell progenitors that co-adhere with BM mMSC cultures [12 –14,21]. We also observed the presence of colonies with various morphologies including less elongated and fibroblastic-like-MSC colonies and cells with cobblestone appearance; confirming the heterogeneity of the primary BM mMSC cultures.
BM mMSC compared to CB nondepleted and depleted mMSC also exhibited low growth kinetics and failed to be maintained in in vitro tissue culture conditions beyond passage 1 (P1), despite the high seeding density to initially establish the BM mMSC cell lines. BM mMSC cultured under atmospheric oxygen (21% O2; normoxia) where shown to induce the expression of p53 and mitochondrial reactive oxygen species in mMSC [44]. These molecules resulted in oxidative stress, increased death and caused growth arrest of BM mMSC [44]. This may provide a plausible explanation to the failure to maintain BM mMSC propagation from whole BM beyond P1, due to long-term atmospheric oxygen exposure that resulted in cell growth arrest. Low oxygen tension (2% or 5% O2) has been shown to support BM mMSC clonogenicity and retain their multipotency [44 –46]. However, data presented in this study and by other groups have shown that BM mMSC compared to their CB counterparts have lower clonogenicity potential and cell yields when established under hypoxia [39,45]. Moreover, studies using Sca-1/Ly-6A null mice suggest that Sca-1 is required for the regulation of mesenchymal progenitor cell self-renewal [47,48]. The very low percentages and levels of Sca-1 expressing cells detected at P1 may also be a confounding factor contributing to the failed self-renewal capacity of BM mMSC in our experiments. Hence, CB-derived mMSC cell lines established under hypoxia represent an alternative source to BM to isolate and enrich for a highly purified population of mMSC.
Studies exploring the use of CB-derived mMSC instead of the conventional BM mMSC use different methods of isolation and culture. These studies generated CB mMSC by the culture of the whole compact bones [23], undigested CB fragments [22], collagenase digestion of CB with or without FACS sorting [37,39], or by the culture of BM-flushed digested CB fragments [43]. All these studies employed either hypoxia or normoxic culture conditions to isolate mMSC from precursor cells. In this study, we aimed to optimize, improvise, and standardize current CB mMSC isolation and culture systems. Hence, we extensively compared the enrichment, growth potential, immunophenotype, functional differentiation capacity, and the immunosuppressive properties of BM-flushed collagenase II digested CB nondepleted and CD45− TER119− (depleted) mMSC that were established under hypoxia and successively passaged under normoxia.
Although lower cell yields were obtained following CD45 and TER119 depletion of CB cells, there was a substantial increase in cell numbers of depleted mMSC as to CB nondepleted cells that were seeded at a 50 × higher seeding density. This was due to the extremely low frequency of CB mMSC progenitors that reside in the CB (0.02%–1%) from whole CB digested fragments. Immunodepletion of mature hematopoietic cells CD45 (lymphocytes) and TER119 (erythroid lineages) eliminated majority of these contaminating cells, particularly CD45 that persisted in early (P2) and long-term (P5) mMSC cultures as shown in this study with nondepleted mMSC. The reduced purity and high heterogeneity of nondepleted mMSC cultures due to contaminating CD45+ cells have been previously shown to persist even above P5 [23]. High frequencies of CD45+ cells that co-adhere with mMSC were also evident in protocols involving the culture of whole bone fragments to enable mMSC to migrate out of the CB [22] and from collagenase digested or undigested nondepleted CB mMSC [22,39,43].
In addition, we report that nondepleted mMSC cultures also have high percentages of CD11b+ cells (monocytes, NK cells, granulocytes, and macrophages), consistent with another report [43]. These innate immune CD11b+ cells were absent in depleted mMSC, further validating the high purity and homogenicity of depleted mMSC. CD11b+ and CD45+ contaminants in nondepleted mMSC can be reduced by subculturing, but they were not eliminated by P5. This may have compromised nondepleted mMSC growth capacity at later passages and their reduced efficacy to functionally differentiate into adipocytes and osteoblasts in response to classical induction conditions. Reduced nondepleted mMSC growth potential also represented a significant impediment to the generation of sufficient number of cells for in vivo infusion. In human studies, we and others have shown that depleting blood cells from the MSC preparations results in higher MSC growth rates and differentiation potential [49] consistent with our observed results with CB depleted mMSC.
Greater enrichment of highly expressing Sca-1+ cells (>90% positivity) were also evident in the depleted CB mMSC compared to nondepleted mMSC at early passages, P1 and P2. Early depleted mMSC cultures (P2) were also highly positive (>85%) for the expression of key mMSC markers CD29 and CD90. Nondepleted mMSC on the contrary exhibited a heterogenous population of cells that were CD29 and CD90 positive (<64%), while also having a negative cell fraction for these markers. These data suggested that CD45 and TER119 immunodepletion of CB progenitors improved CB mMSC enrichment, resulting in highly pure and homogenous cell preparations.
Although adipose-derived mMSC (mASC) isolations can be technically easier compared to harvesting mMSC from the bone in a murine system, MSC-like populations from different sources vary in their immunophenotype, functional differentiation potential, transcriptome, and proteome profiling [50]. Different populations of MSC are also known to respond differentially in an inflammatory milieu and exert different mechanisms of immune modulation [51 –53]. In preliminary studies, we performed immunophenotypic analysis of mASC isolated from epididymal fat of C57B/6 mice. Early mASC cultures established under normoxia or hypoxia expressed lower levels and percentages of Sca-1+ cells (< 22.0%, data not shown) compared to CB mMSC (>90% Sca-1 positivity). Mouse ASC cultures like CB nondepleted mMSC were also contaminated with high percentages of hematopoietic cells CD11b (30.3%) and CD45 (33.4%) (data not shown), while CB depleted mMSC showed absence of these cells. From our experience and reports published by others, a large proportion of contaminating CD34+ cells (30.3%) persist in early mASC cultures [54,55]. Therefore, the heterogeneity of mASC cultures, similar to CB nondepleted mMSC may limit the overall therapeutic efficacy of mASC to control alloimmune responses in preclinical models of allotransplantation rejection.
DC are the most potent antigen-presenting cells of the immune system. We isolated and generated LPS-matured DC from allogeneic BM donors to be used as an antigen-presenting cell to induce robust T cell proliferation. We specifically evaluated and compared the immunosuppressive properties of autologous nondepleted and depleted mMSC on total and T cell subsets (CD4+ and CD8+ T cells). Surprisingly, we observed no differences in the potency of nondepleted and depleted mMSC to suppress in vitro allogeneic DC stimulated T cell proliferation. Nonetheless, the use of a DC stimulated T cell MLR is more indicative of a mechanism by which CB mMSC may modulate T cell responses, previously unidentified in the literature. Previous studies investigating in vitro T cell immunosuppressive properties by CB mMSC have used nonspecific-mitogenic stimulation [43], CD3/CD28 activation of T cells [39], or allogeneic splenocytes (mixed population of stimulator cells) to induce T cell proliferation [43]. In this study, a DC-T cell MLR was used instead to more closely mimic in vivo mechanisms of cell-mediated antigen-specific immune response to the allograft [27]. Evaluating mechanisms by which CB mMSC modulate T cell responses in vitro in the presence of DC warrants further investigation.
Despite the observed similarity of depleted and nondepleted CB mMSC to modulate T cell proliferation in vitro, the administration of CB nondepleted mMSC as a therapy for in vivo allotransplantation rejection maybe limited by several factors discovered by our study. Firstly, nondepleted CB and mMSC tend to have an early decline mMSC proliferative capacity and secondly, nondepleted CB mMSC are heterogenous. The contaminating innate (CD11b+) and adaptive (CD45+) immune inflammatory cells may trigger undesired immune responses in vivo, especially if allogeneic- or third party-derived mMSC are administered to prevent allotransplantation rejection. These innate and adaptive inflammatory cells may amplify immune responses associated with graft rejection, thereby reducing the therapeutic efficacy of mMSC in vivo through mechanisms that we extensively reviewed in a previous publication [27]. For these reasons, we further explored strategies in which we can enhance the immunomodulatory properties of CB depleted mMSC but not nondepleted mMSC.
Given that CB depleted mMSC give rise to a homogenous population with an easy expansion capacity, we conducted further experiments to explore strategies to enhance their immunomodulatory capacity. We have previously shown that proinflammatory cytokines can regulate MSC function to promote a superior immunomodulatory phenotype [33]. For example, IL-17A (MSC-17) or IFN-γ (MSC-γ) preconditioned human BM MSC functioned as superior suppressors of T cells compared to UT-MSC [33]. Interestingly, CB depleted mouse MSC-γ and MSC-17 in this study differed from human MSC-γ or MSC-17 as they were ineffective at enhancing suppression of allogeneically stimulated T cell proliferation compared to UT-MSC. The combination of IL-17A in a dual cytokine cocktail with either IFN-γ (MSC-γ/17) or TNF-α (MSC-TNF/17) also failed to show improvements or have an additive effect on mMSC T cell immunosuppression.
Our data are consistent with another study showing that the treatment of BM mMSC with IFN-γ or IL-17 alone is ineffective at increasing the ability of mMSC to suppress T cell proliferation in vitro [42]. Nevertheless, in a triple cytokine combination cocktail of IFN-γ, TNF-α, and IL-17A (MSC-γ/TNF/17), CB depleted mMSC were shown to be superior at suppressing total T cell proliferation at day 5 MLR, supporting findings from Han et al. (2014). In addition to this previous study, we demonstrated specifically that the enhanced inhibition of CB depleted MSC-γ/TNF/17 compared to UT-MSC was evident only on CD8+ T cells but not CD4+ T cells at days 3 and 5 MLR. We also report that CB MSC-TNF and MSC-TNF/17 relative to UT-MSC exhibited greater inhibition of only CD8+ T cell proliferation at day 5 MLR.
Mechanistically, BM-derived MSC-γ/TNF/17 are known to inhibit in vivo Con-A induced liver injury via the induction of immunosuppressive iNOS [42]. TNF-α is also known to increase MSC adhesiveness and migration in vitro and in vivo with improvement of cardiac function recovery after myocardial infarction [56]. A latter study verified that TNF-α can induce the expression of chemotaxis molecules including CCL2 and CCL5 by MSC [42]. TNF-α in combination with IFN-γ and IL-17A pretreated BM mMSC further upregulated CCL2 and CCL5, while inducing the expression of other chemokines such as CXCL9 and CXCL10 in mMSC [42]. The functional role of immunoregulatory iNOS and chemokines in CB depleted mMSC in suppressing allogeneic DC-stimulated CD8+ T cell proliferation remains to be elucidated.
In summary, immunodepletion and hypoxia preconditioning of mouse CB cells represent a novel protocol to isolate highly purified, homogeneous and immunosuppressive mMSC that would be beneficial in models of allotransplantation rejection and inflammatory diseases. CB depleted mMSC exhibited potent immunosuppressive effect on CD4+ and CD8+ T cells, which are the key effector cells mediating mechanisms of allograft rejection. We have also, for the first time shown that the immunosuppressive properties of CB depleted mMSC on DC-stimulated T cell proliferation can be amplified by preconditioning mMSC with IFN-γ, TNF-α, and IL-17A. A DC-T cell MLR can be a useful system to investigate the immunomodulatory effects of MSC on T cells via the regulation of DC. In the future, we also aim to compare the therapeutic efficacy of CB nondepleted and depleted mMSC to prevent rejection and prolong allograft survival and function in vivo in mouse models of allotransplantation.
Footnotes
Acknowledgments
This work was supported by grants from The Hospital Research Foundation, The Queen Elizabeth Hospital, Adelaide, South Australia. We also thank Svjetlana Kireta for proofreading this article. K.N.S. PhD was financed by the Adelaide Graduate Research Scholarship, University of Adelaide, South Australia and The Hospital Research Foundation Scholarship.
Author Disclosure Statement
The authors declare no competing financial interests.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.