
Abstract
N-methyl-
Introduction
T
Although the number of research articles focusing on the cues, which trigger the NMDAR localization across a developing neuron, and the role of NMDAR in embryonic and adult stem cell proliferation and differentiation have been growing dramatically over the past few years, there is not a single review on this topic. In the proposed review, we will summarize the current understanding of NMDARs in stem cell biology and their involvement in pathophysiological processes, especially during early neuronal development (of immature neurons) and differentiation.
In Vivo NMDAR Modulation and Adult Neurogenesis
The development of early neurons has been attributed to the neuroepithelial cells and the intermediate neuronal progenitor cells in the neocortex region and the dentate gyrus of the brain [13]. Cortical neurogenesis triggers the neuroepithelial cells to undergo cell polarization, increase in cadherin expression and overexpression of the adherens junction, and finally attaining the structure of RGCs. The polarized RGCs undergo interkinetic nuclear migration across the ventricular zone under the influence of retinoic acid (RA) and the interaction of the Notch protein with the notch effector C-promoter binding factor 1 [14,15]. Compartmentalization of the developing brain has been attributed to the RGCs and their rate-limiting regulation of the neuronal migration. It has been observed that the lateral striatum of the brain is localized by retinol binding protein-expressing RGCs, which under the influence of RA undergo differentiation into neurons [16]. The initial neuronal network triggers developmental regulation of ionotropic glutamate receptors across the synaptic soma.
The transitioning of RGCs to mature neurons includes the production of intermediate neuronal progenitor cells. Early neuronal progenitor cells are often less vulnerable to glutamate cytotoxicity and express various glutamate receptors. In the embryonic subventricular zone (SVZ), the transition of the RGCs to the intermediate progenitor cells (IPCs) has been attributed to the downregulation of Pax6 and upregulation of Tbr2 transcription factor (a member of T-box transcription factor) in the cells (Fig. 1). These IPCs express Tbr2 along with Tbr1 to undergo terminal differentiation into neurons [3,4]. However, RGCs undergoing direct differentiation into neurons may or may not express Tbr2 at all. The C-terminal domain of the Trb1 associates with CASK, a peripheral plasma membrane protein, to regulate promoter activity of the GluN1 gene. The mouse, rat, and human GluN2 gene also shown to have a Trb1 binding site in the promoter referred to as GluN2b-2T [17]. Quantitative PCR assay has demonstrated the upregulation of GluN1 and GluN2B mRNA levels in differentiating IPCs [18]. NMDAR-mediated Ca2+ influx further propagates migration of the differentiated cortical neurons [19] in a Rho-GTPase-Rac1-dependent manner. The neuronal network is presumed to fire electrical field signals, which trigger a physical interaction between the NMDAR and activation of Rac1 (Tiam1 and effector Pak1). Tiam1 interacts with the actin cytoskeleton whose remodeling process triggers neural progenitor cell migration. Downstream expressing genes such as HES1, ID2, Cux2, and NeuroD are transcribed, triggering the neural progenitor cells and the dentate gyrus granule cells to mature [2,20].
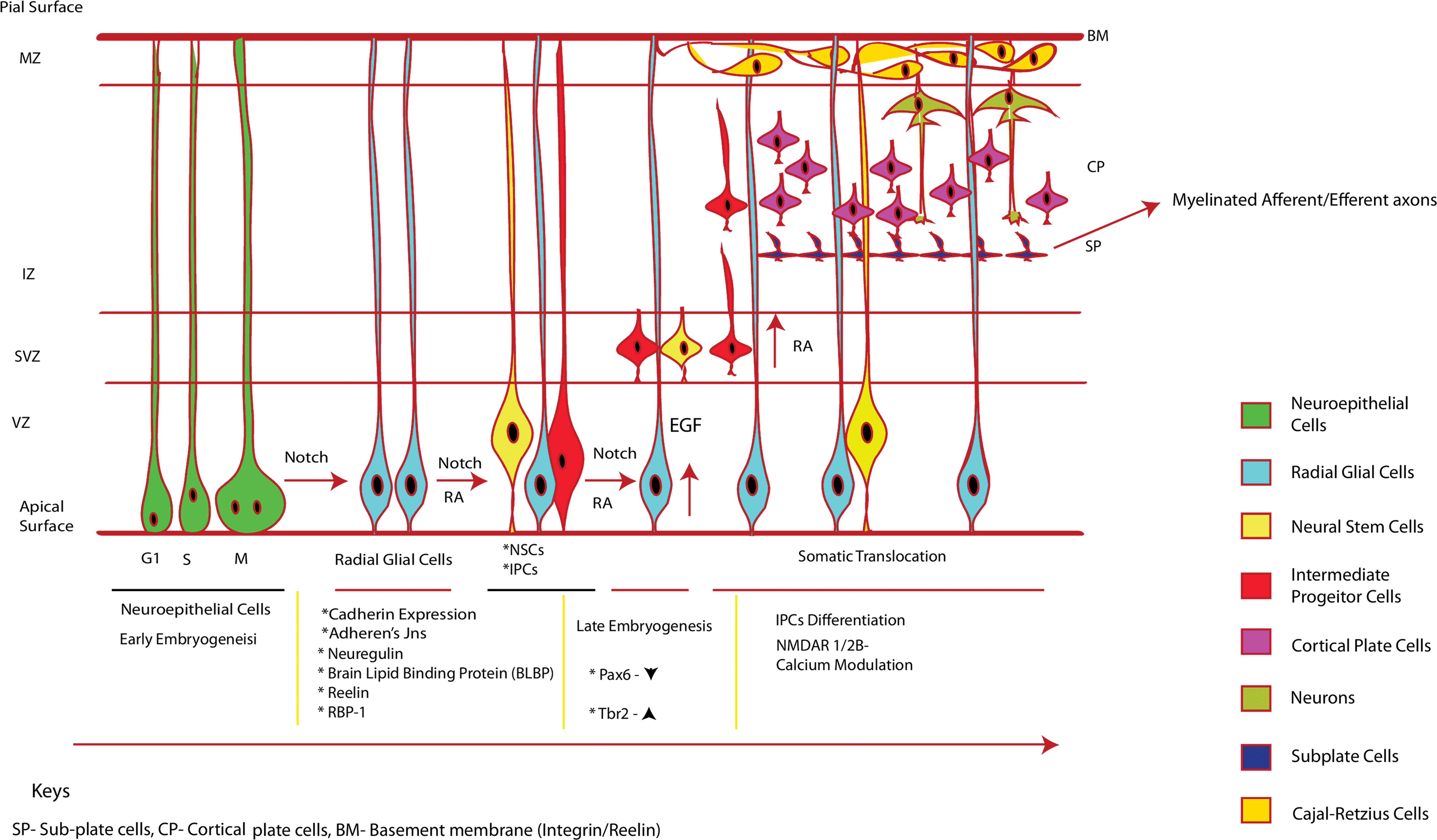
Differential stages of neuroepithelial cells and development into mature neurons: neuroepithelial cells (apical progenitor cells), under the influence of Notch signaling, lose their tight junction and form RGCs. These cells have a short apical process and a longer basal process contacting the pial basement membrane. The RGCs further undergo interkinetic nuclear migration and maintain a proliferative state under the influence of Notch. RA induces cells to divide into radial glia such as NSCs and multiple IPCs. Progressive division of the RGCs further trigger interkinetic nuclear migration of the cells and the NSCs and IPCs stack at the SVZ to form a distinct layer. The IPCs further migrate upward through the IZ to form the MZ and form cell stacks of subplate, cortical plate, and marginal zone basal membrane. The cells loose contact to the subventricular zone/ventricular zone (SVZ/VZ), attach to the basal membrane, start differentiating into immature neurons, attain GluN2B/calcium channel receptors, modulate Ca2+ influx, and finally attain maturity. The cells of the subplate form myelinated afferent/efferent axons. IPC, intermediate progenitor cell; IZ, intermediate zone; MZ, marginal zone; NSC, neuronal stem cell; RGC, radial glial cell; SVZ, subventricular zone. Color images available online at
Finally, terminal differentiation of the neural progenitors (cortical neurogenesis) leads to the formation of either glutaminergic neurons (excitatory) or GABAergic neurons (inhibitory gamma-aminobutyric acid receptor). Adult neurogenesis (an integral aspect of postcortical neurogenesis) occurs in the SVZ and the subgranular zone (SGZ) of the dentate gyrus of the brain, where many signaling pathways culminate to influence the specific differentiation of the neural progenitor cells [21].
Since developing neurons require glutamate for cortical migration during early neurogenesis, early transcription of GluN1/GluN2B and GluN2D indicates potential modulation of neuronal network [22,23]. This has been demonstrated with murine cortical cells, where their migration during corticogenesis is influenced by glutamate/Ca2+-mediated NMDAR modulation [23]. The RGCs of the Xenopus laevis activate NMDAR mediated Ca2+ influx, to trigger NOS signaling-mediated neuronal activity and visual stimulation [24]. Cultured chick radial glial subtypes (Muller glia and Bergman glia) also show expression of NMDAR1 and NMDAR2A/B mRNA transcription within the cells [25]. All these examples indicate that the development of the RGCs into neurons and the induction of synaptic signaling is a stepwise developmental process with targeted localization of NMDARs across the neurons [26].
Notch 1-mediated nuclear factor kappa-light-chain-enhancer of activated B cells receptor (NF-κB)-induced regulation of the genes involved in neurogenesis (adult/embryo) [27] and peripheral neuron myelination [28] is supposed to maintain neural stem cell (NSC) populations in the SGZ by triggering a BCL2/BAX-XL-mediated antiapoptotic effect on the cell population [29]. Among other functions of the NF-κB signaling is the regulation of transcription of the genes FOXO1 and PKA, which regulate axon growth cone development [30]. Axonal induction triggers synaptic complex formation, in which glutamate triggers the NMDA/2-amino-3-(3-hydroxy-5-methyl-isoxazol-4-yl)propanoic acid (AMPA) receptors, and increases the localization of NF-κB subunits in the nucleus of a hyperactive neuron (reviewed in Ref. [30]).
Before the synaptic localization of the NMDARs, the complex assembly is processed in a specialized endoplasmic reticulum (ER) compartment followed by the dendritic Golgi apparatus, where the NMDARs have been shown to interact with the PSD-MAGUK protein complex (Fig. 2). The GluN1/2 subunit has been shown to interact with a MAGUK complex: SAP102, which mediates NMDAR trafficking across the dendrites [31]. SAP102-Sec (exocyst multiprotein complex)-mPins (mammalian homolog of Drosophila melanogaster partner of inscuteable) complex interacts with the GluN1/2 NMDARs in an activity-dependent manner, in which the GluN2B-SAP102 complex predominates in a young developing synapse [32]. PSD-95, another MAGUK protein (part of the PDZ domain containing proteins), predominantly associates with the GluN2A NMDAR complex in the mature neuron [31].
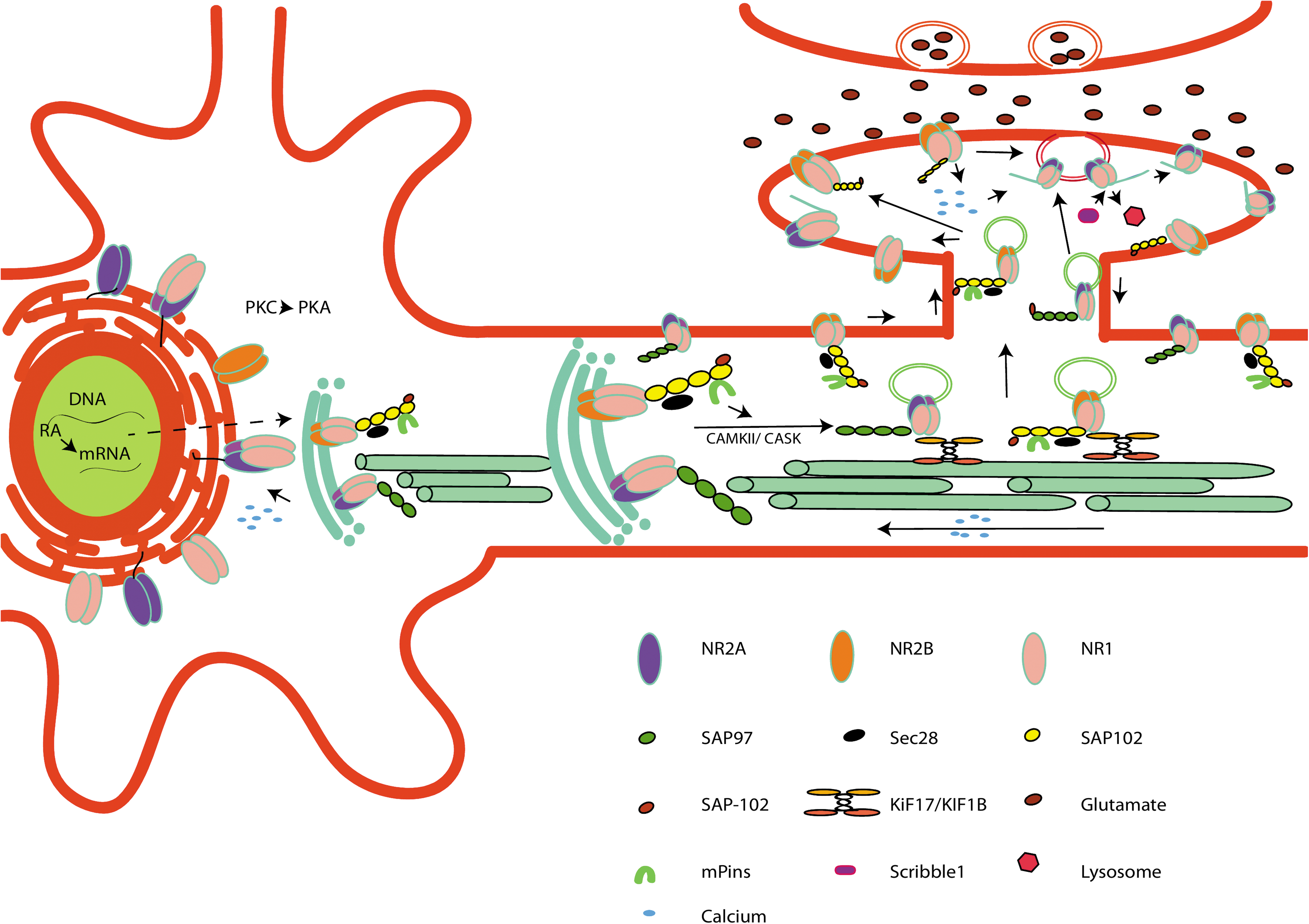
Sequential NMDAR maturation model: RA induces synthesis of GluN1 and GluN2B mRNA and exports from the ER receptors to the Golgi. Compartmentalization of NMDARs takes place at the ER followed by addition of SAP102/SAP97 complex to the subunits, based on receptor composition. The NMDARs are then either inserted into vesicles and transported directly to the plasma membrane or transported into dendrites by associating with the SAP102-Sec8-mPins complex. Alternatively, the NMDARs are transported to the dendritic Golgi outposts using KIF17/KIF1b kinesin motors. The GluN1/2B-PSD-95 complex is first inserted to the synaptic bulb in the developing immature neurons. Glutamate-induced Ca2+ influx through the GluN2B induces a feedback mechanism loop within the neuron, which induces GluN2A mRNA transcription. GluN1-GluN2A/SAP97 are transported to the synaptic and extrasynaptic domains. As the neurons mature, more Ca2+ influx acts as a developmental cue to replace the synaptic GluN2B with the GluN2A complex. GluN2A associates with MKGUK (PSD-95, scribble1), which helps in recycling of the GluN2A complex. ER, endoplasmic reticulum; RA, retinoic acid. Color images available online at
In a developing neuron, the SAP102-Sec8-mPins-NMDAR complex is transported to the synaptic soma with the help of the kinesin motors (KIF17/KIF1b) [33]. As the neurons mature, the switch from the SAP102 to PSD95/93 complex, as the primary PSD-MAGUK, gives an insight about the preferential synaptic clustering of the NMDAR complex in a developing brain [34]. However, there are contradictions in the findings, where SAP102 has been shown to be indiscriminate of the GluN2A/B subunit trafficking [35]. SAP97, another MAGUK protein, mediates NMDAR trafficking from the somatic ER to the dendritic Golgi outpost in a CAMKII/CASK-mediated manner with the help of KIF17 [36 –38]. Overexpression of SAP97 has been shown to increase the NMDARs in the synaptic soma, preferentially GluN2A (SAP97 directly interacts with GluN2A with its PDZ1 domain), and is considered to control the transition between the GluN2A/2B receptors [5,36,38].
RA-mediated ESC differentiation produces neural progenitors that often express early markers such as Sox1 [39], PSA-NCAM [40], nestin [41], and doublecortin [42]. Previous studies have indicated that RA treatment of NSCs leads to an increase in the GluN1 and GluN2B mRNA levels, whereas no significant increase in the GluN2A mRNA level has been observed [18,43]. As previously discussed, during the RA induction of axonal growth cones/neuronal culture, NMDARs undergo selective trafficking and endocytosis leading to a synchronized localization of the receptors. The redistribution of the NMDAR subunits has been accredited to the changes in the Ca2+ levels through the NMDARs in a feedback mechanism loop, increasing or decreasing the GluN2 subunit transcription [44,45]. GluN1 subunit clustering has been observed in axonal cones as early as on day 3 in vitro (DIV3), whereas transfection of the GFP-GluN2A and GFP-GluN2B subunits in DIV3 axons also shows their specific localization and expression at the axonal growth cones [46]. Furthermore, hippocampal culture studies act as a proof of the role of GluN2B-mediated neuroprotection till DIV10 cultures [47].
Striatal NPCs have also been shown to undergo enhanced proliferation in a glutamate-dependent NMDAR-dependent manner [48,49]. Patch-clamp studies indicate the presence of various NMDAR subtypes in the striatal medium spiny neurons eliciting synaptic excitation [50]. Nonetheless, striatal neurons are susceptible to mitochondrial excitotoxicity on an increased accumulation of Ca2+ on NMDA treatment, thus disrupting the homeostasis generally maintained with the cytosolic Ca2+ [51].
NMDAR Subunit Distribution in Neural Progenitor Cells
The physiological implications of the aberrant glutamate uptake or irregular Ca2+ influx by the developing neurons and their association with various neurological diseases have been well documented [6,52,53]. The influence of the NMDAR sublocalization in a developing neuron, preferentially activating downstream ERK signaling [54], activating/deactivating neuronal death pathways [55,56], maintaining LTP/LTD [57,58], and maintaining cell plasticity [59,60], has already been identified. These varied NMDAR functions have been attributed to the selective localization of the NMDARs at the postsynaptic densities and extrasynaptic domains of the neurons [61,62]. However, unlike mature neurons that require AMPA-mediated Mg2+ unblocking of the NMDAR, GABA centric pre-depolarization of the immature neurons is the first step to trigger NMDAR-mediated neural network clustering [63]. Prepolarization persists until the effects of the activated synaptic and extrasynaptic NMDAR trigger the AMPA clustering in the synaptic bulb, leading to an NMDA-mediated pro/antisurvival signaling in developing neurons [64].
The ability of neurons to modulate the surface expression of the NMDARs depending on the functioning of the synaptic networks shows the versatility of the mechanism [65]. The subunit switch from the GluN2B to the GluN2A is a well-documented developmental cue during the postnatal neuron maturation [5]. Hippocampal mossy fiber-CA3 pyramidal neurons have the NMDAR subunits GluN1, GluN2A, GluN2B, GluN2C, and GluN2D localized in postsynaptic membranes, whereas GluN1, GluN2B, GluN3B, and GluN2D are localized at the presynaptic and synaptic membranes [66]. A similar localization of NMDARs can be observed in postnatal day 2 brains, which are predominantly clustered with GluN2B receptors, that switch to GluN2A receptors by day 15 [67]. The switch is an important indicator of neuron maturation stages and establishes their responses to long-term potentiation (LTP) in an Ras-GTP-dependent pathway. That being said, use of various NMDAR-specific antagonists has shed light on their potential role in mediating excitotoxicity and pro/antisurvival signaling in a developing hippocampal neuron. GluN2A is associated with glutamate-mediated neuronal survival. In diseases such as stroke and ischemia, the GluN2A subunit triggers an NMDA-induced toxicity on loss of its C-terminal domain. GluN2B subunit also mediates NMDA toxicity, predominantly in the absence of GluN2A-mediated neuroprotection on phosphorylation of its S-1303 subunit [56,68,69]. The GluN2B antagonist has also been implicated in neuroprotection on aberrant PI3K-mediated cell apoptosis [70]. However, recent studies indicating an equal distribution of the GluN2B in the synaptic and extrasynaptic domains of 3-week hippocampal slices have raised confusion regarding the use of GluN2B antagonists such as ifenprodil or Ro25-6981 as right target therapeutics for the NMDAR-mediated excitotoxicity [71].
The need to understand the functional role of NMDARs in vivo has led to designing models for differentiating neurons from ESCs and induced pluripotent stem cells (iPSCs) [10,11]. The successful differentiation of the hESCs into neurons has already been demonstrated in previous studies [12,72]. However, stringent regulation of the use of hESCs has given rise to alternate neurogenesis models derived from human somatic cells, induced pluripotent stem, and mouse ESCs. These models have been extremely beneficial in differentiating patient-specific diseased cells into neurons and understanding their intrinsic signaling mechanism. The induction of neurons from patient-derived fibroblasts has been performed by the transfection of Oct-4, c-Myc, Nanog, Klf4, Sox2, Brn4, Brn1, and E47 [73 –76]. Alternate sources such as human cortical perivascular cells, mouse Sertoli cells, and human astrocytes have been used as a source for the generation of iPSCs [77 –79]. The use of ES-derived neural progenitors in comparison to other sources has been attributed to their high wingless-int-1 signaling-mediated differential probability [80]. Moreover, a wide array of genes have since been identified, providing an insight about the proliferation, differentiation, and autorenewal of the neuronal progenitors [81]. A deep resequencing method has identified MAP1A, GRIN2B, and CACNA1F to be predominantly susceptible to missense mutations in a large population with neurological disorders [82]. The NMDAR-mediated signaling has also been attributed to various neurological diseases and neural survival. Although most of the neurological disorders have been associated with mature neurons, links between the NMDARs with early onset of diseases during NSC maturation have been an important aspect of research focus.
Diseases such as schizophrenia, ALS, Parkinson's disease, Huntington's disease, congenital mental retardation, spinal muscular atrophy (SMA), and familial dysautonomia have already been shown to be associated with NMDAR malfunction. An onset of collective mutation of high-risk genes such as disrupted-in-schizophrenia-1 (DISC1), catechol-o-methyltransferase (COMT), neuregulin (GluN1G1), and dystrobrevin-binding protein (dysbindin) has been attributed to schizophrenia. DISC1 has been shown to regulate neurogenesis, synaptic organization, axon extension, and maturation and migration of the NPCs in the ventricular zone and SVZ [83]. DISC1 mutation in cultured rat cortical neurons are attributed to an increased GluN2A synaptic localization and calcium influx leading to an increased LTP [84]. In contrast, GluN1 mutation has been attributed to increased BDNF (brain-derived neurotrophic factor)/TrkB (tropomyosin-related kinase receptor B)-mediated hypophosphorylation of the GluN2B subunit, decreased clustering of the GluN1 (erbB4)-PSD-95 complex, and a decreased NMDAR activity in mice, as well as cultured cortical neurons [85 –87]. The same goes for the dysbindin (DTNBP-1) gene mutation whose varied expression level contributes to schizophrenia. Although the exact mechanism of the dysbindin-mediated LTP regulation is not known, increased dysbindin-1 expression has shown a decrease in GluN11 expression in the neuronal surface [88]. Knocking down dysbindin-1 expression has shown an increase in the GluN2A-mediated LTP and a decrease in the extrasynaptic GluN2B expression in neurons [8].
ALS has been associated with mutations in the Cu/Zn superoxide dismutase (SOD-1) gene leading to SOD-1 protein aggregates in the neurons. Enhanced accumulation of the mSOD1 unfolded protein in the ER of the neurons leads to Zn2+- and D-serine-mediated NMDAR, Ca2+ excitotoxicity, and stress-induced caspase-12 activation (reviewed in Refs. [6,89]). Mutation in another gene, TAR DNA binding protein-43, enhances neuronal death independent of the GluN2A-PTEN-TDP43 neuroprotective mechanism [90]. Although mutations in other genes such as C90RF72 and ubiquitin-2 are also the major cause of ALS, not many details have been found [91].
Huntington's disease is associated with partial loss of striatal projection neurons [7]. Mutation of the HTT gene (CAG repeats in exon 1–HTT gene) leads to the addition of polyglutamine repeat to mHtt protein leading to Huntington's disease. This leads to an altered PAX 6 and MMP gene transcription conferring mHTT-induced glutamate excitotoxicity [52,92]. This leads to an aberrant Ca2+ signaling in striatal neurons as seen in cultures [92] along with PACSIN1-mediated endocytosis of the GluN3A receptor during their development. This inhibits the synaptic retrieval of NMDAR GluN3A, preventing clathrin-mediated endocytosis [52]. Also associated is the reduction in active GluN2A receptors in the newly formed synaptic complexes [53]. However, increased expression of GluN11, along with Huntington's disease in the subpopulation of the striatal projection neurons, cholinergic interneurons, and Y-containing interneurons, has been attributed to the ability to survive the disease [29].
Congenital mental retardation is often associated with HCMV infection, which targets NPC targeting numerous genes involved in cellular differentiation and neuronal outgrowth in the NPCs by the downregulation mechanism. One of the primary genes being downregulated is the gene encoding neuronal outgrowth protein SEMA3A (often produced by stressed neurons and mediates NMDA excitotoxicity) and GRIN1 [9,93] leading to the downregulation of NMDAR-mediated Ca2+ influx [93].
iPSC model of neuron culture has given highlights about the NMDAR function and localization in the in vitro SMA model of neuron culture. SMA is associated with the mutation of the survival of motor neuron (SMN) gene. It is already known that the interaction between SMA1-SIP1 (SMN interacting protein) protein complex induces biogenesis of the small ribonucleoprotein core complex (sGluN1NP) [94]. Abolishment of this sGluN1NP spliceosomal complex is often associated with the mutation in the SMN1 gene [95,96], leading to the PI3K/AKT antiapoptotic pathway activation in the neurons [97]. However, use of a NMDA agonist/Ca2+ agonist has been shown to enhance increased cAMP response element-binding protein (CREB)-phosphorylation, leading to CREB-1:CREII-induced SMN gene transcription, and an increased SMA1 protein production in the neurons [97,98].
Targeted therapeutics against the NMDAR subunits, especially in the abovementioned neurological diseases, can be initiated as early as the NSCs or immature neuronal stages of their development. Creation of iPSC disease models to replicate and understand the redistribution of the NMDAR subunits in these diseases can provide insights into the instigation of the disorders [99 –102].
Concluding Remarks
The NMDAR functions and their diverse subunit properties have been the subject of an extensive area of research. Much has been debated on the role of the NMDARs influencing LTP/depression in mature neurons and their activity-dependent localization influencing downstream signaling pathways in the synapses. Both the pro/antisurvival properties of the NMDAR act as a developmental cue during the maturation of a neuron and development of new axonal growth cones. That being said, the lack of information regarding the NMDAR subunit distribution and functions, especially during the early differentiation of neurons (before the formation of neuronal network), has been an area of concern. Evidence continues to accumulate regarding the NMDAR activity in neurons at their immature stage and targeted therapeutics against them in various neurological diseases.
Footnotes
Author Disclosure Statement
No competing financial interests exist.