
Abstract
The role of bone marrow adipocytes (BMAs) in overall energy metabolism and their effects on bone mass are currently areas of intensive investigation. BMAs differentiate from bone marrow stromal cells (BMSCs); however, the molecular mechanisms regulating BMA differentiation are not fully understood. In this study, we investigated the effect of CUDC-907, identified by screening an epigenetic small-molecule library, on adipocytic differentiation of human BMSCs (hBMSCs) and determined its molecular mechanism of action. Human bone marrow stromal cells exposed to CUDC-907 (500 nM) exhibited enhanced adipocytic differentiation (∼2.9-fold increase, P < 0.005) compared with that of control cells. Global gene expression and signaling pathway analyses of differentially expressed genes revealed a strong enrichment of genes involved in adipogenesis, cell cycle, and DNA replication. Chromatin immune precipitation combined with quantitative polymerase chain reaction showed significant increase in H3K9ac epigenetic marker in the promoter regions of AdipoQ, FABP4, PPARγ, KLF15, and CEBPA in CUDC-907-treated hBMSCs. Follow-up experiments corroborated that the inhibition of histone deacetylase (HDAC) activity enhanced adipocytic differentiation, while the inhibition of PI3K decreased adipocytic differentiation. In addition, CUDC-907 arrested hBMSCs in the G0-G1 phase of the cell cycle and reduced the number of S-phase cells. Our data reveal that HDAC, PI3K, and cell cycle genes are important regulators of BMA formation and demonstrate that adipocyte differentiation of hBMSCs is associated with complex changes in a number of epigenetic and genetic pathways, which can be targeted to regulate BMA formation.
Introduction
A
Human BMAs not only share a similar phenotype to those of white and brown adipocytes but also have unique phenotypic characteristics [6,7]. Similar to adipocytes from other depots, lineage-specific differentiation of BMAs is regulated by a number of intracellular signaling pathways induced by hormones and growth factors, which converge on transcriptional regulatory networks that result in changes in DNA landscape, chromatin remodeling, and histone modifications by acetylation, methylation, and phosphorylation [8]. The balance between the opposing activities of histone acetyltransferases and histone deacetylases (HDACs), enzymes involved in histone acetylation and deacetylation, is a determinant of the activation of gene transcription [9].
Histone acetylation is a key mechanism in regulating gene expression and consequently cell differentiation. Inhibition of HDACs causes histone hyperacetylation and activation of target gene transcription, and therefore a number of selective small-molecule HDAC drug inhibitors (HDACi) have been developed with the aim of controlling diverse cellular functions. For example, HDACi exert antiproliferative effects in several human cancer cell models [9,10] and regulate cell differentiation [11], including enhancing osteoblast differentiation of human dental pulp stem cells [12] and BMSCs [13]. Downregulation of HDACs has also been investigated as an approach to regulate adipocytic differentiation of MSCs [14 –16].
Thus, we hypothesized that BMSC differentiation is regulated by an epigenetic mechanism and we employed an epigenetic small-molecule library functional screen to identified compounds, which promoted adipocytic and/or osteoblastic differentiation of human BMSCs (hBMSCs) [14]. In the current study, we focused on CUDC-907 compound, which is a small-molecule dual inhibitor of HDAC and PI3K, as it exerted significant effects on BMA formation and identified its molecular mechanism of action.
Materials and Methods
Compounds
CUDC-907, Abexinostat, and LY294002 (phosphoinositide 3-kinase inhibitors) were obtained from Selleckchem, Inc. (Houston, TX, USA). All compounds were used at a concentration of 500 nM (unless indicated otherwise) and dissolved in dimethyl sulfoxide (DMSO). Control cells were treated with DMSO as a vehicle.
Cell culture
A model for hBMSCs, created by the overexpression of the human telomerase reverse transcriptase gene (hTERT), was used (human mesenchymal stem cell [hMSC]-TERT) [17]. hMSC-TERT expresses known markers of primary hMSCs, exhibits stemness characteristics, and is able to form bone and bone marrow microenvironment when implanted in vivo [17]. Cells were cultured in basal culture medium of Dulbecco's modified Eagle's medium (DMEM) supplemented with
Adipogenic differentiation
Twenty-four hours postincubation with CUDC-907 or the vehicle control, DMEM was replaced with AIM [DMEM supplemented with 10% fetal bovine serum, 10% horse serum (Sigma, St. Louis, MO, USA), 1% Pen-Strep, 100 nM dexamethasone, 0.45 mM isobutyl methyl xanthine (Sigma), 3 μg/mL insulin (Sigma), and 1 μM rosiglitazone (BRL49653; Novo Nordisk, Bagsvaerd, Denmark)]. The AIM was replaced every 3 days. Cells were assessed for qualitative and quantitative adipogenic differentiation on day 7. Cell pellets were collected for total RNA isolation and quantification of messenger RNA expression.
Nile red staining to detect mature adipocytes
Twenty-four hours postexposure to CUDC-907 and on day 7 following adipogenic differentiation, Nile red fluorescence staining and the quantification of mature adipocytes were performed using a stock Nile red solution (1 mg/mL) dissolved in DMSO and stored at −20°C, protected from light. Staining was performed on unfixed cells. Cultured differentiated cells were grown in flat-bottom, 96-well, tissue culture-treated black microplates (Corning, Inc., Corning, NY, USA) and washed once with phosphate-buffered saline (PBS). The dye was added to the cells (5 μg/mL in PBS), followed by incubation for 10 min at room temperature, and washed twice with PBS. The fluorescent signal was measured using the SpectraMax/M5 multimode Spectrophotometer Plate Reader (Molecular Devices Co., Sunnyvale, CA, USA) bottom well scan mode, where nine readings were obtained per well using an excitation wavelength of 485 nm and emission wavelength of 572 nm. Furthermore, fluorescence images were obtained using the FLoid Cell Imaging Station (Life Technologies, Inc., Grand Island, NY, USA). The number of adipocytes was also determined by fluorescence activated cell sorting (FACS) analysis, as previously described [19]. In brief, after trypsinization, the cells were washed with calcium and magnesium-free PBS and fixed with 1% PFA (paraformaldehyde) in 1% BSA (bovine serum albumin). Nile red dye (N3013; Sigma) was added at a final concentration of l00 ng/mL. Following incubation for 5 min at 4°C, the cells were washed, centrifuged, resuspended in 500 μL of PBS, and analyzed using the Navios flow cytometer (Beckman Coulter, Brea, CA, USA). Staining was detected in the green fluorescence channel (FL1). Uninduced cells were used for gating. Data were analyzed using Kaluza flow cytometry analysis software (Beckman Coulter).
Oil Red O staining
Twenty-four hours post exposure to CUDC-907 and on day 7 of adipogenic differentiation, adipogenic differentiation was determined using qualitative Oil Red O staining (ScienCell Research Laboratories, cat No 0843, San Diego, CA, USA) for lipid-filled mature adipocytes. Cells were washed with phosphate-buffered saline (PBS), fixed with fixative solution provided in the kit for 15 min, fixative was removed and cells were washed 3 times with deionized water (diH2O) and subsequently were incubated with freshly-made and filtered Oil Red O staining solution (diluted by adding Oil Red O stock solution 3:2 using diH2O for 15 min at room temperature. Oil Red O solution was removed and cells were washed 5 times with diH2O. Images were acquired using inverted Zeiss microscope (Thornwood, NY, USA).
RNA extraction and complementary DNA synthesis
Total RNA was isolated from cell pellets after 7 days of adipogenic differentiation using a Total RNA Purification Kit (Norgen Biotek Corp., Thorold, ON, Canada) according to the manufacturer's protocol. The concentration and purity of total RNA were measured using the NanoDrop 2000 (Thermo Scientific, Wilmington, DE, USA). Complementary DNA (cDNA) was synthesized from 500 ng of total RNA using the Applied Biosystems™ High-Capacity cDNA Transcription Kit (Applied Biosystems, Inc., Foster City, CA, USA) according to the manufacturer's protocol.
Quantitative real-time PCR
Expression levels of adipocytic-related genes were quantified using the Fast SYBR® Green Master Mix and the Applied Biosystems ViiA 7 Real-Time PCR device (Applied Biosystems, Inc.). Primers are listed in Table 1. Beta-actin, a housekeeping gene, was used for normalization. The ΔΔCT method was used to calculate relative expression and the analysis was performed as previously described [20].
HDAC enzymatic activity assay
HDAC enzymatic activity was measured using the HDAC-Glo™ I/II assay (Promega, Inc., Madison, WI, USA) according to the manufacturer's protocol. This kit measures the relative enzymatic activity for HDAC class I and class II. Briefly, 10,000 cells in a 50 μL volume were seeded per well in a white-walled 96-well plate and incubated with the compound inhibitor at 37°C for 30 min. Trichostatin A (TSA) was used as a positive control (supplied with the kit). HDAC-Glo™ I/II reagent (containing the substrate and the developer reagent) was added and incubated at room temperature for 45 min. Luminescence was measured using the SpectraMax/M5 multimode Spectrophotometer Plate Reader (Molecular Devices Co.).
DNA microarray gene expression profiling
One hundred fifty nanograms of total RNA was labeled using the Low Input Quick Amp Labeling Kit (Agilent Technologies, Santa Clara, CA, USA) and then hybridized to the Agilent Human SurePrint G3 Human GE 8 × 60k Microarray Chip (Agilent Technologies). All microarray experiments were performed at the Microarray Core Facility (Stem Cell Unit, College of Medicine, King Saud University, Riyadh, Saudi Arabia). The extracted data were normalized and analyzed using GeneSpring 13.0 software (Agilent Technologies). Pathway analysis was performed using the Single Experiment Pathway analysis feature in GeneSpring 13.0 (Agilent Technologies), as described previously. The Benjamini–Hochberg false discovery rate multiple testing correction method (P(corr) <0.05) was utilized and a twofold cutoff was used to detect significant differences in transcript abundance.
Western blotting
Total cellular protein was extracted using RIPA lysis solution (Norgen Biotek Corp.). Twenty micrograms of the protein was resolved using Mini-PROTRAN®TGX™ stain-free precast gels and transferred to a polyvinylidene difluoride (PVDF) membrane using the Trans-Blot® Turbo™ Mini PVDF Transfer Pack (Bio-Rad Laboratories, Hercules, CA, USA). Blots were incubated with primary antibodies overnight at 4°C in TBS-Tween (0.05%) with 5% nonfat milk at the designated dilution against acetyl-histone H3 (Lys9) (C5B11) rabbit mAb (1:1,000 dilution; catalog no. 9649); acetyl-histone H4 (Lys8) antibody (1:1,000 dilution; catalog no. 2594); di-methyl-histone H3 (Lys4) (C64G9) rabbit mAb (1:1,000 dilution; catalog no. 9725); total histone 3 (1:2,000 dilution; catalog no. 4499); phospho-Akt (Ser 473) (D9E) XP rabbit mAb (1:2,000 dilution; catalog no. 4060); and against total Akt (1:1,000 dilution; catalog no. 4691). The membrane was subsequently incubated with anti-rabbit IgG-HRP-conjugated antibody (1:3,000 dilution; catalog no. 7074p2). All of the above antibodies were obtained from Cell Signaling Technology (Danvers, MA, USA). Membranes were probed with β-actin mouse mAb at a 1:1,000 dilution (catalog no. 20-372-60072; GenWay Biotech, Inc., San Diego, CA, USA) as a loading control; the membrane was subsequently incubated with anti-mouse IgG-HRP-linked antibody (1:2,500 dilution; GE Healthcare Bio-Sciences, Pittsburgh, PA, USA). Imaging was conducted using the ChemiDoc™ MP Imager (Bio-Rad Laboratories). Band intensity was quantified using the band quantification tool in Image Laboratory 5.0 software (Bio-Rad Laboratories).
Chromatin immunoprecipitation and quantitative PCR validation
hMSC-TERT cells (vehicle [DMSO]-treated or CUDC-907-24-h-treated cells) were fixed with 1% formaldehyde for 15 min and quenched with 0.125 M glycine. Chromatin was isolated by the addition of lysis buffer and disruption with a Dounce homogenizer. Lysates were sonicated and the DNA was sheared to an average length of 300–500 bp. Genomic DNA (Input) was prepared by treating aliquots of chromatin with RNase, proteinase K, and heat for decross-linking, followed by ethanol precipitation. Pellets were resuspended and the resulting DNA was quantified using a NanoDrop spectrophotometer. Extrapolation to the original chromatin volume allowed quantitation of the total chromatin yield. An aliquot of chromatin (30 μg) was precleared with protein A-agarose beads (Invitrogen). Genomic DNA regions of interest were isolated using antibodies against H3K9ac. Complexes were washed, eluted from the beads with sodium dodecyl sulfate buffer, and subjected to RNase and proteinase K treatment. Cross-links were reversed by incubation overnight at 65°C, and ChIP DNA was purified by phenol–chloroform extraction and ethanol precipitation.
For quality assurance, quantitative PCR (qPCR) reactions were carried out in triplicate on specific genomic regions using SYBR Green Supermix (Bio-Rad Laboratories). The resulting signals were normalized for primer efficiency by carrying out qPCR for each primer pair using input DNA.
All chromatin immunoprecipitation-quantitative polymerase chain reaction (ChIP-qPCR) experiments were performed by the Active Motif Epigenetic Service (Active Motif, Carlsbad, CA, USA).
Flow cytometry and cell cycle analysis
hMSCs cells were treated with DMSO control or CUDC-907 (500 nM) for 24 h. Cell pellets were then collected and washed in PBS. Cell pellets were then resuspended in 1 mL of FACS buffer (PBS/0.5% BSA), and then 3 mL of ice-cold 70% ethanol was added to fix the cells for 1 h on ice. Cell pellets were subsequently centrifuged, resuspended in 500 μL of PBS, supplemented with 40 μg/mL RNase A (Sigma) and 50 μg/mL propidium iodide, and analyzed using the Navios flow cytometer (Beckman Coulter). Staining was detected in the fluorescence channel (FL3) and the data were analyzed using Kaluza software (Beckman Coulter).
Time-lapse microscopy
Time-lapse microscopy was performed using hMSC-TERT expressing GFP and an EVOS FL Auto Imaging System equipped with an onstage incubator (Thermo Fisher Scientific Life Sciences, Waltham, MA, USA). Cells were seeded in a Lab-Tek® Chamber Slide (Thermo Fisher Scientific Life Sciences) in the presence of 500 nM CUDC-907 or the vehicle control. Images were captured every 15 min using 40× objectives for a total duration of 48 h.
Statistical analyses
Statistical analyses and the generation of graphs were performed using Microsoft Excel 2010 and GraphPad Prism 6.0 (GraphPad, San Diego, CA, USA). P-values were calculated using the unpaired two-tailed t-test. P-value <0.05 was considered significant.
Results
CUDC-907 promotes adipocytic differentiation of hBMSCs
CUDC-907 is an HDAC and PI3Kα inhibitor, mostly targeting HDAC 1, 2, 3, and 10. It was initially identified by a functional screen of an epigenetic library consisting of 24 compounds based on its ability to promote the BMA differentiation of hBMSCs [14]. To further investigate its mechanism of action, hBMSCs were incubated with CUDC-907 for 24 h at 500 nM and were subsequently induced to adipocyte differentiation. At this concentration, we observed no toxic effects and this dose was used throughout the study. CUDC-907 treatment of hBMSCs enhanced adipocyte differentiation, as evidenced by the greater Oil Red O staining (Fig. 1a–c), and quantitative analysis demonstrated that the number of mature adipocytes identified by Nile red staining was higher (∼2.9-fold increase, P < 0.005, Fig. 1d–f). We also observed a dose-dependent increase in adipogenesis when hBMSCs were treated with CUDC-907, indicating that the increase in adipogenesis is specific to CUDC-907 (Supplementary Fig. S1; Supplementary Data are available online at
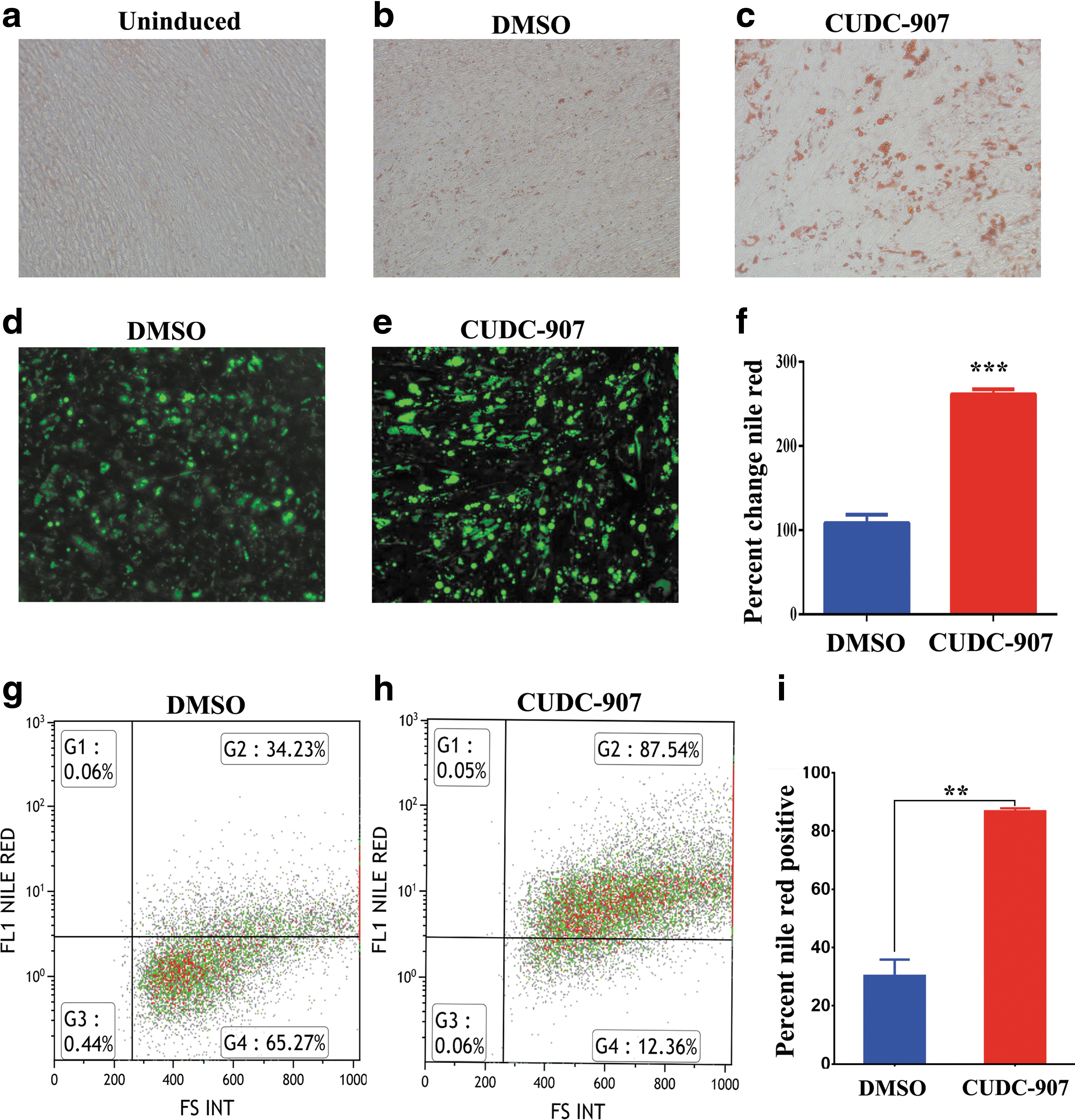
Effects of CUDC-907 on adipocytic differentiation of hMSCs. Representative Oil Red O staining of lipid-filled mature adipocytes on day 7 for uninduced cells
CUDC-907 enhances gene expression in proadipocytic gene networks
To understand the molecular process by which CUDC-907 promotes adipocytic differentiation, global gene expression profiling of hBMSCs exposed to CUDC-907 and induced to adipocytes for 7 days was performed. Hierarchical clustering based on differentially expressed transcripts showed clear separation of the CUDC-907-treated and control cells (Fig. 2a). We identified 1644 upregulated transcripts [>2.0-fold change, P(corr) <0.05; Supplementary Table S1]. A pathway analysis of the differentially expressed genes revealed enrichment for genes associated with several cellular processes of adipocyte differentiation, including cell cycle regulation, DNA replication, and adipogenesis. The pie chart (Fig. 2b) illustrates the top 10 enriched pathways. The adipogenesis pathway is summarized in Figure 2c. The expression of a panel of selected genes from the microarray data based on their involvement in adipogenesis-related processes, that is, AdipoQ, AP2, PPARγ2, ACACB, APOC3, CNTFR, NOG, CDKN1A, and PCK1, was validated by quantitative real-time PCR (qRT-PCR). We found a good concordance between the microarray data and qRT-PCR results (Fig. 2d).
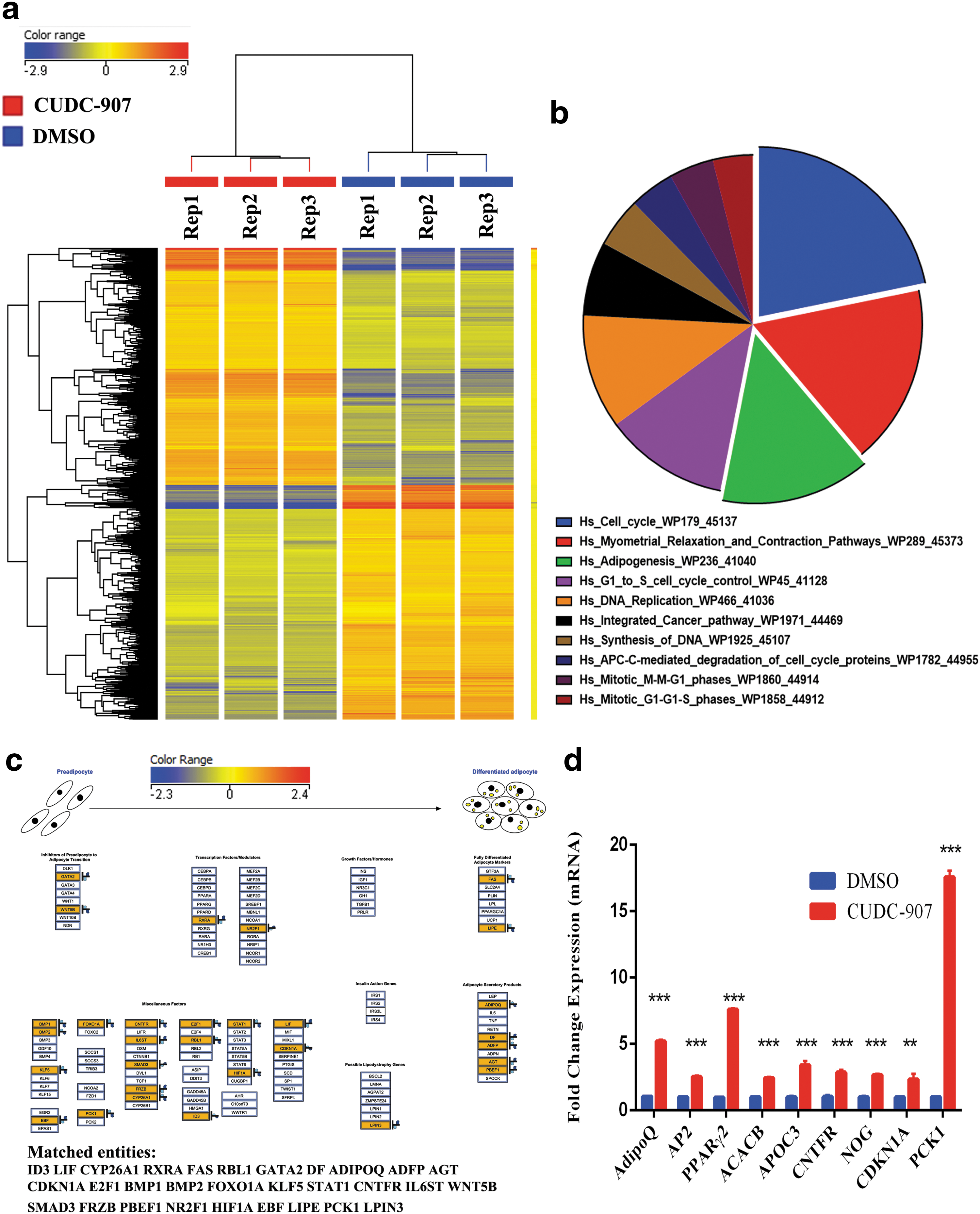
Microarray gene expression profiling of adipocyte-differentiated hMSCs following CUDC-907 treatment.
CUDC-907 promotes adipogenesis through the inhibition of HDAC
To identify the molecular targets of CUDC-907, hBMSCs were exposed to CUDC-907 for 24 h and various histone and PI3K signaling markers were assessed using a western blot analysis. Figure 3a shows a significant increase in H3K9ac, H3K4me2, and H4K8ac histone markers, all known to be associated with actively transcribed genomic regions. There was no significant change in total H3 in CUDC-907-treated hMSCs. In contrast, a decrease in total and phospho-Akt (Ser473) was observed (Fig. 3a). Concordantly, a significant decrease in HDAC activity (∼95% reduction; P < 0.0005) in CUDC-907-treated hBMSCs was observed (Fig. 3b). TSA-treated cells were used as a positive control. We subsequently determined if CUDC-907-associated increase in adipogenesis was mediated by inhibition of HDAC, PI3K, or both. As shown in Figure 3c, a significant increase in adipogenesis was observed in hBMSCs postinhibition with HDAC alone (using Abexinostat), while inhibition of PI3K (using LY294002) diminished adipocytic differentiation. Interestingly, the combination of both HDAC and PI3K inhibitors failed to induce adipogenesis, suggesting additional possible mechanism by which CUDC-907 promotes adipogenesis (Fig. 3c).
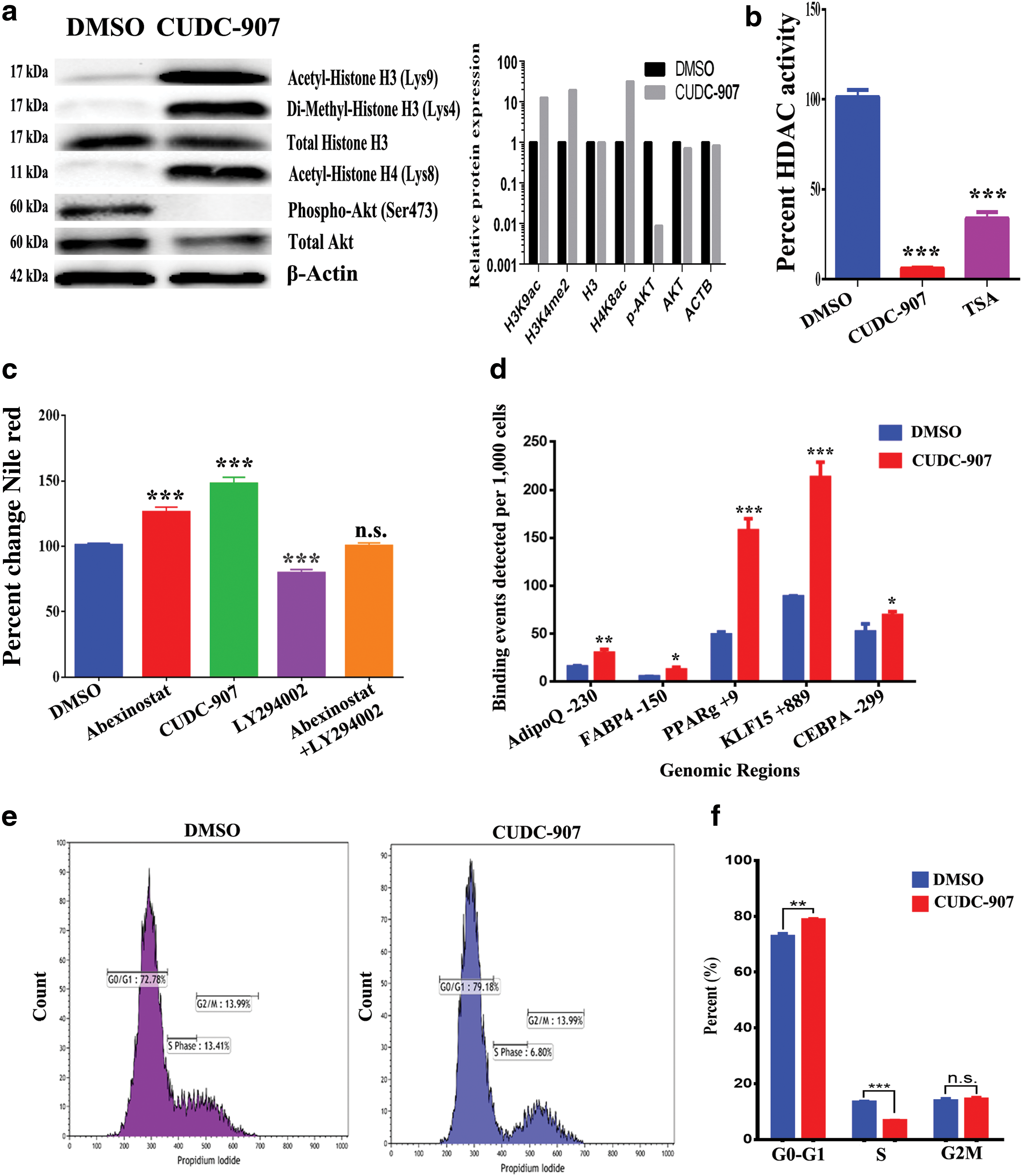
CUDC-907 promotes adipogenesis through the inhibition of HDAC activity.
ChIP-qPCR data reveal significant enrichment in multiple genes related to adipogenesis
We subsequently determined the genomic regions targeted by CUDC-907. hBMSCs were treated with CUDC-907 for 24 h, followed by immunoprecipitation using an antibody recognizing H3K9ac, a histone marker that was markedly increased in CUDC-907-treated hBMSCs (Fig. 3a). The precipitated genomic DNA was subjected to qPCR targeting the promoter regions of AdipoQ, FABP4, PPARγ, KLF15, and CEBPα. As shown in Figure 3d, there was significant enrichment for the H3K9ac marker at the promoter regions of these genes in CUDC-907-treated cells.
CUDC-907 induces cell cycle arrest in hBMSCs
Based on our microarray data and pathway analysis, genes associated with cell cycle were enriched in CUDC-907-treated hBMSCs. A flow cytometry-based cell cycle analysis revealed significant arrest in the G0-G1 phase (78.7% vs. 72.8%) and a reduction in the S phase (6.7% vs. 13.4%) in hBMSCs exposed to CUDC-907 (Fig. 3e, f). Concordantly, time-lapse microscopy indicated a remarkable reduction in cell division in hBMSCs in the presence of CUDC-907 (Supplementary Movies S2) compared with vehicle control (Supplementary Movies S1). Taken together, our data revealed the suppression of cell cycle progression as an additional mechanism by which CUDC-907 promotes adipogenesis.
Discussion
Understanding the molecular mechanism controlling BMA formation is a prerequisite for understanding their biological role in tissue differentiation and overall energy metabolism. In addition, targeting BMA formation represents a novel approach to regulate bone mass and potentially whole body energy metabolism. In the present study, we demonstrated that epigenetic modification employing the small-molecule HDACi CUDC-907 enhances the adipocytic differentiation of hBMSCs through multiple genetic pathways.
The role of epigenetic remodeling in determining differentiation state of stem cells is increasingly recognized and HDACi have been employed as tools to determine the relevance of this mechanism. HDACi have been reported to regulate the self-renewal of embryonic stem cells and MSCs [21 –23]. We have also recently employed functional screening of an epigenetic library of small molecules for potential role in hBMSC differentiation and identified Abexinostat as a potent inducer of adipogenesis through HDAC inhibition and the regulation of transcription of key genes involved in adipogenesis [14]. In the same screen, we identified CUDC-907, a dual inhibitor of HDAC and PI3K, for its role in BMA differentiation.
We observed that CUDC-907 enhanced adipogenesis through the inhibition of HDAC activity. The increase in H3K9ac histone markers likely facilitated the active state of the chromatin structure, increasing the accessibility of transcription factors to adipocyte gene networks and thus inducing adipogenic differentiation [24 –26]. This model is supported by ChIP-qPCR and a gene expression analysis that showed significant upregulation of a number of factors associated with adipogenesis, such as adipoQ, AP2, CNTFR, NOG3, and APOC3 [27 –30], and markers of differentiated adipocytes, for example, phosphoenolpyruvate carboxykinase (PCK1), which is a marker of mature adipocytes and a target gene for PPARγ [31], AP2, and AdipoQ [32].
While our data suggest that the observed effects of CUDC-907 on BMA formation are mediated mainly by HDAC inhibition, inhibition of the PI3K-protein kinase B (PKB)/Akt-PTEN pathway is also involved. PI3K is a principal signaling node associated with regulation of cell proliferation, cell survival, and glucose metabolism. PI3K signaling has been reported to be important for the adipocytic differentiation of adipose-derived hMSCs, and inhibition of PI3K reduces the differentiation of preadipocytes into adipocytes [33]. Hinoi et al. reported a pivotal role for PI3K/Akt and SMAD 1/5/8 signaling during growth differentiation factor-5 (GDF5)-induced brown fat adipogenesis [34]. In addition, PI3K signaling has been linked to several human diseases, including obesity and type 2 diabetes, as it mediates metabolic effects of insulin [35].
We observed that CUDC-907 regulated cell growth due to its effects on cell cycle genes and led to cellular arrest at G0-G1 as well as reduction in S phase. Cell cycle genes have previously been reported to be a target for inducing or inhibiting adipogenesis [36,37] and generally downregulation of cell cycle genes is associated with induction of lineage-specific differentiation. Additionally, CUDC-907 has been reported to regulate proliferation of a number of cancer cell types [38]. In support of this observation, CUDC-907 promoted adipocytic differentiation of the highly proliferative skin-derived stromal cells (SSCs; Supplementary Fig. S3). Therefore, it is plausible that CUDC-907 inhibitory effects on cell cycle contribute to its promoting effects on adipocytic differentiation.
We have employed a well-characterized telomerized cell line as a model for primary human bone marrow MSCs [17]. This cell line exhibits all known characteristics and responses similar to primary MSCs, including the ability to form bone and bone marrow organ in vivo [39]. It is also well suited to perform mechanistic studies performed in the current article due to its stable phenotype, the absence of donor-to-donor variation, and in vitro senescence phenotype that are major confounders when employing primary MSCs [40,41].
Marrow adipose tissue (MAT) is considered a distinct adipocyte compartment compared with white and brown adipose tissue. It is interesting that MAT differentiation induced by CUDC-907 was associated with induction of a network of transcriptional factors similar to those observed in differentiation of extramedullary adipocytes, for example, CEBPα, PPARγ2, and KLF15. Peroxisome proliferator-activated receptor γ (PPARγ2) and CEBPα [42,43] are important regulators of adipogenesis [24,44]. Kruppel-like factors (KLFs) are a family of C2H2 zinc finger proteins that regulate proliferation, differentiation, and development [45]. Mori et al. [46] showed that KLF15 induces adipocytic differentiation through synergy with CEBPα and increases activity of PPARγ2. CEBPα and PPARγ2 act in cooperation with each other to induce a number of adipogenic target genes (eg, AP2 and AdipoQ) that sustain their expression through a positive feedback loop and result in terminal adipogenic differentiation [47 –49]. However, future studies are needed to identify specific factors that are specific for BMA differentiation and to examine the differential effects of CUDC-907 on adipogenesis in different adipocyte compartments. Figure 4 illustrates our working model for the mechanism by which CUDC-907 regulates BMA differentiation, employing an analogy of the current model for extramedullary adipogenesis.
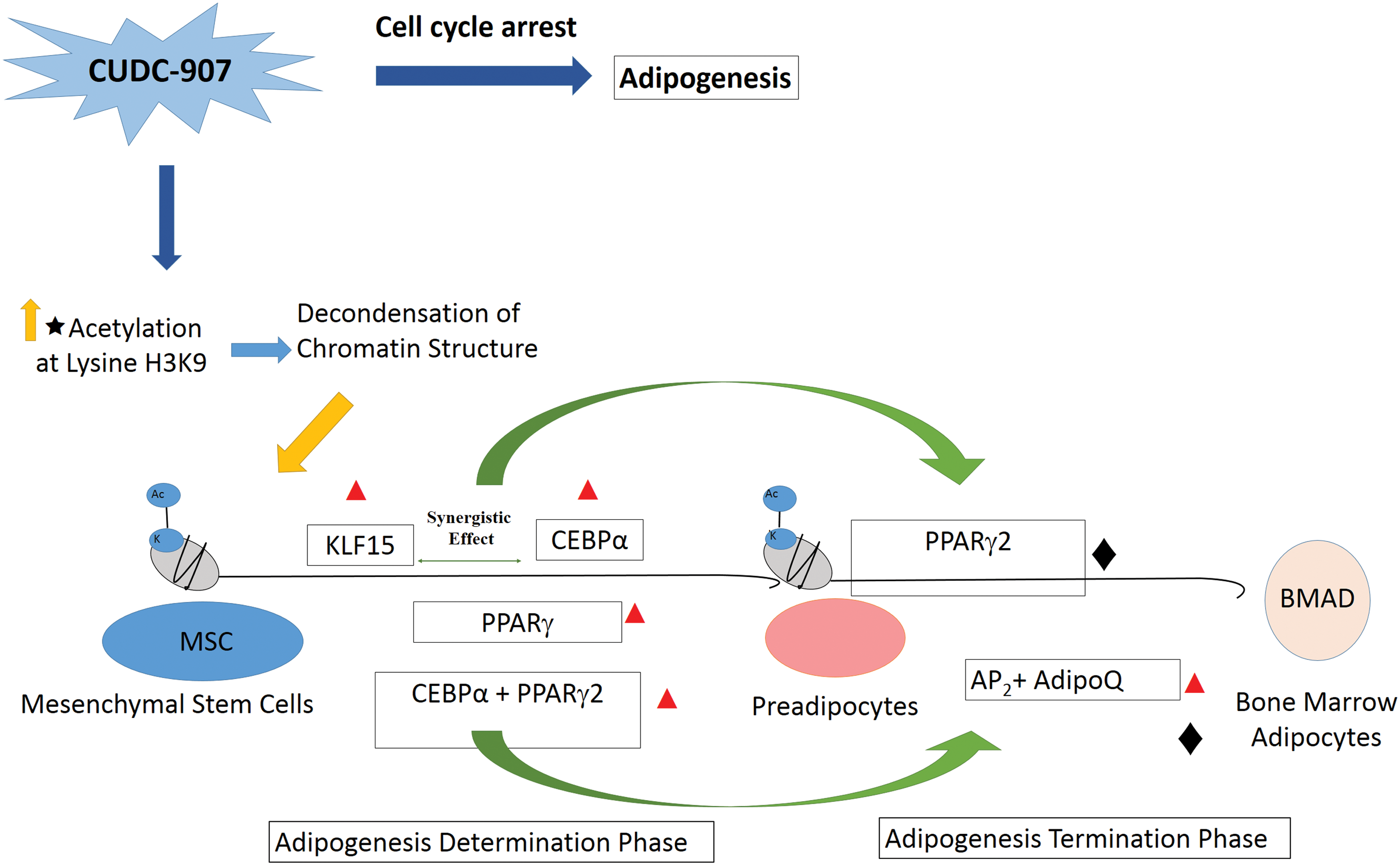
A working model of the molecular mechanisms of CUDC-907 during adipocytic differentiation of hMSCs. In early adipogenesis, the synergistic interaction between KLF15 and CEBPα induces the expression of PPARγ2, which in turn interacts with CEBPα to induce more genes involved in the terminal differentiation of adipocytes, such as AP2
and AdipoQ. Inhibition of the cell cycle promotes adipogenesis. Red triangle identified by ChIP-qPCR; Black diamond differentially expressed in microarray data. BMAD, bone marrow adipocytes. Color images available online at
Footnotes
Acknowledgment
This project was funded by the National Plan for Science, Technology and Innovation (MAARIFAH), King Abdulaziz City for Science and Technology, Saudi Arabia (award no. 11-BIO-1941-02).
Author Disclosure Statement
No competing financial interests exist.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.