
Abstract
Pluripotent embryonic stem cells (ESCs) are unusual in that geminin has been reported to be essential either to prevent differentiation by maintaining expression of pluripotency genes or to prevent DNA rereplication-dependent apoptosis. To distinguish between these two incompatible hypotheses, immune-compromised mice were inoculated subcutaneously with ESCs harboring conditional Gmnn alleles alone or together with a tamoxifen-dependent Cre recombinase gene. Mice were then injected with tamoxifen at various times during which the ESCs proliferated and differentiated into a teratoma. For comparison, the same ESCs were cultured in vitro in the presence of monohydroxytamoxifen. The results revealed that geminin is a haplosufficient gene that is essential for ESC viability before they differentiate into a teratoma, but once a teratoma is established, the differentiated cells can continue to proliferate in the absence of Gmnn alleles, geminin protein, and pluripotent stem cells. Thus, differentiated cells did not require geminin for efficient proliferation within the context of a solid tissue, although they did when teratoma cells were cultured in vitro. These results provide proof-of-principle that preventing geminin function could prevent malignancy in tumors derived from pluripotent cells by selectively eliminating the progenitor cells with little harm to normal cells.
Introduction
M
Teratomas are benign tumors that consist of a solid mass of cells haphazardly organized into tissues representing at least two and usually all three embryonic germ layers. Teratocarcinomas are malignant teratomas from which cancer stem cells (CSCs) have been isolated (termed “embryonal carcinoma cells”(ECCs)). Except for the fact that ECCs are multipotent rather than pluripotent, they are remarkably similar to ESCs [8]. Pluripotent stem cells also have been derived from primordial germ cells (PGCs), the precursor of oocytes, spermatocytes, and germ cell neoplasias, and their characteristics are essentially the same as ESCs [9]. Therefore, as development proceeds, pluripotent cells have a demonstrable probability of becoming CSCs.
In fact, ESCs share many characteristics with CSCs. ESCs are pluripotent, because they can differentiate into cells derived from all three embryonic germ layers (endoderm, mesoderm, and ectoderm). CSCs are multipotent because they can produce all the cell types that comprise the cancer from which they were derived. Both ESCs and CSCs are stem cells because they retain these properties during proliferation (termed self-renewal). Both CSCs and ESCs exhibit rapid proliferation, lack of contact inhibition, genomic instability, unique gene expression signatures, and the ability to form tumors [10 –14]. Therefore, ESCs provide a model for identifying gene targets that might prove useful in selectively killing CSCs with little or no harm to normal cells. One potential target is geminin.
Geminin is a protein unique to the multicellular eukarya that helps restrict genome duplication to once per cell division by preventing assembly of prereplication complexes at DNA replication origins [15 –17]. Geminin also has less well-characterized roles in gene expression and cell differentiation [18,19]. Therefore, it is not surprising that geminin is essential at the beginning of mammalian development. Ablation of geminin alleles (Gmnn) in a mouse zygote results in excess DNA replication and termination of development between the morula and blastocyst stages [20 –22]. Gmnn ablation in newly implanted blastocysts arrests epiblast development [22,23], but the effects of Gmnn ablation at later stages in development are less dramatic, suggesting that the importance of geminin diminishes as development continues [24 –26].
Remarkably, the role of geminin in totipotent and pluripotent cells has not been resolved. Some studies concluded that geminin is required in preimplantation embryos and ESCs to maintain expression of genes necessary for pluripotency [20,27,28]. Other studies concluded that geminin is not required to either maintain or exit pluripotency [22,29], but to prevent aberrant DNA replication from inducing apoptosis [21,22,30]. In one study, depletion of geminin in ESCs undergoing self-renewal in vitro triggered a second round of nuclear DNA replication before the first round is completed (termed DNA rereplication), which resulted in incomplete chromosome duplication, DNA damage, a DNA damage response, and apoptosis, but once ESCs differentiated, they were no longer dependent on geminin for viability [22]. Pluripotent and totipotent cells appear unique in this respect because depletion of geminin in mouse or human embryonic fibroblasts and primary human mammary epithelial cells induces senescence instead of DNA rereplication [22,31 –33], and depletion of geminin in trophoblast stem cells induces terminal differentiation into nonproliferating giant cells [19]. Remarkably, geminin is also essential to prevent DNA rereplication-dependent apoptosis in cells derived from human cancers, but not in cells derived from normal tissues [15,16,34,35] because initiation of DNA replication is restricted by multiple cell cycle events [36].
The issues outlined above led us to examine whether geminin is essential for pluripotent cell viability or for maintaining pluripotency (ie, preventing differentiation) in vivo, and whether or not the requirement for geminin in pluripotent stem cells might be used to selectively eliminate CSCs without harming normal cells. To these ends, nude mice were inoculated with mouse ESCs harboring conditional Gmnn alleles and a tamoxifen-dependent Cre recombinase, and then the effects of tamoxifen on formation and maintenance of teratomas were analyzed. The results confirmed that geminin is essential to prevent DNA rereplication-dependent apoptosis during proliferation in vitro, and extended these studies in vivo by demonstrating that geminin is essential to prevent ESC death as they differentiate into a teratoma. Remarkably, geminin was not essential for either proliferation or viability of nonpluripotent cells within the teratoma, although it remained essential for their cultivation in vitro. Therefore, we conclude that geminin is essential for ESC viability in vivo as well as in vitro, and suggest that selective inhibition of geminin could deplete the CSCs responsible for a germ cell neoplasia with little, if any, harm to normal cells, thereby converting a malignant cancer into a benign tumor.
Materials and Methods
Allografts
Preparation and culture of ESCs harboring either floxed Gmnn alleles [Gmnn(fl/fl)] or floxed Gmnn alleles and a tamoxifen-dependent Cre recombinase [Gmnn(fl/−);ErCre/+] were done as previously described [22]. Dr. Warren Wu and Dr. Todd MacFarlane (NICHD/NIH) provided the ErCre/+ ESCs. Immune-deficient, female Balb/c nude mice 6–8 weeks of age (Charles River Laboratories) were inoculated subcutaneously into each rear flank with 1 × 106 ESCs.
ESCs were harvested by trypsinization (0.05% trypsin with 0.53 mM EDTA) for 5 min at 37°C. The reaction was stopped by diluting the trypsin (1:5) in a fresh ESC culture medium. Cells were pelleted by centrifuging at 600g for 5 min at an ambient temperature. The supernatant was aspirated, and the cells were washed with 20 mL of heparinized saline (10 U/mL) by gently pipetting cells up and down to remove clumps. Cells were pelleted and then resuspended in ice-cold Dulbecco's modified Eagle's medium (DMEM) with high glucose, and supplemented with 10% heat-inactivated fetal bovine serum and 100 U/mL each of penicillin and streptomycin to a final concentration of 107 cells/mL [37]. Final concentration of ESCs in 50% Matrigel/Cultrex was 5 × 106 cells/mL. A syringe was loaded with 0.3 mL ice-cold cell suspension and then a 25-gauge hypodermic needle was attached, air bubbles were released, and 0.2 mL containing 1 × 106 ESCs was injected subcutaneously [6].
Tamoxifen powder was dissolved in 95% sunflower seed oil and 5% ethanol at a final concentration of 10 mg/mL [38], and either 0.2 mg tamoxifen per gram body weight or the tamoxifen vehicle was injected intraperitoneally.
Mice were weighed every day during the inoculations and every 3–4 days thereafter until the end of the study. At the termination of each time course, tumors were excised and their weights recorded. All animal studies were conducted in accordance with accepted standards of humane animal care under protocols approved by the NICHD Animal Care and Use Committee (NICHD Animal Studies Proposal No. 11-056).
Histochemical and immunological staining of tumor tissue
Animals were sacrificed and their tumors excised. Half of the tumor was fixed in 10% neutral-buffered formalin (Sigma Life Sciences), and half was snap frozen on dry ice for use in polymerase chain reaction (PCR) and western immunoblotting analyses. Histochemical and immunological staining of tumor tissue was carried out by the Laboratory Animal Science Program, National Cancer Institute (Frederick, MD). The fixed portions were sectioned serially, so that consecutive sections could be stained with hematoxylin and eosin (H&E) to identify specific tissue types, or stained with primary antibodies against either the OCT4 (C30A3;, Cell Signaling Technology) or Ki67 (ab16667; Abcam) protein. The secondary antibody was anti-rabbit (Leica Biosystems' Bond Polymer Refine Detection Kit DS9800). The sections were then stained with 3,3′-diaminobenzidine.
Quantitative image analysis was done on stained sections that were scanned into a digital format by an Aperio Scanscope XT (Leica, Vista, CA) at 20× magnification. The digital images were first annotated by a board-certified veterinary pathologist using Aperio's ImageScope software (version 12.1.3) and then processed using the Aperio Image Analysis software and an algorithm designed to quantify nuclear staining. The fraction of stained cells was based on the intensity of the stain and its nuclear localization.
Conventional PCR assays
Genomic DNA was extracted from 25 to 50 mg tumor using a Qiagen “DNeasy blood and tissue kit.” DNA concentrations were measured with a Qbit 3.0 fluorometer (ThermoFisher) using the high-sensitivity assay as per the manufacturer's instructions. From 10 to 50 ng DNA was amplified using DNA primers for the Gmnn sense (CTGAAGAGGACCTGAGTTCAGTTC) and antisense (CAACCCCTTTCTCCAGTGATGTTC) strands, as previously described [22]. PCR conditions were 94°C/5 min; then 35 cycles of 94°C/30 s, 59°C/1 min, and 72°C/1.5 min, followed by 72°C/7 min. PCR products were fractionated by electrophoresis in 1.5% agarose gels. DNA amplicons were 1,131 bp for floxed Gmnn alleles, 950 bp for wild-type Gmnn alleles, and 305 bp for ablated Gmnn alleles.
Real-time PCR assays
ThermoFisher TaqMan real-time PCR assays were carried out according to the manufacturer's instructions. Each assay contained 20 ng mouse genomic DNA, either a probe for mouse Gmnn exon 4 (Mm00394575_cn) or Gmnn exon 5 (Mm00394577_cn), and a probe for Tfrc as a reference gene (No. 4458366). The fluorescent signals were quantified using a ViiA 7 PCR machine (ThermoFisher/Applied Biosystems). The number of copies per cell of Gmnn exon 4 and exon 5 in each tumor was calculated using “Copy Caller” software (v2.1; ThermoFisher) and normalized to two copies of the Tfrc gene using the delta/delta CT method.
Western immunoblotting
For ESCs, 5 × 105 cells were resuspended in 50 μL of 2× Laemmli sample buffer (LSB) containing 4% sodium dodecyl sulfate (SDS) and sonicated for 5 s at 4°C. For tumors, frozen tissue was ground up to a fine powder using a mortar and pestle placed on dry ice, and the particles were transferred to a prechilled microfuge tube containing 500 μL LSB/50 mg tissue. The tissue was sonicated six times for 5 s at 4°C. The tube was placed on ice between intervals to prevent heating the sample. One aliquot was subjected to western immunoblotting by fractionating the proteins in 4%–12% gradient NuPAGE gels (Novex/Life Technologies) using NuPage MOPS SDS running buffer and constant 200 V. Transfer of proteins to nitrocellulose membranes was performed as per the manufacturer's instructions. Geminin was detected using rabbit polyclonal antibody raised against amino acids 1–209 (Santa Cruz Biotechnology, Inc.). ECL substrate (Western Bright Sirius; Advansta) was applied to each blot, and the geminin signal was quantified using an Amersham Imager 600 (GE Biosciences) provided with Image Quant software. Separate aliquots were fractionated by electrophoresis in 12% NuPAGE gels, stained with Coomassie Blue, and the fraction of histones quantified using a Typhoon 9600 (GE Biosciences). Fluorescence from histones was quantified using ImageQuant software. The amount of geminin was normalized to the amount of histones in each sample, and the geminin/histones ratios were averaged to obtain the relative amounts of geminin protein per cell from one tumor to the next.
Results
Gmnn is a haplosufficient gene essential for ESC viability in vitro
The ESCs used in this study were derived from blastocysts homozygous for floxed Gmnn alleles [Gmnn(fl/fl)] in which exon 4 was flanked by LoxP sites, and from Gmnn(fl/fl) blastocysts containing one tamoxifen-inducible Cre recombinase gene [Gmnn(fl/fl);ErCre/+] [22]. The ErCre allele allowed Gmnn ablation in vitro by addition of monohydroxytamoxifen (MHT) to cultured cells and in vivo by intraperitoneal injection of tamoxifen. Since these cells were passaged routinely under conditions that support self-renewal, their genotypes were checked before the experiments described below.
Genomic DNA from mice either homozygous or heterozygous for the floxed Gmnn allele, or heterozygous for the wild-type Gmnn allele yielded the expected DNA products in conventional PCR assays (Fig. 1A). As expected, Gmnn(fl/fl) ESCs were homozygous for floxed Gmnn alleles, regardless of the presence or absence of MHT. However, Gmnn(fl/fl);ErCre/+ ESCs contained both Gmnn(fl) alleles and Gmnn(−) alleles in the absence of MHT and exclusively Gmnn(−) alleles in the presence of MHT, suggesting that Gmnn(fl/fl);ErCre/+ ESCs might be heterozygous for Gmnn(fl/fl) alleles. Therefore, real-time PCR was used to quantify the number of copies of Gmnn exon 4, which is deleted by ErCre from floxed Gmnn alleles, and Gmnn exon 5, which remains in both floxed alleles and wild-type alleles. The results revealed that Gmnn(fl/fl) ESCs were homozygous for the floxed Gmnn allele under all conditions and Gmnn(fl/fl);ErCre/+ ESCs were heterozygous for the floxed Gmnn allele in the absence of tamoxifen, but Gmnn nullizygous in the presence of MHT (Table 1). The fact that Gmnn(fl/−);ErCre/+ ESCs accumulated during propagation of Gmnn(fl/fl);ErCre/+ ESCs in the absence of MHT confirmed the presence of tamoxifen-independent activity by ErCre recombinase, which is presumably due to steroids in bovine fetal calf serum [39,40]. The fact that Gmnn(−/−);ErCre/+ ESCs did not accumulate during self-renewal revealed that Gmnn is a haplosufficient gene that is essential for ESC viability in vitro.
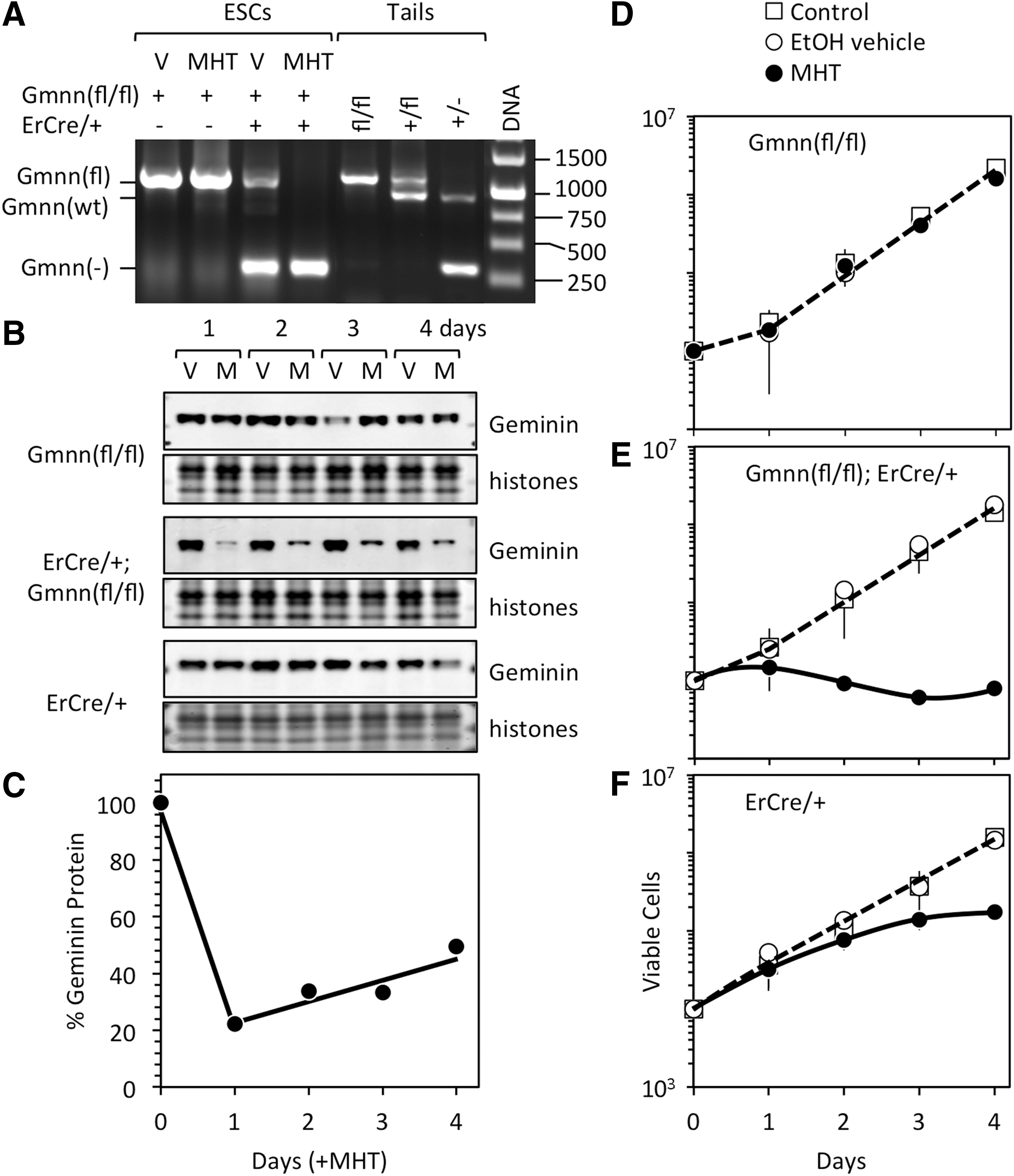
Gmnn ablation in ESCs eliminated geminin expression, terminated cell proliferation, and triggered cell death.
The number of copies of Gmnn exons 4 and 5 are given ±SEM for the number of samples assayed, determined by real-time PCR. Results for both ESCs and tumors were normalized relative to the Tfrc gene (Supplementary Fig. S4).
ESCs, embryonic stem cells; MHT, monohydroxytamoxifen; PCR, polymerase chain reaction; SEM, standard error of the mean.
Geminin prevented DNA rereplication-dependent apoptosis in ESCs in vitro
As previously reported [22], geminin protein levels were not affected in Gmnn(fl/fl) ESCs cultured with either MHT or the ethanol vehicle, whereas they were depleted rapidly in Gmnn(fl/−);ErCre/+ ESCs cultured with MHT, but not with the ethanol vehicle (Fig. 1B). Accordingly, Gmnn(fl/fl) ESCs continued to proliferate (Fig. 1D) under conditions where Gmnn(fl/−);ErCre/+ ESCs rapidly died (Fig. 1E). Nevertheless, as previously shown [22], ESCs that escaped Gmnn ablation began to repopulate the culture (Fig. 1E), which accounted for the increase observed in the level of geminin protein (Fig. 1C).
Fluorescence-activated cell sorting analyses confirmed previous studies [22] where geminin prevented DNA rereplication-dependent apoptosis in ESCs (Supplementary Fig. S1A; Supplementary Data are available online at
Both Gmnn(fl/fl) ESCs and Gmnn(fl/−);ErCre/+ ESCs produced teratomas
To determine whether or not geminin is essential for teratoma formation, nude mice were inoculated subcutaneously with Gmnn(fl/fl) ESCs in their left flank and Gmnn(fl/−);ErCre/+ ESCs in their right flank. Cells from passage 12 were thawed and cultured to passage 16 before allografts were generated to insure that both cell lines proliferated at the same rate. The mice then received intraperitoneal injections of either tamoxifen or the solution in which tamoxifen was dissolved (vehicle) for 4 consecutive days beginning with the day they were inoculated. Thus, each mouse contained its own internal control. A third group of mice remained uninjected. The number of cells in the inoculum was chosen to produce an obvious tumor within 21 days (Fig. 2A). In these and subsequent experiments, mouse body weights increased throughout the experiment (Supplementary Fig. S2), as expected for healthy animals.
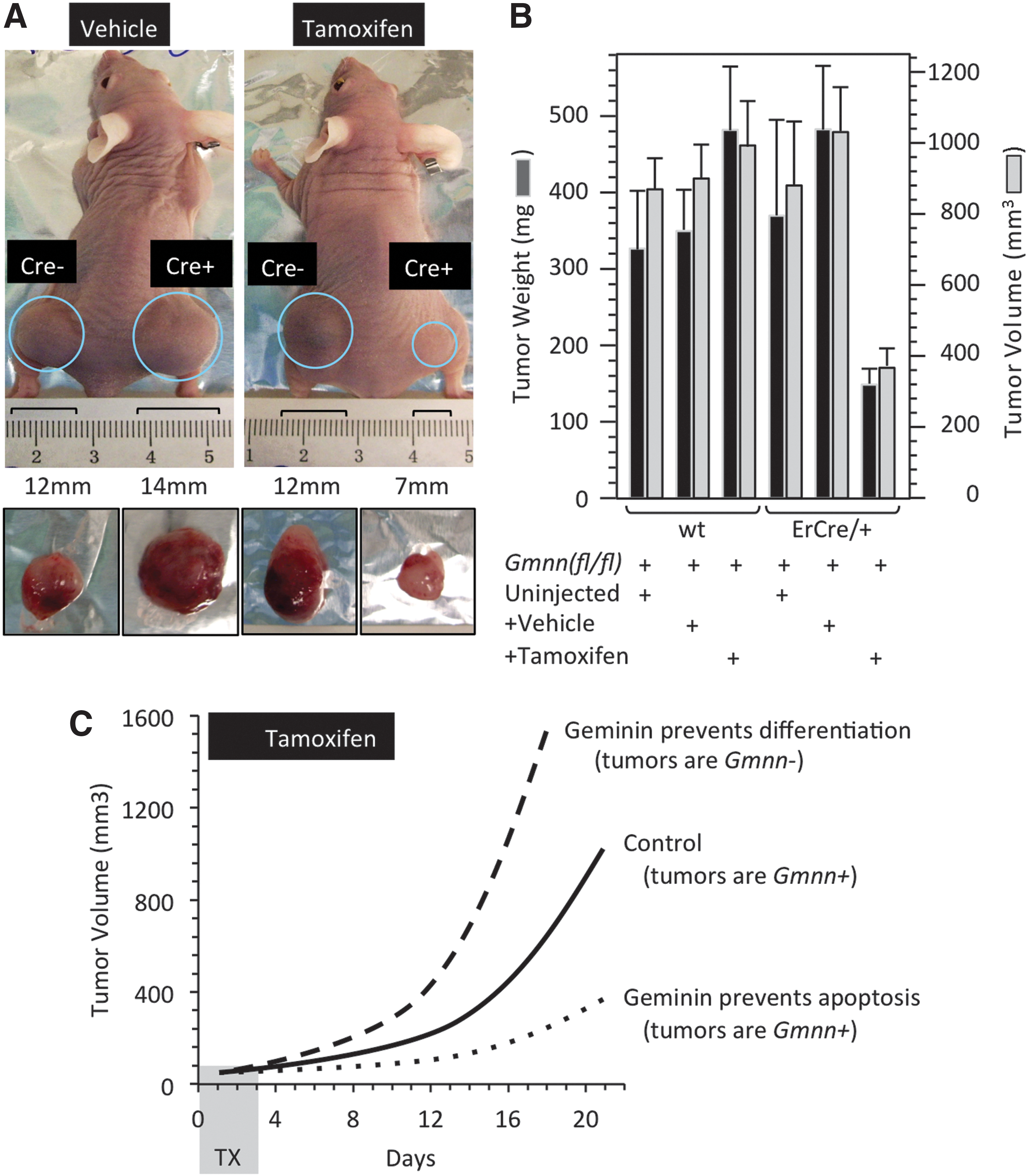
Tumors derived from Gmnn(fl/−);ErCre/+ ESCs were sensitive to TX.
Both natural and ESC-derived teratomas consist of haphazardly arranged mixtures of mature tissues derived from all three germ layers, as expected from the pluripotent nature of their progenitor cells [41]. Therefore, to determine whether or not the tumors produced by Gmnn(fl/fl) ESCs and Gmnn(fl/−);ErCre/+ ESCs were teratomas, tumors were excised at 21 days postinoculation, and slices were stained with H&E to identify specific tissues. Tumors derived from either Gmnn(fl/fl) or Gmnn(fl/−);ErCre/+ ESCs were typical teratomas, regardless of whether mice were injected with tamoxifen, vehicle, or untreated. The tumors contained at least six specific tissues (Supplementary Fig. S3). Neuroepithelium and primitive neuropils are typical of cells derived from the ectoderm. Glands are tissues derived from endoderm. Cartilage is a connective tissue derived from the mesoderm. Patches of collagen confirmed the presence of fibroblasts, the main connective tissue in adults. Myofibers are groups of multinucleated muscle cells that are also derived from the mesoderm. Trophoblast-like cells were not detected.
Tamoxifen inhibited teratoma formation from tamoxifen-sensitive ESCs
If geminin is essential to prevent DNA rereplication-dependent apoptosis in pluripotent cells, as suggested here (Fig. 1 and Supplementary Fig. S1) and elsewhere [22], then early treatment with tamoxifen should reduce the viability of Gmnn(fl/−);ErCre/+ ESCs as they undergo self-renewal. This should delay teratoma formation, and the tumors formed should contain functional Gmnn alleles and express geminin protein. Alternatively, if geminin is essential to prevent differentiation of pluripotent cells by maintaining expression of pluripotent genes, as suggested elsewhere [20,27,28], then tamoxifen should stimulate formation of teratomas derived from Gmnn(fl/−);ErCre/+ ESCs, and the tumors formed should lack functional Gmnn alleles and geminin protein (Fig. 2C).
Both the weight and volume of 21-day teratomas derived from Gmnn(fl/fl) ESCs were unaffected by tamoxifen, whereas teratomas derived from Gmnn(fl/−);ErCre/+ ESCs in mice injected with tamoxifen were 2.5- to 3-fold smaller than those derived from either uninjected mice or mice injected with vehicle (Fig. 2B). This result suggested that geminin was essential for ESC viability in vivo as well as in vitro. To validate this conclusion, the rate of teratoma formation was monitored by changes in tumor volume.
The rate of tumor formation from either Gmnn(fl/fl) ESCs or Gmnn(fl/−);ErCre/+ ESCs, either in uninjected mice or in mice injected with vehicle, were indistinguishable from one mouse to the next (Fig. 3A, C). Moreover, the ratio of tumor volumes between Gmnn(fl/fl) ESCs in the left flank and Gmnn(fl/−);ErCre/+ ESCs in the right flank of the same mouse remained constant over time, confirming that both tumors expanded at the same rate in the same mouse (Fig. 3B, D). In contrast, tumor formation was delayed by 8–10 days in mice injected with tamoxifen (Fig. 3E). Accordingly, the size of the tumor derived from Gmnn(fl/fl) ESCs relative to the size of tumor derived from Gmnn(fl/−);ErCre/+ ESCs within the same mouse increased throughout the time course (Fig. 3F). Thus, the effect of tamoxifen on tumor formation did not vary significantly from one mouse to another, and the difference between tumor sizes did not result from nonspecific effects of vehicle versus tamoxifen injections.
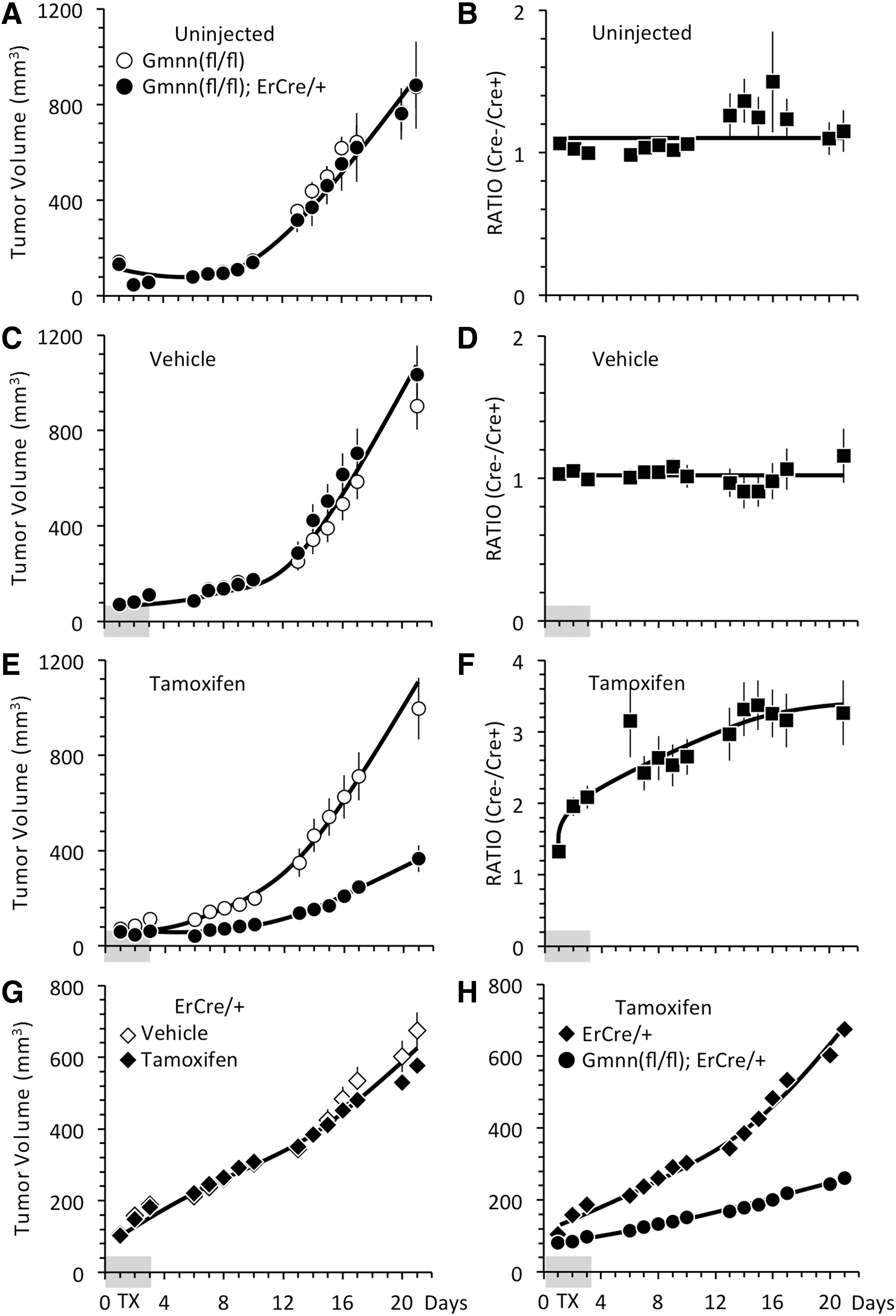
TX delayed formation of tumors derived from Gmnn(fl/−);ErCre/+ ESCs. Mice were inoculated in the left flank with Gmnn(fl/fl) ESCs (◯) or ErCre/+ ESCs (◊◆) and in the right flank with Gmnn(fl/−);ErCre/+ ESCs (●), as in Fig. 2. Some mice were left uninjected
Inhibition of tumor formation in the presence of tamoxifen depended on the presence of both Gmnn(fl/fl) alleles and an ErCre recombinase gene. The rate of tumor formation from ErCre/+ ESCs was unaffected by tamoxifen relative to vehicle (Fig. 3G). Moreover, a direct comparison between ErCre/+ ESCs in the left flank with Gmnn(fl/−);ErCre/+ ESCs in the right flank of the same mouse confirmed that inhibition of tumor formation required the presence of both ErCre and Gmnn(fl/fl) alleles (Fig. 3H).
Tamoxifen eliminated pluripotent progenitor cells from teratomas
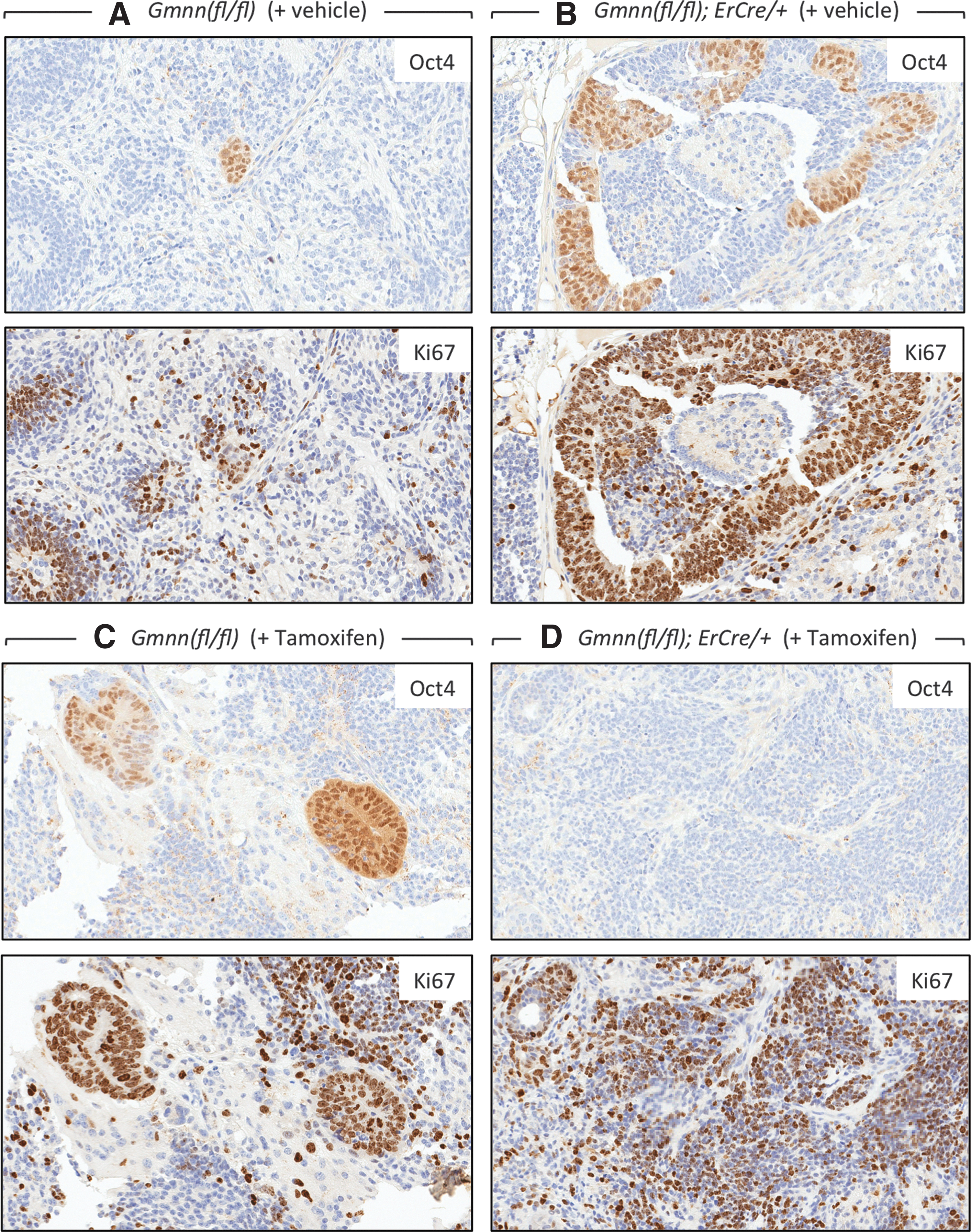
The primary difference between teratocarcinomas and teratomas is that teratocarcinomas contain colonies of morphologically unique pluripotent “embryonal carcinoma cells” that express genes essential for self-renewal, while retaining their ability to differentiate into the multiple cell types that comprise these tumors. ECCs express the same pluripotency biomarkers expressed in ESCs [8]. Among these biomarkers is the OCT4 protein, which can be used clinically to identify human teratocarcinomas [42,43]. Therefore, to identify the pluripotent progenitor cells, tumors were serially sectioned and individual sections were stained with H&E to identify cell types, with OCT4 antibodies to quantify the minimum number of pluripotent cells present (OCT4 is not unique to pluripotent cells), or with Ki67 antibodies to quantify the fraction of proliferating cells. Ki67 is a biomarker for proliferating cells that is present throughout the mitotic cell cycle [44]. Thus, comparison of consecutive serial sections through the same tumor identified pluripotent progenitor cells by three criteria: (1) small tight colonies of undifferentiated cells with large clear nuclei containing prominent nucleoli, and sparse dark cytoplasm in H&E stains, (2) cells that expressed OCT4 protein, and (3) cells that were proliferating.
Remarkably, all of the 21-day tumors contained about 1% OCT4-positive cells, regardless of whether they were derived from Gmnn(fl/fl) or Gmnn(fl/−);ErCre/+ ESCs or they were from mice injected with either tamoxifen or vehicle (Table 2). However, visual inspection of stained tissue slices revealed that tumors derived either from Gmnn(fl/fl) ESCs (Fig. 4A, B) or from Gmnn(fl/−);ErCre/+ ESCs that have not been exposed to tamoxifen (Fig. 4C) contained scattered colonies of OCT4-expressing cells with ESC morphology, whereas colonies of OCT4-expressing cells were absent from tumors derived from Gmnn(fl/−);ErCre/+ ESCs in mice injected with tamoxifen shortly after inoculation (Fig. 4D).
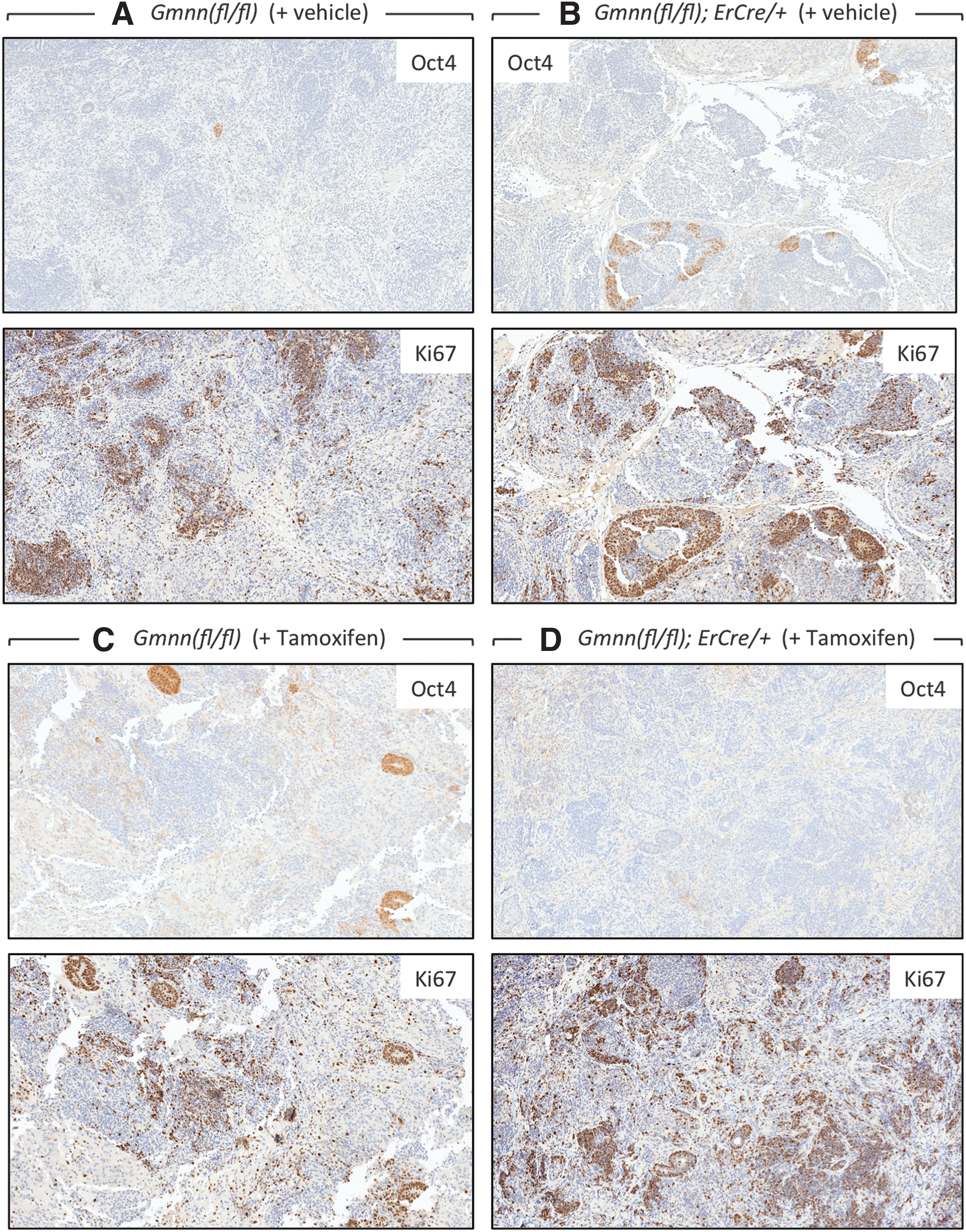
TX selectively eliminated ESCs from tumors. Tumors from the experiments in Fig. 3 were excised on day 21 and consecutive serial tissue sections were immuno-stained for either OCT4 or Ki67 protein, as indicated. Tumors produced from Gmnn(fl/fl) ESCs in mice injected either
The fraction of tumor cells expressing either Ki67 or OCT4 protein is given ±SEM for the number of samples assayed. The number of nuclei scored varied from 20,000 to 900,000.
About 28% of the cells in each tumor were proliferating, since they expressed Ki67 regardless of the genotype of their progenitor cells or their exposure to either tamoxifen or vehicle (Table 2). These cells were distributed in patches throughout each of the tumors with no obvious relationship to specific cell types (Fig. 4). However, direct comparison of consecutive tissue slices revealed that colonies of OCT4-positive cells invariably coincided with Ki67-positive cells (Fig. 5A–C). Therefore, most, if not all, of the colonies of OCT4-expressing cells with pluripotent stem cell morphology were proliferating. However, the OCT4-positive cells in those tumors that were insensitive to tamoxifen represented only 4% of the proliferating cell population, and tumors that were devoid of pluripotent cells were still enriched in proliferating cells (Figs. 4D and 5D). Therefore, tamoxifen treatment had little, if any, effect on the rate of tumor expansion.
All of the OCT4-positive cells also expressed Ki67, but most of the Ki67-positive cells did not express OCT4. Selected areas in Fig. 4 were magnified to reveal the level of coincidence between OCT4-positive cells and Ki67-positive cells, as indicated. Tumors produced from Gmnn(fl/fl) ESCs in mice injected either
These data revealed that, in the absence of either an ErCre allele or tamoxifen, ESCs rapidly differentiated into teratomas that still contained scattered pockets of pluripotent progenitor cells 3 weeks after mice were inoculated with ESCs. In contrast, Gmnn(fl/−);ErCre/+ ESCs exposed to tamoxifen immediately after inoculation apparently died, and only those ESCs that escaped Gmnn ablation continued on to initiate teratoma formation.
Gmnn was ablated in teratomas derived from tamoxifen-sensitive ESCs
To determine whether or not tamoxifen injections from day 0 to 3 had triggered Gmnn ablation, genomic DNA was isolated from tumors at 21 days and subjected to conventional PCR. Tumors derived from Gmnn(fl/fl) ESCs contained only floxed Gmnn alleles (Fig. 6A). Tumors derived from Cre/+ ESCs with wild-type Gmnn alleles contained only wild-type Gmnn alleles (Fig. 6B). However, tumors derived from Gmnn(fl/−);ErCre/+ ESCs appeared to contain only ablated Gmnn alleles, whether they had been treated with tamoxifen or vehicle (Fig. 6A).
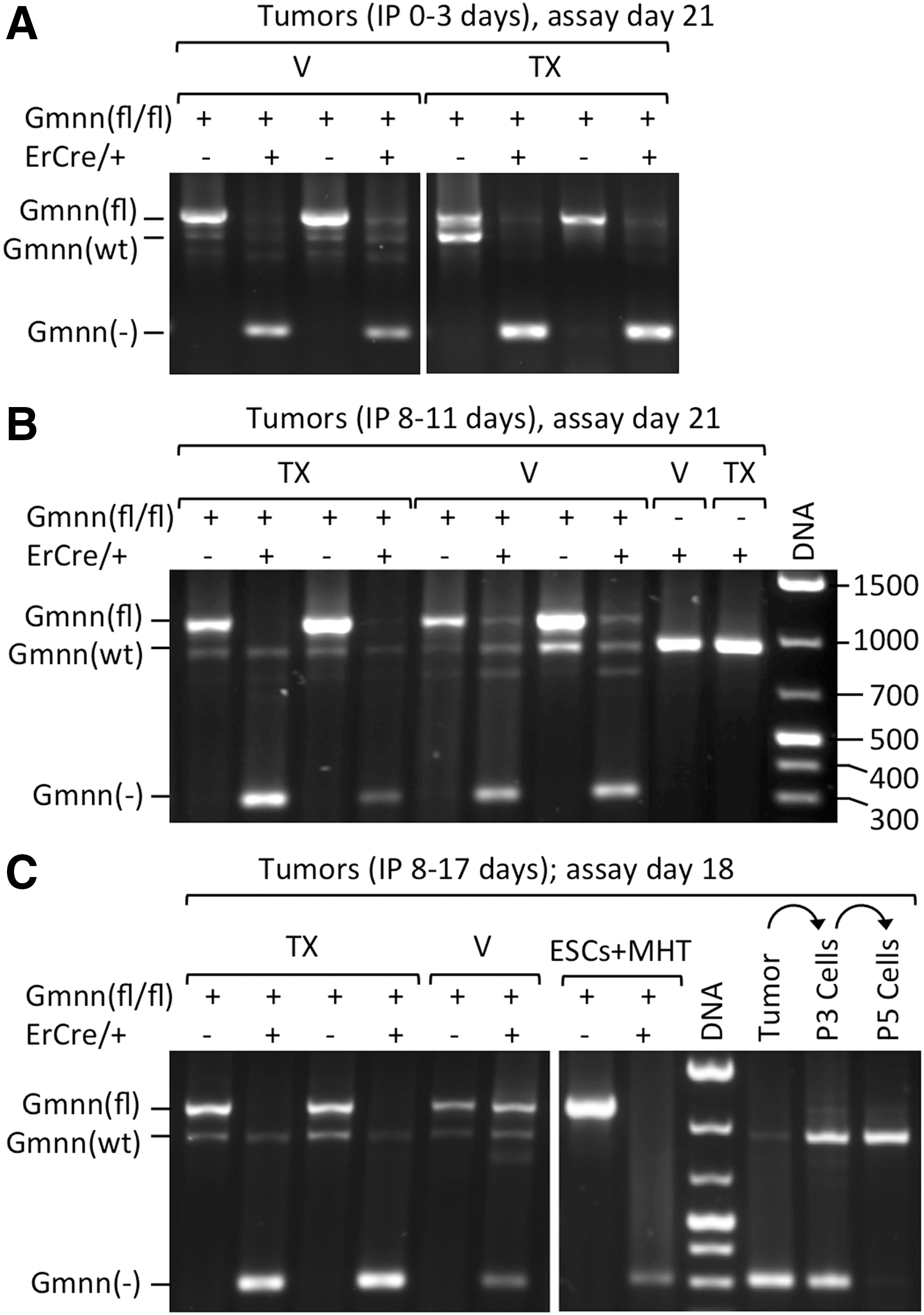
TX triggered Gmnn ablation selectively in tumors derived from Gmnn(fl/−);ErCre/+ ESCs. The status of Gmnn alleles was determined by conventional PCR assays in which wild-type Gmnn alleles [Gmnn(wt)] produced a 950 bp DNA fragment, floxed Gmnn alleles [Gmnn(fl)] produced a 1,131 bp fragment, and exon 4 deleted Gmnn alleles [Gmnn(-)] produced a 305 bp fragment.
Real-time PCR confirmed that both cultured Gmnn(fl/fl) ESCs and the tumors derived from them contained two copies of the floxed Gmnn allele, regardless of whether they were exposed to MHT as cultured cells (Table 1) or tamoxifen during tumor formation in mice (Table 3). In contrast, Gmnn(fl/−);ErCre/+ ESCs were heterozygous for the floxed Gmnn allele. Addition of MHT to these cells rapidly ablated the remaining floxed Gmnn gene. Consequently, tumors derived from Gmnn(fl/−);ErCre/+ ESCs in mice injected with tamoxifen on each of the first 4 days postinoculation were Gmnn(fl/−), because they were derived from the Gmnn(fl/−);ErCre/+ ESCs that escaped the action of tamoxifen. Thus, real-time PCR extended the results of conventional PCR by providing an accurate ratio of Gmnn(fl) to Gmnn(−) alleles in each tumor.
Real-time PCR as in Table 1.
Gmnn ablation in teratomas depleted geminin protein
The results from PCR analyses were confirmed by quantifying the ratio of geminin protein in tumors derived from Gmnn(fl/fl) ESCs inoculated into the left flank with the amount in tumors derived from Gmnn(fl/−);ErCre/+ ESCs inoculated into the right flank of the same mouse. Geminin protein was identified by western immunoblotting as an ∼30-kDa protein that reacted with geminin antibodies. The average amount of geminin per cell was determined by normalizing the geminin signal to the histone signal in each tumor sample. The results confirmed that geminin protein levels in tumors reflected the number of Gmnn alleles, as determined by real-time PCR (compare Fig. 7A with Fig. 6A and Table 3). Although geminin protein is present only during S, G2, and early M phase of cycling cells [34], the fraction of cycling cells in these tumors was constant (Table 2, % Ki67). Therefore, the amount of geminin protein observed in these tumors reflects the fraction of functional Gmnn alleles.
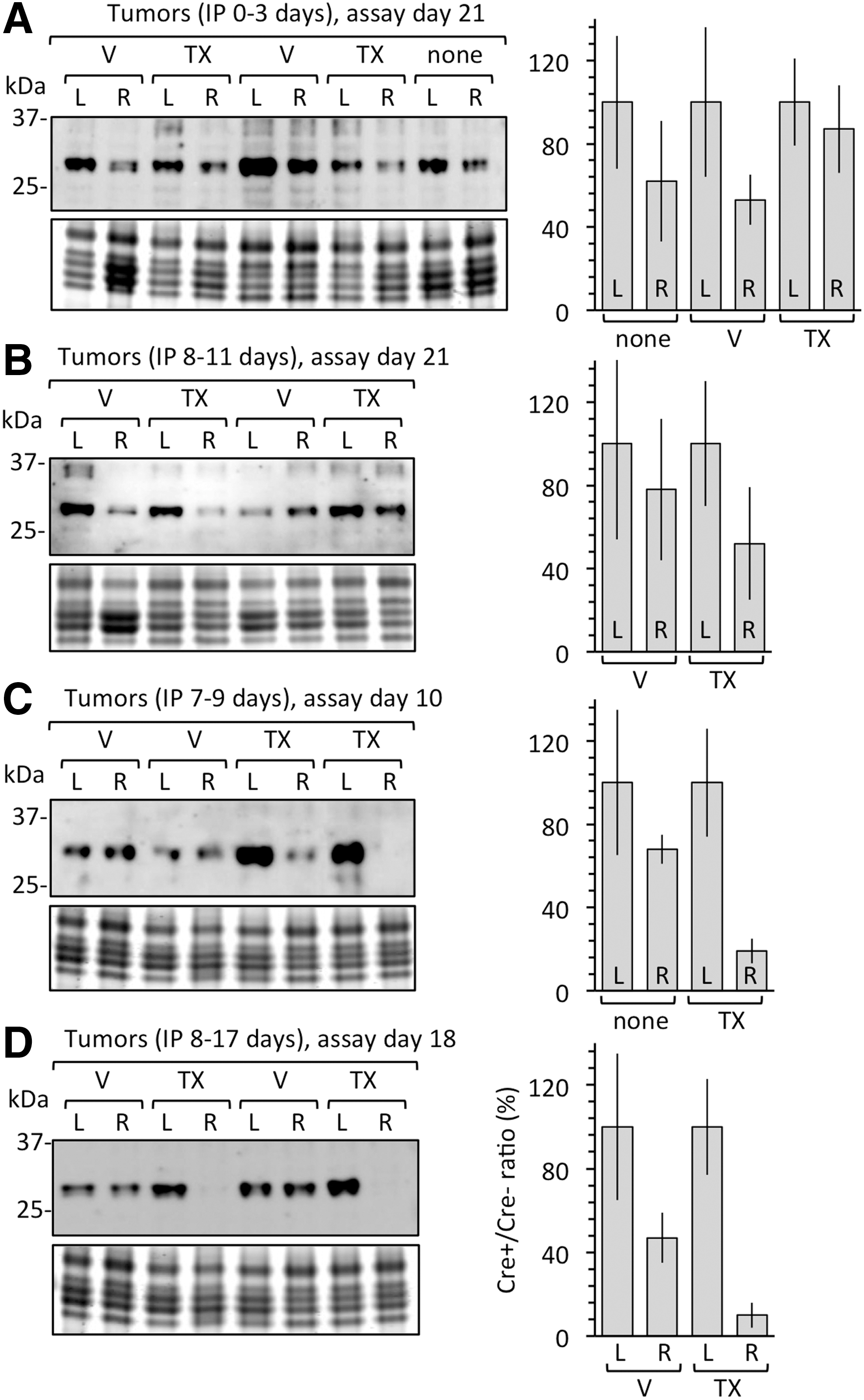
TX eliminated geminin expression selectively in tumors derived from Gmnn(fl/−);ErCre/+ ESCs. Samples from the same tumors used for PCR analysis of Gmnn alleles in Fig. 6 were also used to quantify the relative amounts of geminin protein by western immunoblotting. The amount of geminin protein detected in each sample was normalized to the amount of histone protein detected in an aliquot of the same sample fractionated on a different gel. Bar graphs adjacent to each gel pattern indicate the mean differences between the amount of geminin in tumors from the left flank (L) [derived from Gmnn(fl/fl) ESCs and defined as 100%] and the amount of geminin in the tumor from the right flank (R) of the same mouse [derived from Gmnn(fl/−);ErCre/+ ESCs].
Taken together, the preceding results demonstrated that tumors formed in mice treated with tamoxifen from day 0 to 3 were teratomas derived from ESCs that escaped tamoxifen-dependent ablation of floxed Gmnn alleles, as observed in studies on cultured ESCs (Fig. 1; [22]); they produced geminin protein at levels consistent with the genotype of their ESC progenitors. Therefore, the role of geminin in ESCs was to prevent DNA rereplication-dependent apoptosis, rather than to prevent ESC differentiation by maintaining a pluripotent state.
Tamoxifen delayed expansion of teratomas by selectively eliminating pluripotent progenitor cells
The preceding studies revealed that all of the ESCs inoculated into nude mice either died or differentiated into teratoma cells within 3 weeks, suggesting that the proportion of pluripotent progenitor cells is inversely related to the age of the teratoma. Therefore, to determine whether or not tumor expansion was dependent on pluripotent progenitor cells, mice were inoculated with ESCs, injected with tamoxifen from day 7 through 9, and their tumors isolated on day 10.
Tumors derived from Gmnn(fl/fl) ESCs contained an average of 15% OCT4-positive cells (Table 4, 10-day tumors) that displayed large areas of OCT4-positive cells, which also expressed Ki67 (Fig. 8A) with pluripotent stem cell morphology (Fig. 8C). These tumors contained, on average, 12% OCT4-positive cells and 83% Ki67-positive cells, 11 times the population of OCT4-positive cells in 21-day-old tumors and thrice the population of Ki67 cells (compare 10-day tumors in Table 4 with 21-day tumors in Table 2). In contrast, tumors derived from Gmnn(fl/−);ErCre/+ ESCs in mice injected with tamoxifen were enriched in Ki67-positive cells, but they contained only one third as many OCT4-positive cells and very few colonies of putative progenitor cells (Table 4 and Fig. 8B). Moreover, real-time PCR assays on DNA from 10-day teratomas revealed a 50% decrease in the number of cells with at least one functional Gmnn allele (Table 5), as well as a similar decrease in the amount of geminin protein per cell (Fig. 7C). Thus, tamoxifen selectively eliminated pluripotent progenitor cells from young teratomas.
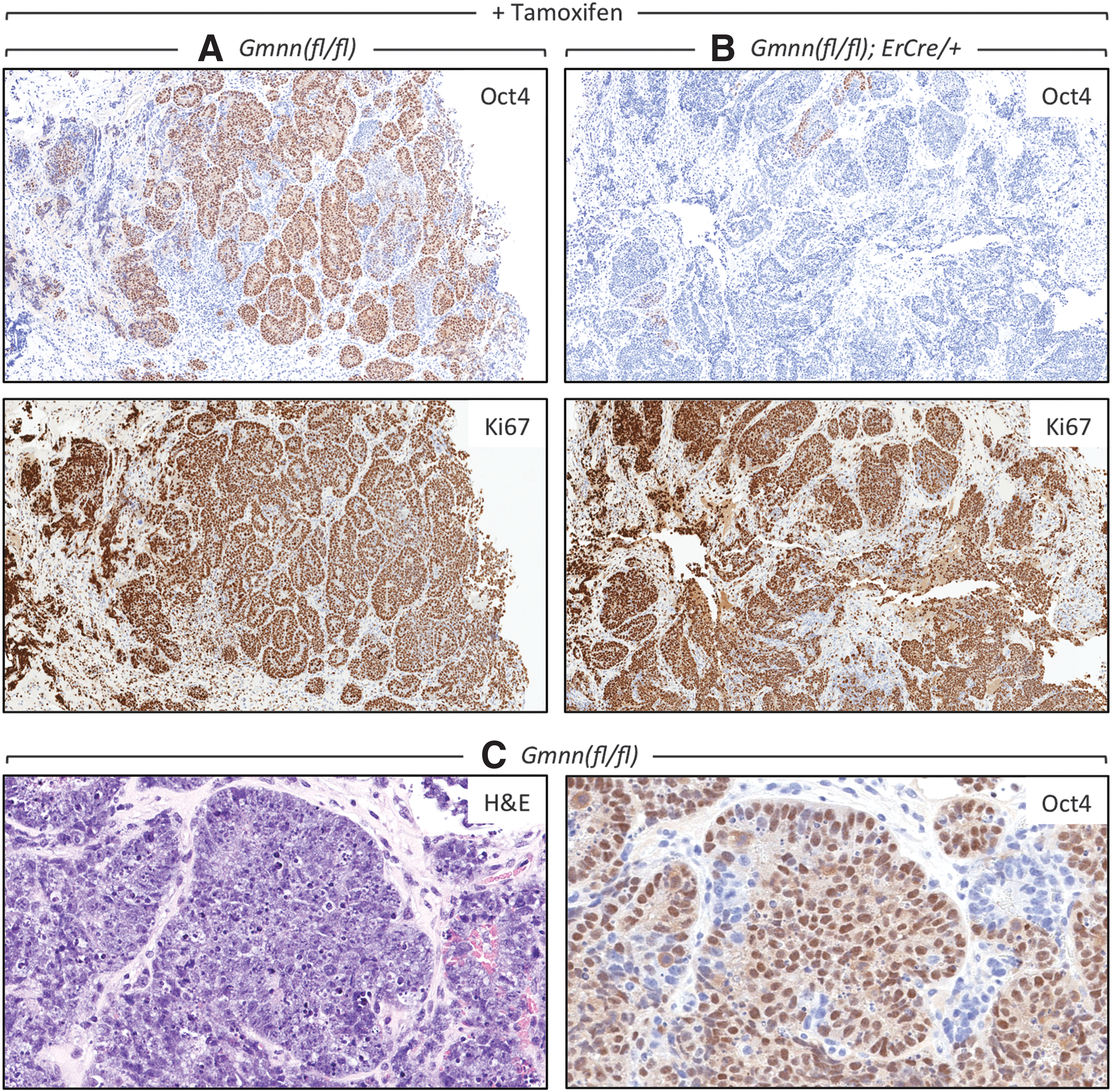
Tumors at day 10 postinoculation were enriched for pluripotent cells that were sensitive to Gmnn depletion.
The fraction of tumor cells expressing either Ki67 or OCT4 protein is given ±SEM for the number of samples assayed. The number of nuclei scored varied from 20,000 to 900,000.
Real-time PCR as in Table 1.
To determine whether or not Gmnn ablation inhibited expansion of an established teratoma, mice were inoculated with ESCs and then allowed to produce palpable tumors before receiving either tamoxifen or vehicle. Injections of vehicle on days 8, 9, 10, and 11 postinoculation had no effect on the expansion of tumors derived from either Gmnn(fl/fl) ESCs or Gmnn(fl/−);ErCre/+ ESCs (Fig. 9A), and the ratio of the size of tumors derived from Gmnn(fl/fl) and Gmnn(fl/−);ErCre/+ ESCs in each mouse remained constant (Fig. 9B). Similarly, the expansion of tumors derived from Gmnn(fl/fl) ESCs was unaffected by tamoxifen (Fig. 9C). However, the expansion of tumors derived from Gmnn(fl/−);ErCre/+ ESCs was arrested temporarily by tamoxifen and then continued growing at the same rate as Gmnn(fl/fl)-derived tumors. This was evident when the same tumors in different mice were compared (Fig. 9C) and the ratios of tumor volumes in the same mouse were compared (Fig. 9D). The sharp inflection in the ratios began within 24 h of the injection on day 8 as the left flank tumor continued to expand, while expansion of the right flank tumor was arrested temporarily. When the right flank tumor recovered from the tamoxifen and resumed its normal expansion rate, the ratios decreased with time until once again the two tumors were expanding at the same rate, and the ratios between Cre− and Cre+ tumors in the same mouse remained constant with time.
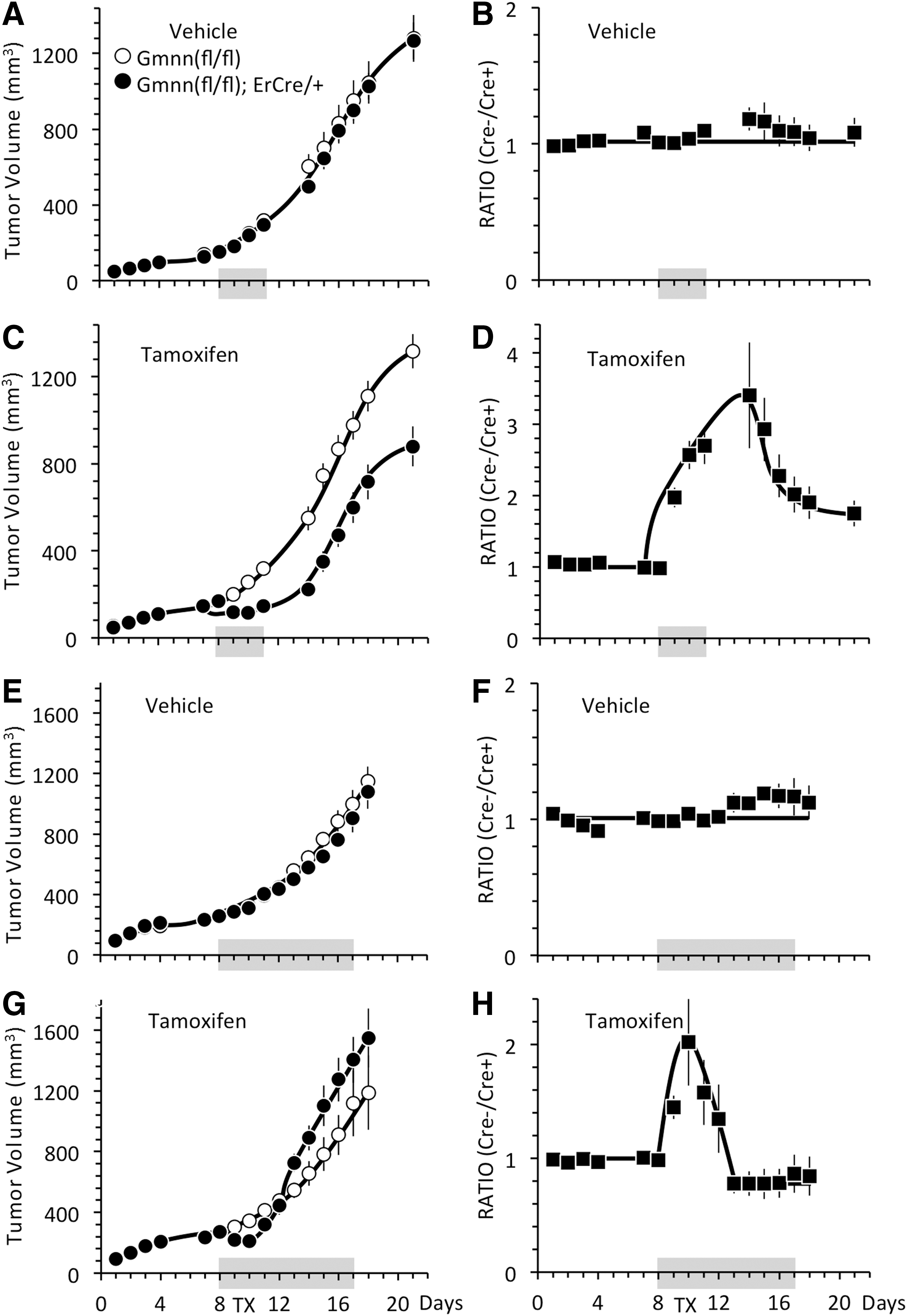
TX did not prevent teratoma expansion. Mice were inoculated in the left flank with Gmnn(fl/fl) ESCs (●) and in the right flank with Gmnn(fl/−);ErCre/+ ESCs (◯), as in Fig. 2.
When mice received tamoxifen for 4 consecutive days after tumors were palpable (days 8–11), the tumors at 21 days postinoculation contained the same percentages of Ki67- and OCT4-positive cells as 21-day tumors from mice treated for 4 consecutive days after inoculation (Table 4, 21-day tumors). About 28% of the cells in all tumors were Ki67 positive and 1%–2% were OCT4 positive, suggesting that those ESC-derived progenitor cells that were not eliminated by tamoxifen treatment simply differentiated into teratoma tissues. Moreover, conventional PCR (Fig. 6B), real-time PCR (Table 5, 21-day tumors), and western immunoblotting assays (Fig. 7B) from these tumors gave results indistinguishable from the 21-day tumors from mice treated with tamoxifen on days 0–3. Thus, the temporary delay imposed on established teratomas derived from tamoxifen-sensitive ESCs resulted from depletion of the ESC-derived OCT4-positive progenitor cells present in early teratomas.
Teratomas continued expansion in the complete absence of geminin and pluripotent progenitor cells
In an effort to permanently arrest tumor expansion, tamoxifen was administered for 10 consecutive days beginning on day 8. Remarkably, tumors derived from Gmnn(fl/−);ErCre/+ ESCs were again arrested temporarily, but then they continued to expand at the same rate as tumors derived from Gmnn(fl/fl) ESCs. As in previous experiments, injection of the tamoxifen vehicle did not affect tumor expansion (Fig. 9E, F), and injection of tamoxifen did not affect the expansion of tumors derived from Gmnn(fl/fl) ESCs (Fig. 9G, -◯-). The rate of expansion of tumors derived from Gmnn(fl/−);ErCre/+ ESCs, however, paused for about 3 days and then continued to expand rapidly (Fig. 9G, -●-). This transition was also evident in the ratios of Cre− tumors to Cre+ tumors in the same mouse (Fig. 9H).
Tumor histology, PCR analysis, and geminin protein analysis revealed that 10 consecutive injections of tamoxifen had effectively eliminated geminin expression throughout the teratoma without reducing the fraction of proliferating cells. The fraction of OCT4-positive cells in 18-day-old tumors derived from Gmnn(fl/−);ErCre/+ ESCs in mice treated with tamoxifen was, on average, fourfold less than control tumors (Table 6) and clones of OCT4-positive cells not detected in these tumors. Nevertheless, the fraction of Ki67-positive cells in all 18-day tumors was, on average, 1.8-fold greater than tumors isolated at 21 days postinoculation (compare Table 6 with Tables 2 and 4).
Real-time PCR revealed that teratomas derived from Gmnn(fl/−);ErCre/+ ESCs in mice injected with tamoxifen for 10 consecutive days after teratomas were established were Gmnn(−/−) (Table 7). Teratomas derived from Gmnn(fl/fl) ESCs contained two Gmnn alleles, whereas teratomas derived from Gmnn(fl/−);ErCre/+ ESCs in mice injected with vehicle contained one Gmnn allele and those in mice injected with tamoxifen contained none. Conventional PCR detected only floxed Gmnn alleles and wild-type Gmnn alleles in tumors lacking a Cre gene and only ablated Gmnn alleles and wild-type Gmnn alleles in tumors with a Cre gene (Fig. 6C).
Real-time PCR as in Table 1.
Tumors formed in mice treated with tamoxifen from day 8 to 17 were composed primarily of cells depleted of Gmnn alleles, most of which were proliferating, and therefore produced only 10% as much geminin (Fig. 7D). Efforts to culture Gmnn(−/−) cells from teratomas were unsuccessful. By passage five, cultures contained only fibroblasts with wild-type geminin genes that had infiltrated the tumor from the host (Fig. 6C). Therefore, although cells lacking Gmnn alleles proliferated in solid tumors as efficiently as cells with Gmnn alleles, Gmnn(−/−) cells were at a distinct disadvantage in vitro.
The continued expansion of teratomas completely depleted of functional Gmnn alleles, geminin protein, and pluripotent progenitor cells, and their large population of proliferating Ki67-positive cells, demonstrated that the differentiated cells that comprise a teratoma can continue to proliferate in the absence of geminin as long as they are part of a solid tissue mass.
Discussion
Geminin is essential for pluripotent cell viability
The role of geminin at the beginning of mammalian development has been enigmatic. Some studies concluded that geminin is essential in totipotent blastomeres and pluripotent cells to maintain expression of “pluripotency genes” that prevent differentiation into specialized cell lineages [20,27,28], whereas other studies concluded that geminin is not required to regulate pluripotency [22,29]. Still, others concluded that geminin is essential at the beginning of mammalian development to prevent aberrant DNA replication from inducing apoptosis [21,22,30]. To resolve this conundrum, we utilized the ability of pluripotent stem cells to produce teratomas as an established model for identifying genes essential for cell differentiation and embryonic development [3,7,45].
The results presented in Fig. 1 and Supplementary Fig. S1 confirm the previous report [22] that geminin is essential in vitro to prevent DNA rereplication-dependent apoptosis in pluripotent stem cells during self-renewal. Analysis of teratoma formation in mice extends these results to show that this characteristic of pluripotent cells is not an in vitro artifact, but occurs under in vivo conditions as well. If geminin is essential to maintain pluripotency, then Gmnn ablation should increase the rate of teratoma formation and the tumors should be Gmnn nullizygous (Fig. 2C). Alternatively, if geminin is essential for ESC viability, then Gmnn ablation should delay teratoma formation and the tumors should have the same genotype as their ESC progenitors. The results demonstrated clearly that ESCs containing tamoxifen-sensitive Gmnn floxed alleles as well as a Cre allele responded as expected if Gmnn was essential to prevent DNA rereplication-dependent apoptosis in vivo as well as in vitro.
Gmnn alleles were easily ablated in ESCs with a Gmnn(fl/−);ErCre/+ by MHT in vitro, which resulted in DNA rereplication-dependent apoptosis, or by tamoxifen in vivo, which delayed formation of a teratoma by those ESCs that escaped Gmnn ablation and therefore produced teratomas in which the average cell was Gmnn hemizygous. In contrast, neither MHT nor tamoxifen affected geminin expression in either Gmnn(fl/fl) ESCs or ErCre/+ ESCs. Thus, when tamoxifen was administered immediately after inoculating mice with ESCs, the resulting teratomas had the same number of functional Gmnn alleles as their ESC progenitor cells. In the absence of tamoxifen, ESCs simply continued to differentiate into the multitude of normal nonmalignant cells and tissues that comprised the teratomas at 21 days postinoculation, in which only ∼1% of the cells were colonies of pluripotent progenitor cells.
To confirm that tamoxifen induced ablation of Gmnn selectively eliminated pluripotent progenitor cells from teratomas, mice were injected with tamoxifen for 3 days just before isolating tumors at 10 days postinoculation. These early teratomas were enriched with pluripotent progenitor cells, but prior treatment with tamoxifen depleted them from tumors derived from tamoxifen-sensitive ESCs, but not from tumors derived from tamoxifen-insensitive ESCs.
Geminin is essential for the viability of totipotent blastomeres in preimplantation embryos and pluripotent stem cells in the epiblast. Gmnn ablation following fertilization arrested development as embryos entered the morula stage, presumably through depletion of maternally inherited geminin [20 –22]. In some cases, the resulting abnormal embryos appeared to be undergoing DNA damage-dependent apoptosis [21,22], whereas in other cases, they appeared to be undergoing premature differentiation into trophoblast giant cells [20]. However, if the amount of maternally inherited geminin was greater in some embryos than others, then the outer blastomeres would have differentiated into trophoblast cells in those embryos with sufficient geminin to sustain development to the early morula stage. In that case, depletion of maternally inherited geminin would eliminate the remaining totipotent blastomeres, while triggering terminal differentiation of the trophoblast cells into giant cells [19]. Gmnn ablation in the postimplantation epiblast causes neural tube defects through disrupted progenitor specification and neuronal differentiation, which terminate development [23], but Gmnn ablation at later embryonic stages does not eliminate neural progenitor cells [24,26]. One explanation is that the epiblast, from which ESCs and epiblast stem cells are derived [46,47], contains pluripotent progenitor cells that require geminin for viability, whereas later developmental stages do not, and therefore, Gmnn ablation in the epiblast eliminates the pluripotent progenitor cells required for neural tube development, whereas Gmnn ablation at later stages does not eliminate the stem cells.
Geminin as a therapeutic target in the treatment of germ cell neoplasias
Although geminin was essential for pluripotent cell viability during self-renewal in vitro and during teratoma formation in vivo, it was not essential for proliferation of the differentiated cells within a teratoma. Extensive tamoxifen treatment eliminated all of the Gmnn(fl/fl) alleles in cells derived from ESCs harboring an ErCre recombinase gene, thereby eliminating geminin protein as well as all of the cells that depended on geminin for either viability or efficient proliferation. Remarkably, these Gmnn nullizygous teratomas, devoid of pluripotent progenitor cells, but rich in proliferating Ki67-positive cells, continued to expand at the same rate as control teratomas. Thus, geminin was not essential for proliferation of most, perhaps all, of the normal nonmalignant cells within a compact tissue. However, the Gmnn nullizygous cells in these teratomas could not be cultured in vitro, consistent with the effects of Gmnn depletion in mouse primary embryonic fibroblasts and immortalized fibroblasts [22]. Therefore, the requirement for geminin is not only cell-type dependent but also context dependent. The differential effects of geminin depletion on pluripotent stem cells compared with their differentiated progeny suggest that a drug that selectively prevents geminin function in mammals could, in principle, convert a germ cell cancer into a benign tumor by eliminating the stem cells responsible for malignancy and metastasis.
Teratoma and teratocarcinoma are generic names for a variety of benign and malignant germ cell neoplasias. They account for 95% of testicular cancers and 70% of ovarian tumors (3% malignant) that occur principally during adolescence and early adulthood [41,48]. Remarkably, about 10% of germ cell neoplasias occurs at nongonadal sites such as inside the cranium, mouth, neck, thoracic cavity, and pelvis. One explanation for the midline distribution of nongonadal tumors is that PGCs, the precursors for both oocytes and spermatogonia, get lost as they migrate along the midline of the fetus to descend into the pelvis as ovarian cells or into the scrotal sac as testicular cells. [49]. Another possibility is that some of the ESCs that give rise to PGCs in the epiblast of postimplantation embryos [9], as well as to all other tissues in the fetus, might simply remain in a quiescent state as development proceeds, thereby becoming dispersed among various tissues until environmental signals trigger their differentiation [50]. ESCs activated at ectopic sites in either postgastrulation embryos or in adults efficiently induce teratoma or teratocarcinoma formation [5,51].
PGCs are closely related to pluripotent cells because PGCs can be derived from ESCs [52] and ESCs can be derived from PGCs [53 –55]. Therefore, PGCs would be expected to be sensitive to Gmnn ablation. In fact, geminin is essential for proliferation and viability of sperm stem cells (spermatogonia), but not for differentiation of spermatids into mature sperm [56], suggesting that geminin would also be essential for the formation of male germ cell neoplasia, and presumably other germ cell neoplasias as well.
Germ cell neoplasias are identified clinically by the presence of colonies of OCT4-expressing ECCs [42,43,57,58]. Not surprisingly, the histology of OCT4-positive ECC colonies in these human teratocarcinomas are quite similar to the OCT4-positive colonies in the 10-day tumors produced in nude mice from Gmnn(fl/fl) ESCs (Fig. 8). ECCs express all the biomarkers of ESCs, exhibit a pattern of differentiation in vitro that is essentially the same as for ESCs, and like ESCs can proliferate indefinitely without losing their ability to differentiate into a variety of different cell types, either during normal mammalian development or during the formation of a tumor [8,59,60]. The primary difference is that ECCs are multipotent stem cells, whereas ESCs are pluripotent stem cells. Therefore, since teratocarcinogenesis is determined primarily by the genetic background of the stem cell progenitor [61], it is likely that the progenitor cells derived from a teratocarcinoma will be sensitive to geminin inhibition.
Geminin is not essential in adult tissues
The fact that ablation of geminin in established teratomas did not prevent the normal tissues, which comprise a teratoma, from continued expansion mirrored the response of tissues in adult animals to geminin ablation. Therefore, chemotherapy directed at geminin should eliminate the progenitors of germ cell neoplasias with little, if any, harm to other cells in the organism. Geminin is essential for preimplantation development of the blastocyst and postimplantation development up to day 10.5 [20 –23], but beyond that, some tissues are sensitive to geminin levels, whereas others are not. Geminin is essential for hematopoietic stem cell (HSCs) viability during embryogenesis [62], but Gmnn ablation in adult HSCs does not result in excess DNA replication, apoptosis, or cell-cycle arrest, although it does result in anemia due to changes in proliferation rates of various differentiated cell types [25]. Adult HSCs survive indefinitely when transplanted into irradiated hosts [25]. Gmnn ablation in the lymphoid lineages and peripheral T cells did not alter the proliferation or the differentiation potential of these cells in vivo [36,63]. White blood cells have a normal DNA content in vivo [25]. Neural stem cells continue to proliferate normally in the absence of functional Gmnn alleles [24,26]. The resistance to geminin depletion in the cells of adult tissues reflects the fact that multiple concerted pathways exist, which restrict genome duplication to once per cell division [36].
Targeting geminin might be useful against other cancers as well. Cells derived from cancers of the colon, breast, lung, kidney, and bone, all require geminin to prevent DNA rereplication-dependent apoptosis, whereas cells derived from the normal tissues do not [35,64]. Geminin is overexpressed in many tumors, and the prognosis for recovery is inversely related to the level of geminin expression [65,66]. In fact, suppressing geminin expression can inhibit tumor formation by metastatic human breast carcinoma cells [67]. The results presented demonstrate, in principle, that selective inhibition of geminin activity can prevent formation of at least some tumors with little or no harm to normal cells.
Footnotes
Acknowledgments
This project was supported, in part, by the National Institute for Child Health and Human Development and, in part, by the National Cancer Institute, National Institutes of Health, under contract no. HHSN261200800001E. The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services, nor does mention of trade names, commercial products, or organizations imply endorsement by the U.S. Government. The Scientific Director, NICHD/NIH, approved this research for publication (BMGR/SEDR-MD/MD-16-07729-R).
Author Disclosure Statement
No competing financial interests exist.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.