
Abstract
Glucocorticoid-induced osteoporosis (GIOP) is a widespread clinical complication due to the common use of glucocorticoids. Excess glucocorticoids induce apoptosis of bone marrow-derived mesenchymal stem cells (BMSCs), which have been shown to play an increasingly important role in the pathogenesis and therapy of osteoporosis. Tetramethylpyrazine (TMP), an extract from one of the most recognized herbs in traditional Chinese medicine (Chuanxiong), has been reported to have antiapoptotic properties. In this study, we tested whether TMP protects rat BMSCs following exposure to glucocorticoids in vitro and in vivo. We treated BMSCs with different concentrations of TMP (50, 100, or 200 μM) and exposed them to 10−6 M dexamethasone (Dex) for 48 h in vitro. Our data showed that TMP inhibited Dex-induced cytotoxicity and protected BMSCs from apoptosis. Interestingly, further results demonstrated that TMP prevented apoptosis in BMSCs by promoting autophagy in an AMP-activated protein kinase (AMPK) and mammalian target of rapamycin (mTOR) pathway-dependent manner. In addition, calcein fluorescence double labeling and microcomputed tomography scanning indicated that 12 weeks of TMP administration augmented bone formation and protected trabecular bone mass in GIOP rats. We also discovered that first-passage BMSCs isolated from the TMP treatment group had a lower rate of apoptosis and a higher light chain 3 (LC3)-II/LC3-I ratio than the GIOP group. Our findings demonstrate for the first time that TMP can protect BMSCs from exposure to excess glucocorticoids by promoting autophagy through AMPK/mTOR pathway and might be an effective agent for the prevention and treatment of GIOP.
Introduction
G
Bone marrow-derived mesenchymal stem cells (BMSCs) are crucial in maintaining the dynamic homeostasis of bone tissue, and much attention has been paid to the role of BMSCs in the pathogenesis of osteoporosis in recent years [6,7]. It is reported that the in vitro effects of glucocorticoids on BMSCs are dose dependent. Low doses of glucocorticoids are able to induce BMSC differentiation into osteoblasts [8,9], whereas high doses lead to apoptosis and cell death [10]. Given that the formation of new bone is primarily dependent on BMSCs and osteoblasts that arise from BMSCs, the negative effects on BMSCs caused by high doses of glucocorticoids is undoubtedly responsible for bone loss in GIOP [11]. Therefore, the recovery of BMSCs in GIOP is essential for the clinical treatment of osteoporosis.
Ligusticum wallichii Franchat (Chuanxiong) is one of the most recognized herbs in traditional Chinese medicine and is widely used for the treatment of ischemic stroke, cerebral infarction, and degenerative diseases of the central nervous system [12]. The bioactive component that is extracted from Chuanxiong, tetramethylpyrazine (TMP), has been reported to have anti-inflammatory, anticancer, antioxidative, and anti-apoptotic effects in many cell types [13 –15]. However, whether TMP can enhance the survival of BMSCs, protect BMSCs from glucocorticoid-induced apoptosis, and be used for the prevention and treatment of GIOP has not been established. Therefore, in this study, we aimed to determine whether TMP could protect BMSCs from the effects of high-dose glucocorticoids and elucidate the underlying mechanisms.
Autophagy is a conserved catabolic process that degrades and recycles dysfunctional organelles, protein aggregates, and intracellular pathogens [16]. During this process, cytoplasmic targets are sequestered within autophagic vacuoles (also called autophagosomes) and fused with lysosomes to form autolysosomes, which are degraded [17]. This physiological process is essential for cell growth, survival, development, and homeostasis and is widely implicated in many pathophysiological disorders [18]. Autophagy can also be activated when the cell is under stressful conditions [19]. The relationship between autophagy and apoptosis is complex and important for cell survival. Previous studies have shown that autophagy can preserve cell viability and protect the cells from apoptosis as a prosurvival mechanism [20,21]. However, autophagy has also been shown to lead to programmed cell death and serve as a self-destructive process in some circumstances [22]. Interestingly, AMP-activated protein kinase (AMPK), which has been shown to be activated by TMP [23], is also an important positive regulator of autophagy. The AMPK and mammalian target of rapamycin (mTOR) pathway is known to be activated in hypoxia, starvation, or many other stresses [24]. Our previous data demonstrated that autophagy in rat BMSCs was induced both in vitro and in vivo by exposure to glucocorticoids [25]. However, whether autophagy and the AMPK/mTOR pathway are involved in the protective effects of TMP on GIOP–BMSCs remains unclear.
In this study, we investigated whether TMP has a protective effect on BMSCs following exposure to glucocorticoids both in vitro and in vivo. Our findings showed that TMP inhibited apoptosis of BMSCs by promoting autophagy through the AMPK/mTOR pathway and rescued the bone loss caused by excess glucocorticoids, which suggests that TMP is a potential target drug for the prevention and treatment of GIOP.
Materials and Methods
Cell culture and treatment
Isolation and primary culture of BMSCs were performed based on previously described methods [26], and cells were characterized using mesenchymal stem cell minimal criteria [27]. Briefly, the bone marrow was flushed out of the tibia and femur of Sprague-Dawley (SD) rats and seeded into 75 cm2 culture flasks with α-minimum essential medium (Thermo Fisher Scientific, Waltham, MA), containing 10% fetal bovine serum and 1% penicillin–streptomycin (all from Gibco Life Technologies, Carlsbad, CA) under conditions of 5% CO2 and 37°C. The medium was changed every 2–3 days to remove nonadherent cells. When the adherent cells were confluent, they were detached using 0.25% Trypsin-ethylenediaminetetraaceticacid (EDTA; Gibco Life Technologies) and passaged at a ratio of 1:2. BMSCs were incubated using a regular culture medium, which contained different concentrations of TMP (0, 50, 100, or 200 μM) for 48 h after exposure to 10−6 M dexamethasone (Dex; Sigma-Aldrich, St. Louis, MO) for 24 h. The doses of TMP were chosen based on previous studies [14,15]. For each experiment, Dex administration continued after TMP treatment. To measure the effects and mechanisms of autophagy in BMSCs, 10 nM of the autophagy promoter rapamycin (Cell Signaling Technology, Danvers, MA), 5 mM of the autophagy inhibitor 3-methyladenine (3-MA; Sigma-Aldrich), 100 nM of the lysosomal fusion inhibitor bafilomycin A1 (Baf; Sigma-Aldrich), and 10 mM of the AMPK inhibitor compound C (Sigma-Aldrich) were added.
Cell viability assay
BMSCs were treated with different concentrations of TMP (0, 10, 50, 100, 200 μM, or 1 mM) for 24 or 72 h to test the toxicity of TMP. A Cell Counting Kit-8 (CCK-8; Dojindo Molecular Technologies, Kumamoto, Japan) was used to measure cell viability in accordance with the manufacturer's instructions, and the cells were measured at a wavelength of 450 and 630 nm using a Thermo Labsystems Multiscan MK-3 enzyme-linked microplate reader (Thermo Fisher Scientific).
Annexin V-fluorescein isothiocyanate/PI double staining
Cell apoptosis was assessed using Annexin V/propidium iodide (PI) double staining. The cells were collected and incubated with fluorescein isothiocyanate (FITC)-conjugated Annexin V in a binding buffer (0.01 M HEPES, 0.14 M NaCl, and 2.5 mM CaCl2, pH 7.4) for 30 min at 37°C in the dark. After incubation, the cells were washed and resuspended in 200 mL of phosphate-buffered saline with 1% fetal calf serum and were incubated with 10 mL of 1 mg/mL PI solution. The Annexin V-positive cells were detected using a FACS Calibur flow cytometer (BD Biosciences, San Jose, CA), and the results were analyzed using the Cell Quest software (BD Biosciences). Annexin V-FITC conjugates were detected with the FL1 channel of the FACS Calibur machine. PI was read on the FL2 channel.
Quantification of caspase-3 activity
The caspase-3 activity was measured spectrophotometrically by quantifying pNA cleavage by caspase-3-specific substrates using a Caspase-3 Assay Kit (Beyotime, Shanghai, China). After the cells were lysed and the samples were incubated with Ac-DEVD-pNA for 2 h at 37°C according to the manufacturer's instructions, the protein concentrations were read at 405 nm.
Transmission electron microscopy detection
Cells were detached from the plates using 0.25% Trypsin-EDTA (Gibco Life Technologies) and fixed with 2% paraformaldehyde/2% glutaraldehyde (Sigma-Aldrich) in 0.2 M of sodium cacodylate buffer (pH 7.4; Sigma-Aldrich). Cell pellets were postfixed with 1% (v/v) osmic acid (Sigma-Aldrich) in sodium cacodylate buffer and stained with 1% uranyl acetate (Amresco, Solon, OH). Following dehydration, the pellets were embedded in Durcupan (Sigma-Aldrich). Ultrathin sections (50 nm) were prepared using an Ultrotome Ultracut S (Leica Microsystems, Wetzlar, Germany), and images were captured using a JEM-1230 transmission electron microscope (JEOL Ltd., Tokyo, Japan).
Immunostaining
Cells were fixed in 4% paraformaldehyde for 15 min, permeabilized with 100% methanol (Sigma-Aldrich) for 10 min, and then incubated with the primary antibody anti-light chain 3 (LC3B; 1:200; Cell Signaling Technology) overnight, followed by the secondary antibody anti-DyLight 594 (1:200; Abcam, Cambridge, MA) for 1 h. Subsequent to incubation with 4′,6-diamidino-2-phenylindole (DAPI; 0.5 μg/mL; KeyGEN, Nanjing, China) for 5 min, the cells were analyzed using a FluoView FV1000 confocal laser scanning microscope (Olympus Corporation, Tokyo, Japan). The percentage of positively stained cells was calculated in three random fields.
Terminal deoxynucleotidyl transferase dUTP nick end labeling assay
Cells were cultured in four-well chamber slides (Thermo Fisher Scientific). Apoptotic cells were identified using terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) staining with an In Situ Cell Death Detection Kit, Fluorescein (Roche Diagnostics, Indianapolis, IN), according to the manufacturer's instructions. The cells were incubated with DAPI (0.5 μg/mL; KeyGEN) for 5 min and analyzed using a FluoView FV1000 confocal laser scanning microscope (Olympus Corporation). Green indicated TUNEL-positive cells, and the percentage of positive cells was calculated in three random fields.
Western blot analysis
After the cells were collected, the proteins were extracted using a lysis buffer (Beyotime); the cell lysates were resolved using sodium dodecyl sulfate–polyacrylamide gel electrophoresis (Bio-Rad Laboratories, Inc., Hercules, CA) and were electrophoretically transferred to nitrocellulose membranes (Bio-Rad Laboratories, Inc.). After blocking with 5% non-fat milk for 1 h at room temperature, the membranes were incubated with primary antibodies, including β-actin (1:10,000; Sigma-Aldrich), anti-LC3B (1:1,000), anti-p62 (1:1,000), anti-phosphorylated (Thr172) and total AMPKα (1:1,000), and anti-phosphorylated (serine 2,448) and total mTOR (1:1,000; all from Cell Signal Technology), followed by horseradish peroxidase-conjugated secondary antibodies (1:2,000; Beyotime). Proteins were visualized using SuperSignal West Dura Chemiluminescent Substrate (Pierce Biotechnology, Inc., Rockford, IL).
Animals and experimental procedures
Fifty 4-month-old female SD rats, weighing 223 ± 18.5 g, were obtained from the Experimental Animal Center at the Fourth Military Medical University in Xi'an, China, and were housed under specific pathogen-free conditions (20°C, 12-h light/12-h dark cycles and 50%–55% humidity) with free access to food and water. Ten female SD rats were used to isolate the BMSCs. Forty female SD rats were intraperitoneally administered either distilled water (as the control group, n = 10) or 2.5 mg/kg prednisolone (Sigma-Aldrich; as the GIOP group, n = 30) daily for 12 weeks. One week after the first administration, the 30 rats in the GIOP group were randomly divided into three experimental groups of 10 rats per group. The rats were injected intraperitoneally with sesame oil (as a vehicle control), 5 or 20 mg/kg body weight of TMP (Sigma-Aldrich) daily for 12 weeks. The doses of TMP we used were chosen based on previous in vivo studies [12,13]. Then, BMSCs were isolated from the control, GIOP, and TMP-treated GIOP groups. There was no significant difference in total body weight among all the groups before the rats were sacrificed. All experimental procedures in animals were approved by the Ethics in Animal Research Committee of the Fourth Military Medical University (permission code 20110405-5).
Calcein double labeling
Calcein double labeling of the bones was performed by intraperitoneally injecting rats with calcein (5 mg/kg) 12 and 2 days before sacrifice. Next, the left femurs were fixed, dehydrated, embedded, and sliced (10 μm). The double calcein green labels were measured on cortical bone near the proximal metaphysis using florescence microscopy (Olympus BX-60, Tokyo, Japan). The mineral apposition rate (MAR), mineralizing surface/bone surface, and bone formation rate (BFR) were measured according to standardized protocols using the Osteomeasure Histomorphometry System (Osteometrics, Decatur, GA). All of these parameters were in accordance with the histomorphometric nomenclature and definitions of the American Society of Bone Mineral Research.
Microcomputed tomography assessment
The distal femurs were scanned using explore Locus SP Pre-Clinical Specimen micro-computed tomography (CT; GE Corp., Fairfield, CT) with an 8 mm resolution, a 50 kV tube voltage, and a 0.1 mA tube current. Reconstruction and three-dimensional (3D) quantitative analyses were determined using the software provided by a desktop micro-CT system (GE Corp.). Similar settings for scans and analyses were used for all of the samples. The scanning regions were confined to the distal metaphysis and extended 2.0 mm proximally from the proximal tip of the primary spongiosa. The following 3D indices in the defined region of interest were analyzed: BMD, structure model index (SMI), trabecular number (Tb.N), trabecular thickness (Tb.Th), trabecular separation (Tb.Sp), and relative bone volume over the total volume (BV/TV, %). The operator who conducted the scan analyses was blinded to the procedure associated with the specimens.
Statistical analyses
Statistical analyses were performed using the SPSS software, version 15.0 (SPSS, Inc., Chicago, IL). Quantitative data are presented as the mean ± standard deviation and compared using a one-way analysis of variance with a post hoc test to determine the significance among the different groups.
Results
TMP inhibits Dex-induced cytotoxicity in BMSCs
Previous studies have shown that high doses of Dex, which is a potent synthetic glucocorticoid, decreased BMSC proliferation and induced apoptosis in vitro [10,28]. In this study, we exposed BMSCs to 10−6 M Dex. Before testing whether TMP had a protective effect on BMSCs in vitro, we first examined the cytotoxic effects of TMP on BMSC viability using a CCK-8 assay. The results showed that TMP (10, 50, 100, and 200 μM, or 1 mM) treatment alone did not affect cell survival (Fig. 1B). Subsequently, we tested the effect of TMP on BMSCs following exposure to excess glucocorticoids. We showed that exposure to 10−6 M Dex alone significantly reduced the number of BMSCs (Fig. 1C). When different concentrations of TMP (50, 100, or 200 μM) were administered to BMSCs for 48 h after exposure to 10−6 M Dex, the survival of BMSCs was significantly improved compared to Dex treatment alone. These data show that TMP inhibits Dex-induced cytotoxicity.
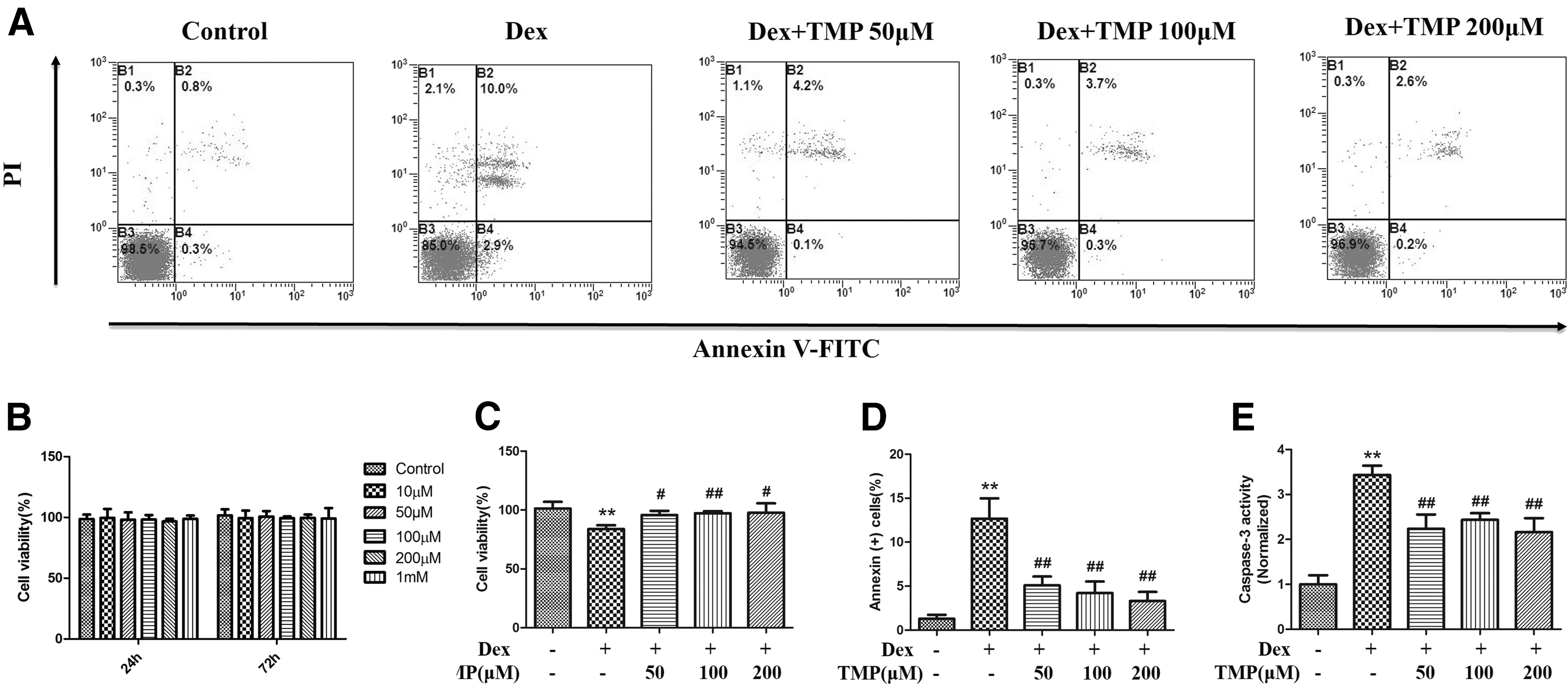
TMP inhibits Dex-induced cytotoxicity and apoptosis in BMSCs. Cells were exposed to 10−6 M Dex for 24 h before treatment with TMP (50, 100, or 200 μM) for 48 h.
TMP protects BMSCs from Dex-induced apoptosis
To investigate the underlying mechanism of TMP on BMSC viability, we examined apoptosis in BMSCs. BMSCs were cultured with 10−6 M Dex in the presence or absence of different concentrations of TMP, and Annexin V-FITC/PI double staining was used to detect cell apoptosis using flow cytometry. We also assessed the caspase-3 activity, which is an executor caspase that is activated by both intrinsic and extrinsic pathways in apoptosis. Our results showed that 10−6 M Dex significantly induced apoptosis in BMSCs; however, TMP treatment significantly decreased the number of annexin-positive cells (Fig. 1A, D) and inhibited the caspase-3 activity in BMSCs (Fig. 1E). These data suggest that TMP partially protects BMSCs from Dex-induced apoptosis.
Autophagy is enhanced by TMP in BMSCs and protects against Dex-induced apoptosis
It has been reported that glucocorticoids can induce autophagy in BMSCs [25]; however, whether TMP can improve the survival of Dex-treated BMSCs by modulating autophagy is unclear. To investigate whether TMP can modulate autophagy in BMSCs, we exposed BMSCs to 10−6 M Dex and treated them with 50 μM of TMP for 48 h. Autophagy was morphologically assessed by the formation of autophagic vacuoles, which are also called autophagosomes. Thin-section electron microscopy analysis identified that there was more autophagic vacuoles in the 50 μM TMP group than the Dex group (Fig. 2A). When autophagy occurs, microtubule-associated protein LC3 is recruited and aggregates in the cytoplasm; therefore, the expression of LC3 and an increased level of LC3-II are important markers of autophagy [29]. Immunofluorescence analysis showed that 50 μM TMP group increased the percentage of LC3-positive cells compared with the Dex group (Fig. 2B). Western blot analysis also showed that there was an increased LC3-II:LC3-I ratio in the 50 μM TMP group compared with the Dex group (Fig. 2D). These results demonstrate that TMP enhances the autophagic activity in BMSCs exposed to Dex.
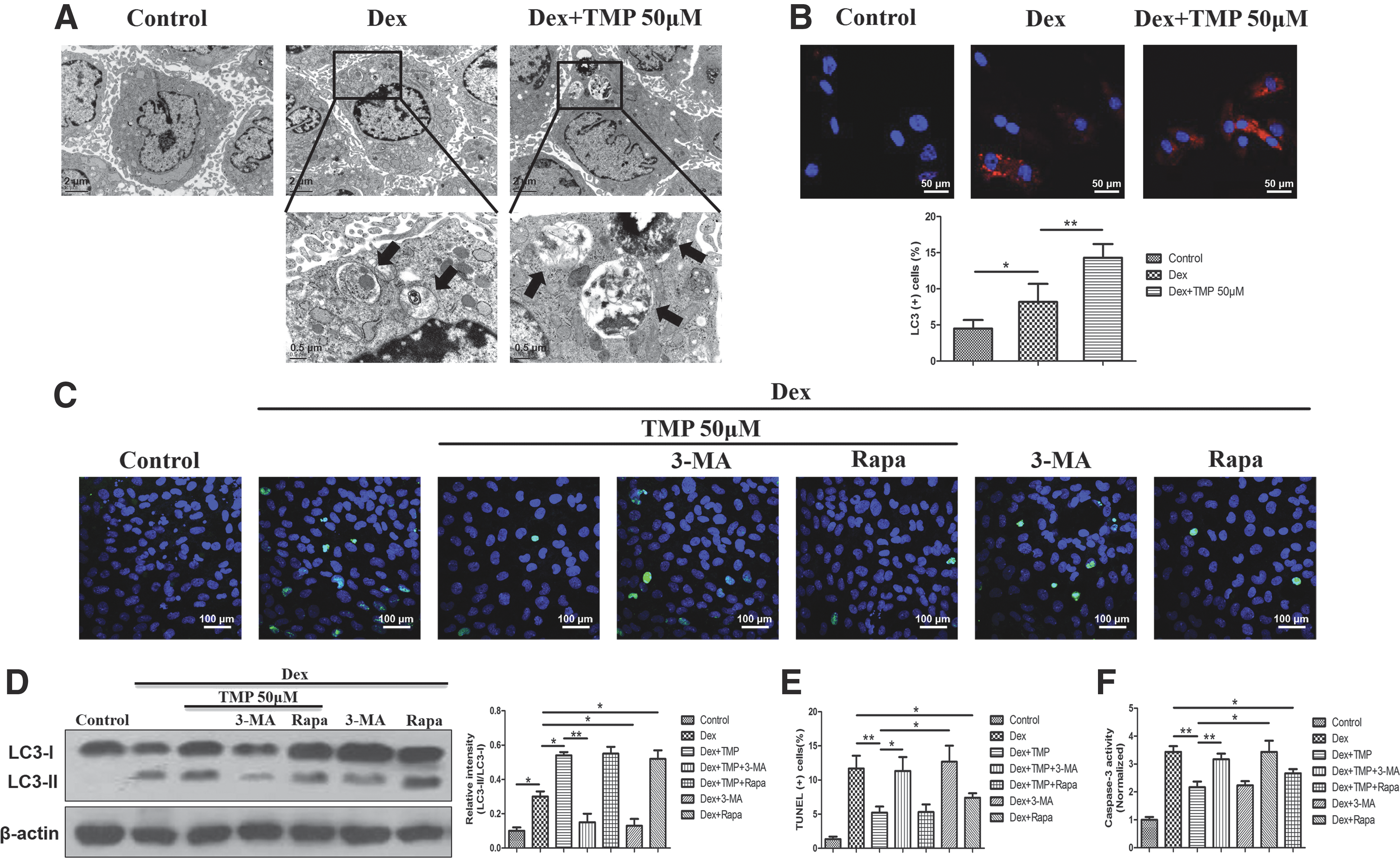
TMP inhibits apoptosis by promoting autophagy in Dex-treated BMSCs.
To further determine whether autophagy was involved in the protection of TMP against apoptosis in Dex-treated BMSCs, 3-MA (inhibitor of autophagy) or rapamycin (activator of autophagy) was used to observe autophagy and apoptosis in BMSCs. BMSCs were treated with 50 μM TMP for 48 h after exposure to 10−6 M Dex in the presence or absence of 3-MA and rapamycin. Then, we performed western blot, TUNEL, and caspase-3 assays. We also detected the effect of 3-MA and rapamycin alone in the presence of Dex. The results displayed that 3-MA significantly reduced the LC3-II:LC3-I ratio and abrogated the antiapoptotic ability of TMP-treated BMSCs as demonstrated by an increase in TUNEL-positive cells and higher caspase-3 activity compared with the 50 μM TMP group (Fig. 2C–F). Furthermore, rapamycin did not further enhance autophagy or attenuate Dex-induced apoptosis in the 50 μM TMP-treated BMSCs. Also, rapamycin alone could significantly promote autophagy and inhibit Dex-induced apoptosis in BMSCs, which indicated that the TMP-enhanced autophagy in BMSCs under Dex exposure might be mediated in the same way as rapamycin did. These results suggest that TMP-induced autophagy is a possible mechanism for the protective effects of TMP against Dex-induced apoptosis in BMSCs.
TMP promotes autophagic activity through AMPK/mTOR pathway
Given that our previous study has shown that 10−6 M Dex can activate autophagic flux in BMSCs [25], we want to know whether TMP alone is able to induce autophagy without exposure to Dex in BMSCs and whether the autophagosome accumulation and LC3 induction promoted by TMP are due to autophagic flux impairment. We used Baf, which blocks the fusion of autophagosomes with lysosomes, and assessed the protein levels of LC3 and p62 to evaluate autophagic flux. Cells were treated with or without 10−6 M Dex and 50 μM TMP in the presence or absence of 100 nM Baf. Western blot analysis showed that TMP alone was not able to induce autophagy without Dex as TMP treatment could not markedly increase the LC3-II:LC3-I ratio or decrease the p62 level compared with the control group. Baf could significantly increase the LC3-II:LC3-I ratio and decrease the p62 level no matter TMP was added or not. The TMP + Dex + Baf-treated group showed a higher ratio of LC3-II:LC3-I and a lower p62 level than the TMP + Dex-treated group (Fig. 3A). These results indicate that TMP only promotes autophagy when BMSCs are exposed to Dex and this effect is not caused by impairing autophagic flux.

TMP promotes autophagic flux through AMPK/mTOR pathway.
To investigate which pathway mediated TMP-induced autophagy activation under exposure to Dex, we concentrated on the AMPK/mTOR signaling pathway because of its essential role in regulating autophagy [24,30]. We determined the expression level of phospho-AMPK, total AMPK, phospho-mTOR, and total mTOR in BMSCs following administration of the AMPK inhibitor compound C. The western blot results showed that AMPK phosphorylation was upregulated and mTOR phosphorylation was downregulated in the 50 μM TMP group compared with the Dex group, whereas the expression of AMPK and mTOR did not significantly change among the groups. Compound C attenuated the effects of 50 μM TMP on both AMPK phosphorylation and mTOR phosphorylation. Moreover, the LC3-II/LC3-I ratio decreased following compound C treatment in the 50 μM TMP-treated BMSCs, which suggested that the autophagic activity was blocked (Fig. 3B). These results demonstrate that the AMPK/mTOR pathway mediates TMP-induced autophagy in BMSCs following exposure to Dex.
TMP augments bone formation and improves bone mass in GIOP rats
To determine whether TMP exerted protective effects on GIOP BMSCs in vivo, a GIOP rat model was used to test the biological function of TMP. Dynamic bone histomorphometry parameters were calculated using calcein double labeling to evaluate the effect of TMP on bone formation in GIOP rats. Calcein was injected twice (12 and 2 days) before the rats were sacrificed. As shown in Fig. 4A, the GIOP group exhibited a significant decrease in interlabel distance compared with the control group. MAR, mineralizing surface/BS and BFR were significantly increased in the TMP-treated groups (5 or 20 mg/kg) compared with the GIOP group (Fig. 4B).
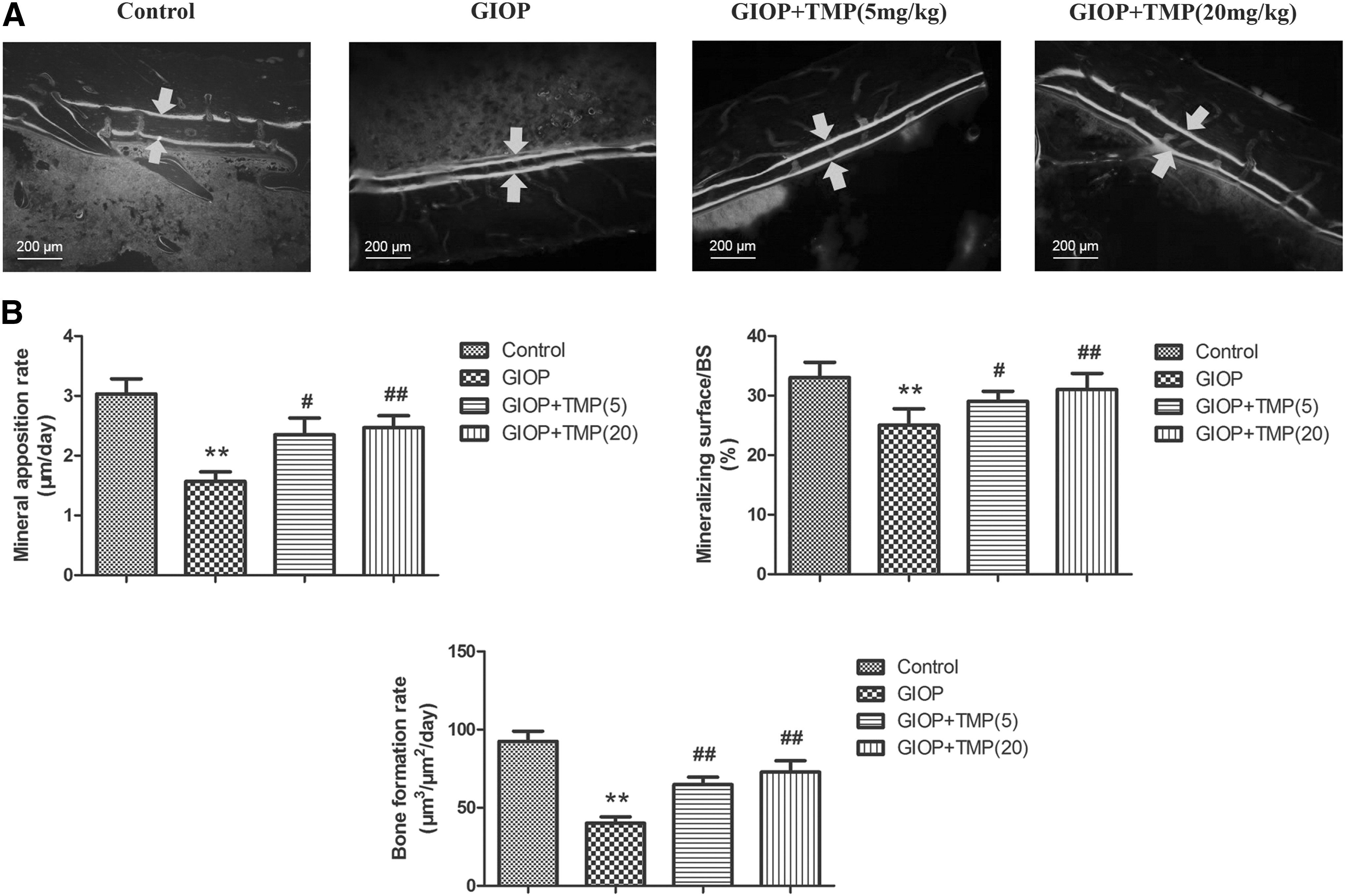
TMP increases the MAR and BFR in GIOP rats.
To further evaluate the effect of TMP on bone mass and microarchitecture in the GIOP rats, distal femurs were collected and scanned using micro-CT assessment. The analyses of the trabecular bone of the distal femur showed that excess glucocorticoids significantly reduced the bone mass and deteriorated the bone microarchitecture, as indicated by a decrease in BMD, Tb.N, Tb.Th, and BV/TV and an increase in SMI and Tb.Sp in the GIOP rats (Fig. 5A, B). Treating the GIOP rats with 5 or 20 mg/kg of TMP partially reversed the effects on these bone parameters and improved the microarchitecture of the trabecular bone in the distal femur. These results demonstrate that TMP can augment bone formation and protect the trabecular bone mass from excess exposure to glucocorticoids.

TMP displays protective effects on trabecular bone mass in GIOP rats.
TMP protects GIOP-BMSCs undergoing apoptosis and induces autophagy
To determine whether TMP could affect BMSC apoptosis and autophagy in GIOP state, we isolated BMSCs from GIOP rats with or without 12 weeks of TMP treatment, and investigated apoptosis and autophagy in the first passage cells. The GIOP model was evaluated using micro-CT, and as shown in Fig. 5, BMD was decreased and Tb.Sp was significantly increased compared with the untreated control group. The results showed that the apoptosis rate of BMSCs in the GIOP rats was increased compared with the untreated control group. However, 5 or 20 mg/kg of TMP significantly decreased the number of apoptotic BMSCs (Fig. 6C–E). As shown in Fig. 6A, we observed more autophagosomes in the 5 and 20 mg/kg TMP treatment groups than in the GIOP group. Furthermore, the LC3-II:LC3-I ratio was significantly higher in both TMP groups compared with the GIOP group (Fig. 6B). These observations are partially consistent with our in vitro data, which reveal that BMSCs respond to TMP in vivo, and TMP treatment promotes autophagy and partially rescues BMSCs from glucocorticoid-induced apoptosis.

The effects of TMP on the BMSC autophagy and apoptosis in GIOP state. BMSCs were isolated from GIOP rats with or without 12 weeks of 5 or 20 mg/kg TMP treatment and analyzed at the first passage.
Discussion
BMSCs have self-renewal properties and give rise to osteoblasts during the process of bone formation; therefore, they are essential for the maintenance of bone tissue homeostasis [31]. Our previous study revealed that the proliferative ability of both BMSCs derived from GIOP rats and Dex-treated BMSCs was impaired [25]. Excess glucocorticoids induce negative effects on BMSC survival and function. These defective BMSCs result in a reduction of osteogenesis and bone formation, and finally contribute to bone loss in GIOP [32]. In this study, we used a small molecular compound to protect the defective BMSCs to find a new target drug for preventing and treating GIOP.
Dex is a potent synthetic glucocorticoid steroid drug and is widely used in cell culture protocols to differentiate BMSCs into mature cells [33]. However, the direct effects of Dex on BMSC proliferation and apoptosis have been reported to be diverse, depending on the dose and species used. Gao et al. showed that high doses of Dex induced apoptosis in mice BMSCs and this effect plateaued at 10−6 M [10]. Our previous study also suggested that 10−6 M Dex decreased proliferation in rat BMSCs in vitro [25]. Therefore, in this study, we used 10−6 M Dex to generate an in vitro model of excess glucocorticoid exposure stress. TMP has been demonstrated to suppress arsenic nephrotoxicity by protecting against apoptosis in human renal proximal tubular epithelial cells and protect dopaminergic neurons against methyl-4-phenyl-1,2,3,6-tetrahydropyridine/1-methyl-4-phenylpyridinium induced neurotoxicity by inhibiting apoptosis [15,34]. Our data showed that Dex-induced cytotoxicity and apoptosis were partially inhibited by TMP treatment in BMSCs, but not in a dose-dependent manner in the range of doses we used. Based on the significant antiapoptotic effects of TMP in our study, one possible reason we assume is that the lowest dose of TMP we used inhibits apoptosis to a very large degree that higher doses cannot be more effective in this condition. These results demonstrate that TMP, in part, improves BMSC survival following high-dose Dex treatment. This finding suggests that TMP can potentially relieve the damage from glucocorticoid exposure and rescue the bone loss in GIOP.
The autophagic process occurs at basal levels in all cell types and is important both for regular cellular quality control and for responding to environmental and internal stressors. There is growing consensus that autophagy can protect cells and help cells overcome stressor challenges. Furthermore, autophagy has recently been implicated in skeletal growth and homoeostasis maintenance [35,36]. Autophagy deficiency in osteoblasts could increase oxidative stress and the secretion of the receptor activator NFKB1, which favors the generation of osteoclasts and reduces mineralization capacity [37]. Onal et al. reported that the conditional deletion of Atg7 in osteocytes, which suppressed autophagy in a cell-specific manner, resulted in a significant decrease in cortical bone mass in mice [38]. Compared with terminally differentiated bone cells, autophagy in BMSCs has not been extensively studied. Recently, high levels of basal autophagy have been reported in human mesenchymal stem cells, which are significantly reduced during differentiation to osteoblasts, suggesting that autophagy is also important for the maintenance of stem cell populations [39]. For BMSCs, the ability to survive under stressful conditions, such as exposure to excess glucocorticoids, may be critical. Autophagy serves as a stress adaptation that suppresses apoptosis under certain circumstances. Sanchez et al. reported that activation of autophagy provides needed energy and secretes antiapoptotic factors to promote survival in mesenchymal stem cells during long periods of serum deprivation [40]. Our previous data also showed that glucocorticoid-induced autophagy could protect BMSCs against apoptosis [25]. In addition, Zhang et al. reported that Atorvastatin effectively promoted autophagy and reduced apoptosis in mesenchymal stem cells during both hypoxia and serum deprivation, which suggests that certain drugs can benefit cells by activating autophagy [41]. This study indicates that TMP promotes an autophagic activity in BMSCs exposed to Dex and prevents glucocorticoid-induced apoptosis at the same time, and the antiapoptotic effect of TMP is partially mediated by its ability to modulate autophagy. However, TMP is not able to increase basal autophagy in BMSCs and its action seems strictly linked to stress situations like glucocorticoid treatment. These findings imply that TMP may potentially be a safe and specific drug for relieving side effects of glucocorticoids such as GIOP. On the other hand, current researches demonstrate that autophagy can be a double-edged sword in the pathological process of diseases. Uncontrolled excessive promotion of autophagy may contribute to cell death. Thus, without doubt, we should try to control the autophagic activity within a moderate range for therapeutic benefit very carefully. It is a challenge and requires further investigation.
Many signaling pathways are involved in autophagy regulation, including PtdIns3K-Akt-mTORC1, AMPK, p53, and Bcl-2 protein family [42]. It has been reported that AMPK could be activated by TMP. So we detected the expression levels of the AMPK/mTOR pathway to further clarify the mechanisms underlying TMP-induced autophagy. We used a pharmacological inhibitor of the pathway, and the results suggested that autophagy promoted by TMP in BMSCs was partially dependent on the AMPK/mTOR pathway. However, other mediators involved in the TMP regulation of autophagy activation could not be excluded.
GIOP rats share similar features as GIOP patients who receive long-term glucocorticoid therapy and experience significant bone loss due to excess glucocorticoid exposure, which primarily results from trabecular bone loss [43,44]. Therefore, trabecular bone microarchitecture and the BFR are appropriate predictors of glucocorticoid-induced bone loss and bone quality deterioration. We calculated both dynamic and static bone histomorphometry parameters and confirmed that TMP could ameliorate trabecular microarchitecture and prevent bone mass decrease in GIOP rats and TMP might be a good candidate for the prevention and treatment of GIOP. In addition, we discovered that the first-passage BMSCs isolated from the GIOP rats after 12 weeks of TMP treatment had a lower rate of apoptosis and a higher LC3-II/LC3-I ratio than the GIOP group, which suggests that the BMSCs respond to TMP under a disease state in vivo. The promotion of autophagy and the protection of BMSCs from apoptosis are the possible mechanisms of TMP treatment in GIOP rats.
In conclusion, we showed, for the first time, that TMP could prolong BMSC survival following exposure to excess glucocorticoids by preserving cell viability and inhibiting apoptosis through the AMPK/mTOR-dependent promotion of autophagy in vitro. In vivo administration of TMP increased bone formation and prevented bone mass decrease in GIOP rats. Autophagy enhancement and protection of BMSCs against apoptosis in a GIOP state may be responsible for the antiosteoporosis effects of TMP. Our findings suggest that TMP treatment and regulation of BMSCs may be a promising strategy to prevent and treat GIOP.
Footnotes
Acknowledgments
This work was supported by the Program for Changjiang Scholars and Innovative Research Team in University (no. IRT13051) and National Natural Science Foundation of China (81572192 and 81472043).
Author Disclosure Statement
No competing financial interests exist. No benefits in any form have been or will be received from a commercial party directly or indirectly by the authors of this article.