
Abstract
Human bone marrow-derived mesenchymal stem cells (BMSCs) are clinically promising to repair damaged articular cartilage. This study investigated TWIST1, an important transcriptional regulator in mesenchymal lineages, in BMSC chondrogenesis. We hypothesized that downregulation of TWIST1 expression is required for in vitro chondrogenic differentiation. Indeed, significant downregulation of TWIST1 was observed in murine skeletal progenitor cells during limb development (N = 3 embryos), and during chondrogenic differentiation of culture-expanded human articular chondrocytes (N = 3 donors) and isolated adult human BMSCs (N = 7 donors), consistent with an inhibitory effect of TWIST1 expression on chondrogenic differentiation. Silencing of TWIST1 expression in BMSCs by siRNA, however, did not improve chondrogenic differentiation potential. Interestingly, additional investigation revealed that downregulation of TWIST1 in chondrogenic BMSCs is preceded by an initial upregulation. Similar upregulation is observed in non-chondrogenic BMSCs (N = 5 donors); however, non-chondrogenic cells fail to downregulate TWIST1 expression thereafter, preventing their chondrogenic differentiation. This study describes for the first time endogenous TWIST1 expression during in vitro chondrogenic differentiation of human BMSCs, demonstrating dynamic regulation of TWIST1 expression whereby upregulation and then downregulation of TWIST1 expression are required for chondrogenic differentiation of BMSCs. Elucidation of the molecular regulation of, and by, TWIST1 will provide targets for optimization of BMSC chondrogenic differentiation culture.
Introduction
F
The twist family basic helix-loop-helix transcription factor 1 (TWIST1) is expressed in developing skeletal mesenchyme, where it regulates mesenchymal cell fate [1 –5]. Consistent with this, some skeletal dysplasia, characterized by abnormal limb development and premature osteogenesis, occurs as a result of TWIST1 haploinsufficiency [6 –9]. This latter observation, differentiation of osteoprogenitor cells triggered by decreased TWIST1, has led to an investigation of the potential of TWIST1 as a target to facilitate osteogenic differentiation of MSCs for clinical application [10]. Notably, the initiation of osteoblast differentiation is determined by the removal of direct TWIST1 inhibition on RUNX2, the master transcriptional regulator of osteogenic differentiation [11]. That TWIST1 is similarly capable of directly binding to and inhibiting SOX9 [12], the master transcriptional regulator of chondrogenesis, poses the question of whether TWIST1 could likewise be an in vitro target for optimization of MSC chondrogenic differentiation.
Several studies have previously demonstrated TWIST1 repression of chondrogenesis and chondrocyte maturation downstream of β-CATENIN and TGF-β signaling in murine and chick models [13 –15]. Little is known, however, of the role of TWIST1 during human chondrogenic processes; a single study demonstrating significant downregulation of SOX9 expression and glycosaminoglycan (GAG) production in human MSCs overexpressing TWIST1[16] is, to our knowledge, the only available data. The aim of this study was, therefore, to analyze the endogenous expression profile of TWIST1 during chondrogenesis. We hypothesized that downregulation of TWIST1 expression is necessary and sufficient for chondrogenic differentiation of adult human bone marrow-derived MSCs (BMSCs). In this study, we demonstrate dynamic regulation of TWIST1 expression, with both upregulation and downregulation of TWIST1 that are necessary for in vitro BMSC chondrogenic differentiation.
Materials and Methods
Isolation and culture
Primary human articular chondrocytes (HACs) from three donors (67–83 years) undergoing total knee arthroplasty were harvested from the femoral condyles with the approval and according to the local ethical committee's rules, and with the implicit consent of the patients (MEC2004-322: Erasmus Medical Centre, Rotterdam). A single-cell suspension was obtained after digestion with 2 mg/mL protease XIV (Sigma-Aldrich, St. Louis, Mo), followed by 1.5 mg/mL collagenase B (Roche Diagnostics, Mannheim, Germany). HACs were seeded (4,000 cells/cm2) and expanded (de-differentiated) in Coon's modified Ham's-F12 (Biochrom A.G., Berlin, Germany) containing 10% fetal calf serum (FCS; Invitrogen, Paisley, Scotland, United Kingdom). The medium was renewed three times per week, and HACs were used at passage 2 for all experiments.
After obtaining informed consent and with approval of the local ethical committees, bone marrow aspirates from the iliac crest were collected from 12 patients (15–74 years) undergoing total hip replacement (MEC 2004-142: Erasmus Medical Center, Rotterdam; MEC 2011.07: Albert Schweitzer Hospital, Dordrecht). Primary human adult BMSCs were selected by plastic adherence and cultured at a density of 2,300 cells/cm2 at 37°C, 5% CO2, 21% O2 in α-MEM (Gibco, Carlsbad, CA) containing 10% FCS (Lonza, Verviers, Belgium), 1 ng/mL fibroblast growth factor-2 (FGF2; AbD Serotec Kidlington, United Kingdom), 10−4 M ascorbic acid-2-phosphate (Sigma-Aldrich), 1.5 μg/mL fungizone, and 50 μg/mL gentamicin (Gibco). The medium was renewed twice weekly, and BMSCs were used at passages 3–5 for experiments throughout this study.
To assess the effect of transforming growth factor-β1 (TGFβ1), FGF2, tumor necrosis factor-α (TNFα), and wingless-related integration site-3A (WNT3A) on TWIST1 expression in monolayer, normal expansion medium was replaced 24 h after seeding (22,000 cells/cm2) with α-MEM containing 10−4 M ascorbic acid-2-phosphate and either 1 ng/mL or 10 ng/mL TGFβ1 (R&D systems, Minneapolis, MN), FGF2 or TNFα (Peprotech, Rocky Hill, NJ), or 25 ng/mL or 250 ng/mL WNT3A (purified from conditioned medium of Drosophila S2 cells modified with a WNT3A vector and obtained as 50 μg/mL solution in CHAPS as previously described [17]) for 24 h.
TWIST1 silencing
Cells were seeded (4,000–5,700 cells/cm2; day 0) and cultured for 24 h in the expansion medium mentioned earlier. The medium was then replaced (day 1) with expansion medium containing either 3.5 nM siRNA against TWIST1 (siTWIST1; Ambion) or 3.5 nM non-targeting negative control siRNA (scramble, scTWIST1; Ambion). Treatment was repeated after 72 h (day 4). All conditions, including control conditions (Ctrl), were also treated with Lipofectamine RNAiMAX carrier (1:2300; Invitrogen, California) and OptiMEM (Gibco) to improve transfection efficiency. After 24/48 h (day 5/6), cells were harvested for chondrogenic culture. The medium of cells for mRNA and protein analysis was replaced with expansion medium only, 24 h before harvesting.
Chondrogenic differentiation
After expansion or 24/48 h after the second silencing treatment, HAC pellets and BMSC pellets containing 2 × 105 cells were formed by centrifugation at 250 g and cultured for 1 or 21 days in chondrogenic induction medium consisting of DMEM-HG supplemented with Glutamax, ITS +1 (B&D Bioscience, Bedford, MA), 40 μg/mL
Gene expression analysis
For mRNA analysis, both HAC (1 well/donor 24 h after last refresh) and BMSC monolayer cells (3 wells/donor 24 last refresh) were washed with PBS and treated on ice with RLT Plus or RLT lysis buffer (Qiagen GmbH, Hilden, Germany) containing 1% β-mercaptoethanol; HAC pellets (2 pellets/donor 24 h after the last refresh) and MSC pellets (2–3 pellets/donor 24–96 h after the last refresh) were manually homogenized in RLT Plus lysis buffer, or RNA-Bee (TEL-TEST, Friendswood, TX) with RNA extracted by addition of 20% chloroform. Further RNA isolation and purification was performed by using the RNeasy PLUS or RNeasy MicroKit (Qiagen). cDNA was prepared by using the RevertAid First-Strand cDNA Synthesis Kit (MBI Fermentas, St. Leon-Rot, Germany) according to the manufacturer's instructions, and quantitative real-time polymerase chain reaction was performed with TaqMan Universal PCR MasterMix (Applied Biosystems, Capelle a/d Ijssel, The Netherlands) or SybrGreen (Eurogentec, Seraing, Belgium). Primer sequences (Applied Biosystems) are listed in Table 1. After a comparison of housekeeping genes (GAPDH, RPS27a, HPRT1, B2 M, 18S), GAPDH for chondrocytes and RSP27A for BMSCs were chosen for their stability across all conditions. Relative gene expression was calculated according to the 2−ΔΔCt formula [18].
Western blot
Cells were washed with PBS, and on ice, incubated in M-PER containing 1% Halt™ Protease Inhibitor Cocktail and 1% Halt Phosphatase Inhibitor (Thermo Scientific). Cell lysate was harvested, and total protein content was determined by using a BCA assay (Thermo Scientific). For western blot analysis, 8 μg of total protein was electrophoresed on an 8%–16% sodium dodecyl sulfate-polyacrylamide gel. Proteins were then transferred to a nitrocellulose membrane (Millipore, Billerica, MA). After blocking with TBS-0.1% Tween 20 (TBS-T) and 5%–8% bovine serum albumin (BSA) for 2 h, the membrane was probed with one of the following antibodies: mouse anti-human TWIST1 (1:400; Santa Cruz Biotechnology, CA) or mouse anti-human α-tubulin (1:1000; Santa Cruz Biotechnology) in 5% BSA. After washing with TBS-T, the membranes were incubated with peroxidase-conjugated anti-mouse antibody (1:2000; Dako, Glostrup, Denmark) in 2.5% nonfat dried milk.
Quantification of blots was carried out by using free ImageJ software, with measurements recorded by 2 independent researchers.
(Immuno)histochemistry analysis and quantification
After chondrogenic differentiation for 1 day or 3 weeks as described earlier, 3 pellets/donor were collected for histochemical analysis. Pellets were fixed for 24 h in 4% formalin before paraffin embedding. Sections (6 μm) were stained to assess GAG content with 0.4% thionine solution. BMSCs were divided into “chondrogenic” and “non-chondrogenic” categories based on their positivity for thionine. All BMSCs categorized as “chondrogenic” formed pellets >70% positive for thionine, and all “non-chondrogenic” BMSCs gave rise to pellets negative for thionine (Supplementary Fig. S1; Supplementary Data are available online at
For collagen type II immunostaining, antigen retrieval was performed by using 0.1% pronase, followed by incubation with 10 mg/mL hyaluronidase (both Sigma-Aldrich). Thereafter, sections were incubated for 2 h with the collagen type II antibody (II-II/II6B3; Developmental Studies Hybridoma Bank, University of Iowa) and staining was developed by using an alkaline phosphatase-labeled secondary antibody in combination with Neu Fuchsin substrate. An isotype IgG1 monoclonal antibody served as the negative control. To determine proliferative activity, antigen retrieval was performed at 95°C in 10 mM citrate buffer (pH6) with 0.05% TWEEN followed by incubation with a monoclonal mouse anti-human KI67 (Clone MIB-1) primary antibody (1:40; Dako, Netherlands bv, Heverlee, Belgium). Staining was developed by using biotinylated anti-mouse Ig (LINK; Biogenex, ZA-000UM) and alkaline phosphatase-conjugated streptavidin (LABEL; Biogenex, ZA-000UM) in combination with Neu Fuchsin substrate. Negative controls were treated with an isotype IgG1 mouse antibody. Quantification of KI67 staining was performed by manual counting of cells (>900 cells/condition; 2–3 pellets/donor) across multiple randomly taken images.
Murine articular cartilage development
Microarray analysis was performed as previously described [19]. Briefly, embryos were recovered on gestational day 11.5 (E11.5) and E13.5 from three timed pregnant outbred CD-1 IGS mice (Charles River Laboratories, Sulzbach, Germany & Margate, United Kingdom; AREC-P-10-47). Gestational stage E0.5 was considered as the noon of the day that the vaginal plug was first detected. Cryosections of hindlimbs stained with Cresyl Violet were prepared for harvesting of the intermediate layer (II) and the outer interzone (OI) of the femorotibial interzone (IZ) that gives rise to the stable articular cartilage, and the femoral and tibial transient embryonic cartilage (EC) that is the cartilage template for future long bones, by laser capture microdissection (Carl Zeiss Microscopy GmbH). Three biological replicates, each from a different litter, were harvested. Total RNA was extracted, amplified, and labeled by using the Agilent Low Input Quick Amp Labelling Kit (Agilent Technologies) and cRNA was hybridized to Agilent Whole Mouse Genome Oligo Microarrays (Agilent SurePrint G3 Mouse 8 × 60 L Microarray, Agilent Technologies). Fluorescence signals of the hybridized microarrays were detected by using Agilent's Microarray Scanner System, which were read and processed by Agilent Feature Extraction Software (FES). The use and the treatments of the animals for this work were approved by the institutional animal research ethics committee of University College Dublin (AREC-P-10-47).
Data analysis
Data were analyzed with PSAW statistics 20 software (SPSS, Inc., Chicago, IL) or in the case of linear correlation analysis, GraphPad Prism Software 5.00 (GraphPad Prism Software, La Jolla, CA). Normality and variance were determined by the Shapiro–Wilks test and Levene's Test of Homogeneity. In the case of normally distributed data with equal variance, a Student's t-test or one-way ANOVA with Bonferroni post hoc analysis was performed. A Mann–Whitney U test or Kruskal–Wallis test with pairwise comparisons was performed in the case of non-normally distributed data. In cases where the assumption of variance assumptions was not met, a one-way ANOVA (Welch's F test) with Games–Howell post hoc analysis was performed. Data were deemed statistically significant for P < 0.05.
Results
Downregulation of TWIST1 expression occurs with chondrogenic differentiation
To understand whether TWIST1 is involved in cartilage development and formation, we investigated the pattern of Twist1 gene expression in the mesenchyme of developing embryonic murine limbs. Comparing Twist1 expression in mesenchymal cells of the joint interzone (13.5II and 13.5OI) and transient epiphyseal cartilage (13.5EC) with cells of the mesenchymal condensation from which they arise (11.5 M; Fig. 1A), a significant downregulation in Twist1 expression was observed in all tissues (Fig. 1B). Despite this, and consistent with its hypertrophic phenotype (Fig. 1A), only 13.5EC mesenchymal cells displayed a significant corresponding upregulation of all chondrogenic differentiation markers examined: Col2a1 (P < 0.01), Sox9 (P < 0.01), and Runx2 (P < 0.05; Fig. 1B). Of the interzone layers from which the articular cartilage arises, mesenchymal cells of 13.5II failed to significantly upregulate any of the same differentiation markers whereas mesenchymal cells of 13.5OI upregulated Col2a1 (P < 0.01) and Sox9 (P < 0.001). This led to an investigation of the involvement of TWIST1 expression in the chondrogenic program of stable articular cartilage chondrocytes (Fig. 1C). After 3 weeks of HAC redifferentiation, significantly lower TWIST1 expression was observed than in HACs after expansion (P < 0.05), along with a significant upregulation of COL2A1 (P < 0.05), SOX9 (P < 0.05), and RUNX2 (P < 0.05).

TWIST1 expression decreases during chondrogenic differentiation.
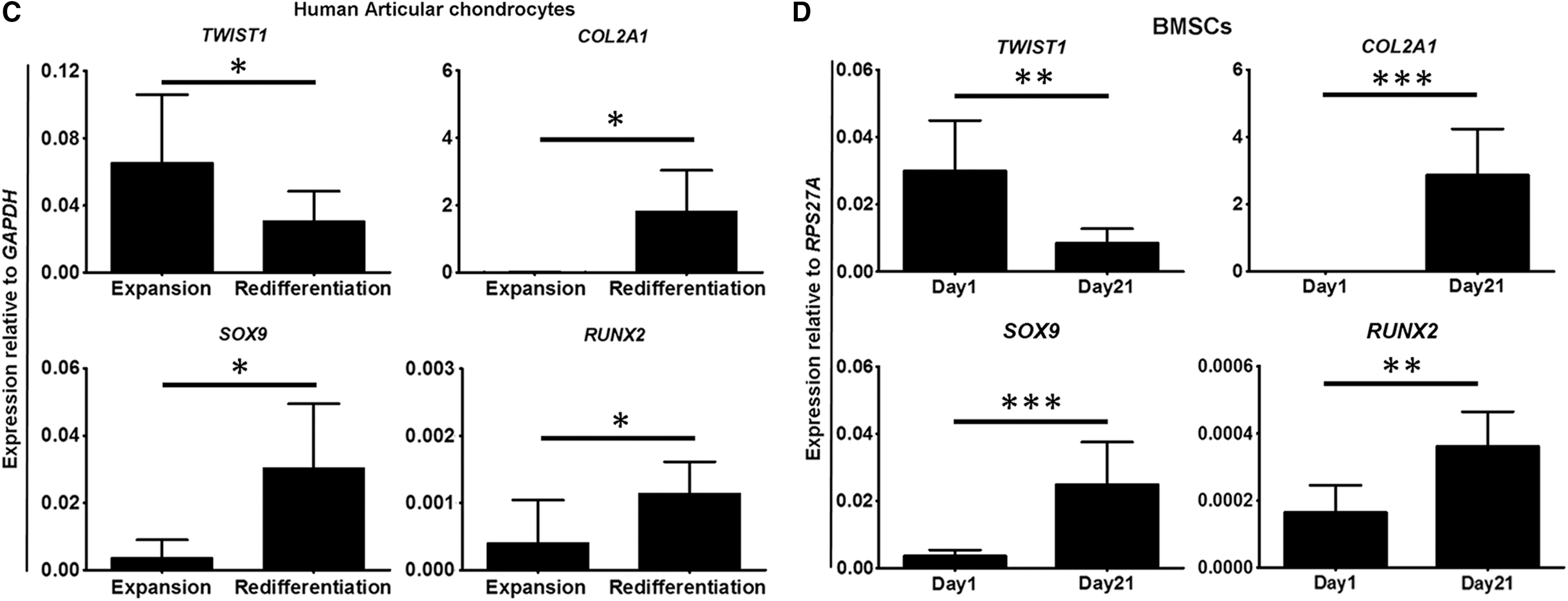
Encouraged by this, TWIST1 expression levels were then investigated in BMSCs in chondrogenic culture. As hypothesized, BMSCs undergoing chondrogenic differentiation, confirmed by COL2A1 (P < 0.001), SOX9 (P < 0.001), and RUNX2 (P < 0.01) expression, significantly downregulated TWIST1 expression (P < 0.01; Fig. D). These data are, therefore, supportive of a role for TWIST1 in the inhibition of chondrogenic differentiation of mesenchymal cells.
Silencing of TWIST1 expression in BMSCs does not improve chondrogenic differentiation
Having observed downregulation of TWIST1 with chondrogenic differentiation, we attempted to improve in vitro chondrogenic differentiation of BMSCs through inhibition of TWIST1 expression by using siRNA technology (Fig. 2A). Despite lower TWIST1 expression levels (P < 0.001; Fig. 2B) and protein levels (Fig. 2C) in TWIST1-silenced BMSCs, which appeared morphologically distinct (Fig. 2D), no improvement, or even a reduction, in BMSC chondrogenic differentiation was observed (Fig. 2E, F). Indeed, after 21 days of chondrogenic culture, the GAG and collagen type II content (Fig. 2E) of TWIST1-silenced BMSCs appeared to be less, with no improvement in COL2A1 (P = 0.857), SOX9 (P = 0.286), or RUNX2 (P = 0.082) expression (Fig. 2F). These findings suggest an important role for TWIST1 in not only inhibition of chondrogenic differentiation but also promotion of BMSC chondrogenic differentiation.
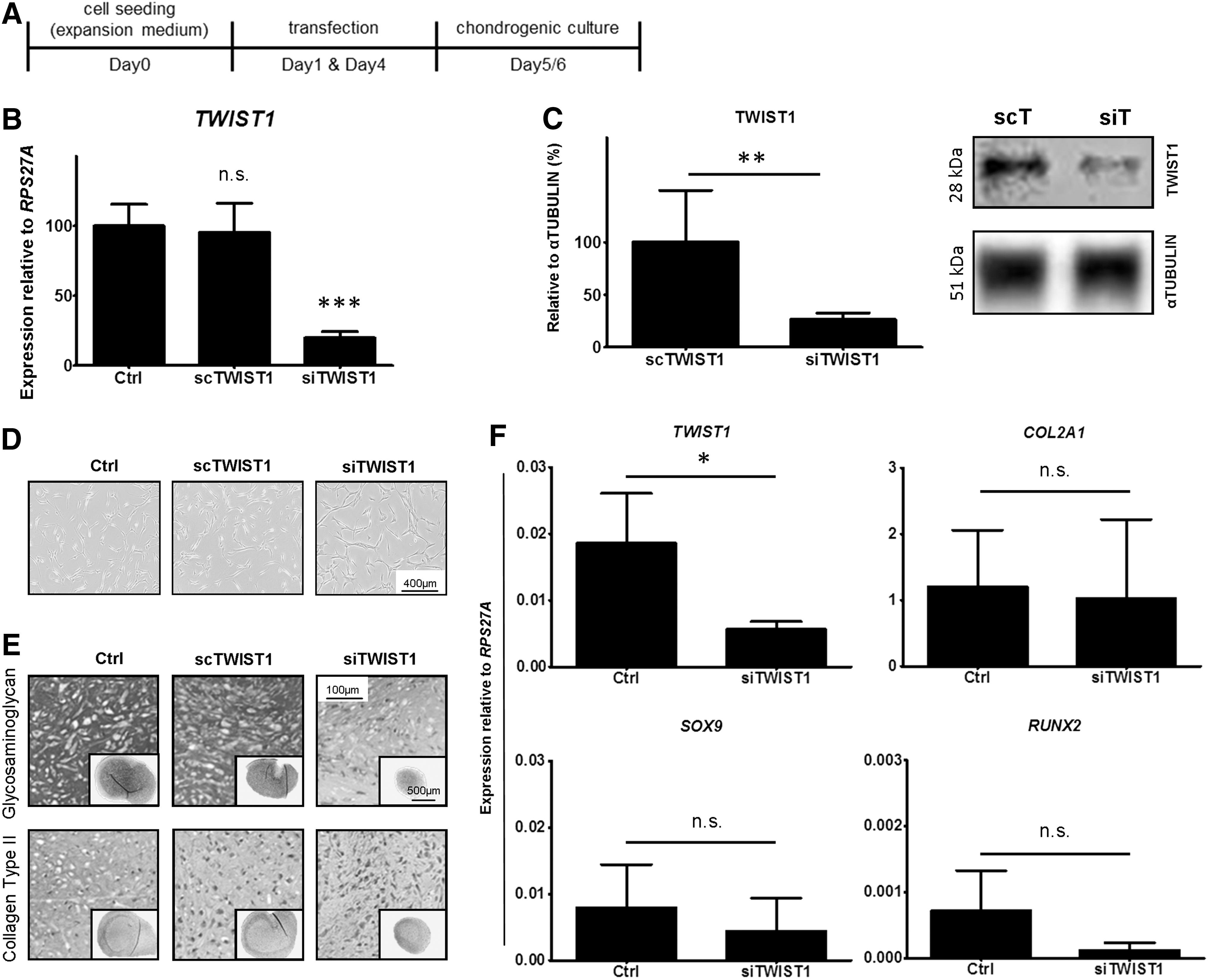
TWIST1-silenced BMSCs display no improvement in chondrogenic differentiation potential.
Initial upregulation of TWIST1 expression is required for chondrogenic differentiation of BMSCs
To gain an understanding of why TWIST1 silencing negatively impacted BMSC chondrogenic differentiation, we further investigated TWIST1 expression during in vitro culture, including non-chondrogenic BMSCs. Unexpectedly, between the end of monolayer expansion and day 1 of chondrogenic pellet culture, both chondrogenic (P < 0.05) and non-chondrogenic (P < 0.01) BMSCs upregulated TWIST1 expression (Fig. 3A). In non-chondrogenic BMSCs, however, upregulation was not followed by a downregulation of expression during chondrogenic differentiation culture (day 1 vs. day 21; P = 0.672). To evaluate whether the observed upregulation of TWIST1 expression occurred as a direct result of exposure to TGFβ1 in the chondrogenic induction medium, we assessed the effect of the presence or absence of TGFβ1 on BMSCs in monolayer (Fig. 3B) and in a chondrogenic pellet culture (Fig. 3C). After 24 h in monolayer, TGFβ1 significantly downregulated TWIST1 expression levels (P < 0.001; Fig. 3B), whereas a comparison of TWIST1 expression levels in BMSCs after 1 day of chondrogenic pellet culture revealed no direct effect of TGFβ1 (Fig. 3C), implying that initial upregulation in chondrogenic culture is not a consequence of TGFβ1 exposure.
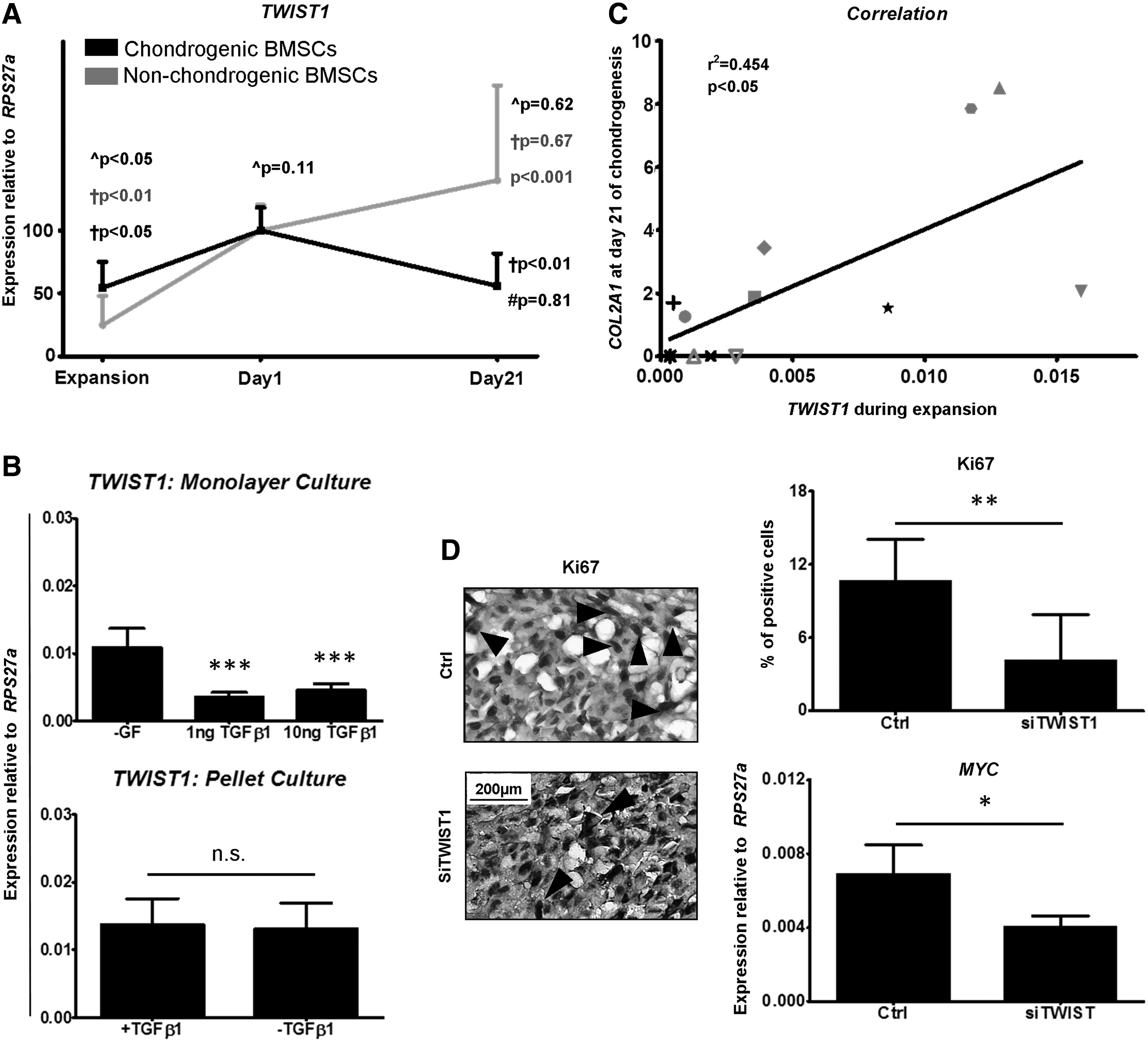
TWIST1 expression increases during BMSC chondrogenic culture.
After monolayer treatment, TWIST1-silenced BMSCs appeared to reach subconfluence more slowly (Fig. 2D). We, therefore, next investigated whether this upregulation in TWIST1 expression and failure of TWIST1-silenced BMSCs to undergo chondrogenesis were linked to the timing of treatment and proliferation. Indeed, after expansion, higher TWIST1 expression levels correlated with greater COL2A1 expression levels after 21 days of chondrogenic differentiation (r 2 = 0.454, P < 0.05). Furthermore, KI67 staining (P < 0.01) and MYC gene expression analysis (P < 0.05) revealed significantly less proliferating cells in pellets formed from TWIST1-silenced BMSCs after 1 day of chondrogenic culture (Fig. 3D), suggesting that failure of TWIST1-silenced BMSCs to chondrogenically differentiate may be due to a lack of proliferation during chondrogenic induction. In summary, these data demonstrate for the first time that during in vitro BMSC chondrogenic differentiation, an initial upregulation of TWIST1 expression followed by downregulation of expression is required for chondrogenesis (Fig. 4).

Proposed model of TWIST1 expression during BMSC chondrogenic differentiation. TWIST1 is expressed during chondrogenic induction where it is involved in proliferation. Thereafter, chondrogenic BMSCs downregulate TWIST1 expression whereas non-chondrogenic BMSCs fail to do so, preventing differentiation.
Discussion
The TWIST1 transcription factor is a recognized inhibitor of mesenchymal cell differentiation and is a novel target for improvement of in vitro engineering of MSC-based bone tissue [10,11,14,20]. A similar target function of TWIST1 for in vitro MSC chondrogenic differentiation is envisioned; however, although it has been established that TWIST1 inhibits chondrogenic processes in murine and chick mesenchymal cells [12,14,15], a role for TWIST1 in human chondrogenic differentiation processes still requires establishment. In this study, we provide the first evidence of dynamic regulation of TWIST1 expression during in vitro human BMSC chondrogenic differentiation and we link the observed difference with donor variation. In addition, our data support the important role of TWIST1 expression in sustaining early chondrogenesis, and therefore the entire chondrogenic differentiation process.
During skeletal development, Twist1 expression is present and restricted to limb bud mesenchyme from initiation through to the early stages of chondrogenesis [21,22]. In our embryonic model of chondrogenesis, Twist1 expression was present and observed to decrease as mesenchymal cells progressed in development. Similarly, HACs and BMSCs undergoing chondrogenic differentiation downregulated TWIST1 expression, whereas non-chondrogenic BMSCs failed to do so. Consequently, we considered TWIST1 an important inhibitor of chondrogenesis and chondrocyte gene expression, and we believed that removal of TWIST1 inhibition would improve in vitro BMSC chondrogenic differentiation. We, however, failed to foresee an upregulation of TWIST1 expression in the first days of our chondrogenic culture set-up and subsequently, TWIST1-silenced BMSCs actually displayed worse chondrogenic differentiation potential.
That TWIST1 expression increases during this initial phase of chondrogenic culture is not completely unexpected. One of the first steps of cartilage formation in vivo is condensation of mesenchymal cells, a step that is equally important in in vitro chondrogenesis [23,24]. Twist1 expression is known to correlate with regions under the influence of FGF signaling, and TWIST1 itself has been shown to be active during FGF/FGFR signaling [4,5,7,25 –27]. Previously, we demonstrated that FGFR2, important for the condensation phase during development [28 –30], is present in BMSCs during chondrogenic induction [31]. Furthermore, extensive work by the Tuan group has revealed that condensation of adult human MSCs occurs downstream of TGFβ1-induced β-CATENIN expression, both of which are known upstream signaling pathways of TWIST1 [23]. Therefore, upregulation of TWIST1 in our culture may occur as a result of condensation processes. Absence of TGFβ1 signaling, however, had no direct effect on TWIST1 expression levels, and for all but one donor TWIST1-silenced BMSCs appeared to condense normally during chondrogenic induction. Thus, TWIST1 expression does not appear crucial to this process and was not likely the cause of impaired chondrogenic differentiation of these cells.
Indeed, we believe that failure of TWIST1 silencing to improve chondrogenic differentiation may be linked to proliferation. Higher proliferation rate in monolayer expansion culture is often linked to better chondrogenic differentiation potential [17]. Moreover, Dexheimer et al. demonstrated a requirement for proliferation during chondrogenic induction culture [32]. Freshly isolated adult BMSCs express high levels of TWIST1, a known promoter of MSC proliferation [16], which is downregulated during expansion culture [16,17], as too is the chondrogenic differentiation potential of BMSCs [17]. In our system, TWIST1 silencing could impact both monolayer expansion proliferation and pellet culture proliferation. Indeed, TWIST1-silenced BMSCs seemed slower to reach confluence during expansion culture compared with control or scramble-treated BMSCs [data not shown]. In addition, TWIST1-silenced BMSCs displayed fewer proliferating cells after 1 day of chondrogenic pellet culture. It is, therefore, possible that decreased proliferation in our TWIST1-silenced BMSCs resulted in their failure to undergo chondrogenesis. More extensive studies interfering with proliferation are required to confirm this.
Investigation of endogenous TWIST1 expression during chondrogenic differentiation culture raised another interesting question: Why do non-chondrogenic BMSCs, unlike chondrogenic BMSCs, fail to downregulate TWIST1 expression after this initial upregulation? WNT signaling is a known upstream regulator of TWIST1 expression that promotes MSC proliferation [14,15,17], which is transiently upregulated during chondrogenic induction [23] and which must be downregulated during chondrogenic differentiation [17]. Hence, its potential expression profile, upregulation followed by downregulation, mirrors that observed for TWIST1 in chondrogenic BMSCs. It is possible, therefore, that failure of WNT signaling downregulation in these cells is responsible for their undesired differentiation outcome. Treatment of BMSCs with WNT3A, however, revealed no consistent effect on TWIST1 expression levels. Similarly, treatment with two other known regulators of chondrogenesis and TWIST1 expression, FGF2 and TNFα [10,17,33 –35], failed to have an impact on TWIST1 expression levels (Supplementary Fig. S2). Only TGFβ1 treatment demonstrated a clear and significant effect, an effect that could not be observed during chondrogenic induction. In addition, non-chondrogenic BMSCs had lower expression levels of TWIST1 after expansion, which correlated with lower COL2A1 expression after chondrogenic differentiation. Failure of MSCs to undergo differentiation is often proposed to occur as a result of these cells being more committed and less able to respond to differentiation stimuli. That TWIST1 expression is associated with an uncommitted state in adult MSCs [16,17] lends support to this concept.
In our proposed model (Fig. 4), an upregulation of TWIST1 during chondrogenic induction followed by downregulation during differentiation is required to promote chondrogenic differentiation of adult human BMSCs. As an upregulation of TWIST1 occurs during induction, likely occurring in association with proliferation, silencing of TWIST1 expression before this induction phase may not prove a successful strategy for improvement of in vitro BMSC chondrogenic differentiation due to its interference with BMSC proliferation, as was demonstrated in this study. To overcome the lethality of Twist1 null models, conditional Twist1 gene allele models have been generated [36]. A similar conditional inactivation strategy may be required for BMSCs undergoing chondrogenic differentiation. The exact timing of the inactivation, however, would require extensive research and is almost certainly to vary between donors. In actuality, Twist1 expression is required by immature chondrocytes to prevent their progression toward hypertrophy, through inhibition of Runx2 expression [15]. Chondrogenically differentiated BMSCs, redifferentiated HACs, and mesenchymal cells of the transient EC all fail to sustain a stable articular chondrocyte phenotype, but they show signs of hypertrophy and share a similar expression profile: decreased TWIST1 expression with increased RUNX2 expression. Thus, maintaining moderate levels of TWIST1 expression during differentiation may help in acquiring and maintaining a more stable articular chondrocyte phenotype.
In this study, we demonstrate for the first time the dynamic regulation of TWIST1 expression during chondrogenic differentiation of BMSCs. Furthermore, we highlight a distinct difference between chondrogenic and non-chondrogenic BMSCs during chondrogenic culture: their regulation of TWIST1 expression. After initial upregulation of TWIST1 expression during induction of chondrogenic differentiation, non-chondrogenic BMSCs fail to downregulate TWIST1 expression, unlike chondrogenic BMSCs. Elucidation of TWIST1 upstream and downstream molecular activity will further identify targets to facilitate optimization of in vitro BMSC chondrogenic differentiation for clinical purposes.
Footnotes
Acknowledgments
The authors would like to acknowledge Dr. Andrea Lolli and Prof. Dr. Roberta Piva (University of Ferrara) for assistance in setting up and optimizing the silencing protocol, and Dr. Derk ten Berge (Erasmus MC) for provision of WNT3A. This research was financially supported by Science Foundation Ireland (SFI; grant no. 11/RFP/BMT/3150), the Netherlands Institute for Regenerative Medicine (NIRM; grant number FES0908), and the Netherlands Organisation for Health Research and Development (ZonMw) Translational Adult Stem Cell Research programme (TAS; grant no. 116005009). R.N. was further supported by a VENI grant from the Dutch Technology Foundation (STW, grant no. 13659). None of the aforementioned had any involvement in the design or interpretation of data of this study.
Author Disclosure Statement
No competing financial interests exist.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.