
Abstract
Mesenchymal stem cells (MSCs) have been proved to be an important element in cell-based therapy. Photobiomodulation used extremely low-level lasers (LLLs) to affect the behavior of cells. The effect mechanism of LLLs on MSCs from human remained to be discovered. In this study, cell viability was assessed using MTS assays and cell cycle was evaluated by fluorescence-activated cell sorting (FACS). The influence of LLLs on mitochondrial biogenesis (fission or fusion) and function (ATP, reactive oxygen species [ROS], nitric oxide [NO]) was evaluated by transmission electron microscope, FACS, quantitative real time polymerase chain reaction (q-PCR), and immunocytochemistry. Cell migration and cytoskeleton alteration (actin and tubulin) were evaluated using transwell assay, immunocytochemistry, enzyme-linked immunosorbent assay, and western blotting. Cell apoptosis was evaluated using FACS, immunocytochemistry, and western blotting. We investigated that certain influence of LLLs on MSCs in vitro 6 or 24 h after 1 h of LLL irradiation. The mechanism of the effects included proliferation rate increase mediated by increased S phase proportion; mitochondrial biogenesis and function alteration mediated by fusion (Mfn1, Mfn2, and Opa-1) and fission (Fis1, Drp-1, and MTP18)-related proteins, NRF1, TFAM, PGC-1a, and upregulated intracellular ROS and NO concentration; migration acceleration through the ERK1/2 and FAK pathway and upregulation of growth factors such as HGF and PDGF; and resistance to apoptosis with increased Bcl-2 and decreased Bax, or through tunneling nanotube formation between LLL-treated MSCs and 5-fluorouracil-induced apoptotic MSCs. These observations suggested that LLLs enhanced stem cell survival and therapeutic function, which could appear to be an innovative pretreatment in the application of MSCs.
Introduction
S
Photobiomodulation affected the activity of endogenous photoreceptors by light, altering cell proliferation and metabolism through mitochondrial retrograde signaling pathways. Low-level lasers (LLLs) were a common light source used in photobiostimulation therapy. Light in this region of the spectrum can penetrate tissue and lacks the carcinogenic properties [4]. LLL has been considered to be a noninvasive physiotherapy in clinical application, for example, osteoporosis, with changes of the state of bone MSCs [5]. Since light therapies are widely used in medicine, combination of stem cell therapy and light therapy appears to be a wise choice.
Mitochondria play central roles in energy metabolism and apoptosis. With vital functions, mitochondria usually become the target of extracellular stimuli and seem closely related to those biological effects of LLL [6 –8]. Microarray technology discovered a significant upregulation of gene expression in pathways involved in energy production and cellular antioxidant protection [9].
In our study, we chose human adipose-derived mesenchymal stem cells (hAD-MSCs) to investigate the effects of 50 Hz LLL on mitochondria and identify the underlying mechanisms of biological effects such as alteration in proliferation, migration, and apoptosis. With exploration in mechanism and manipulation, it will help develop more accurate studies about MSCs and might provide a novel therapeutic strategy for precondition of MSCs.
Materials and Methods
Cell culture and treatment
MSCs were isolated from human adipose tissues obtained from patients undergoing tumescent liposuction with informed consent and all experiments were approved by the Ethics Committee at the Chinese Academy of Medical Sciences and Peking Union Medical College. After isolation, cells were plated in a 75-cm2 culture flask in 12 mL of regular growth medium and incubated at conditions of 37°C and 5% CO2. Cultured MSCs were detected by flow cytometric analyses of expressed surface antigens as comprising a unique phenotypic population. MSCs cultured to the third passage were used in the experiments [10].
LLL apparatus
The exposure system used in the present experiment was designed by the medical apparatus company (No. LXW660-II, Jixing, China). The working conditions of the therapeutic apparatus were as follows: power source 220 ± 22 V, 50 ± 1 Hz; light output wavelength 660 ± 20 nm, and single point light output power 3–4.5 MW. All irradiation experiments were performed on the aseptic bench at room temperature and the control groups were processed under the same conditions without laser irradiation.
MTS assay
A standard MTS assay protocol was used to assess cell viability. MSCs were seeded into 96-well plates at 104 initial numbers per well in stem cell culture solution after various irradiation times. The cells were cultured for 24, 48, 72, or 96 h. Three hours before each of the desired time points, 20 μL/well of Cell Titer 96® Aqueous One Solution Reagent was added. After 3 h at 37°C in a humidified 5% CO2 atmosphere, the absorbance was detected at 490 nm using an enzyme-linked immunosorbent assay (ELISA) plate reader. Each point represents the mean ± standard deviation of three replicates.
Automated cell counting
MSCs were grown in growth medium after 1 h of irradiation with about 104 initial cells for 6 days. Detachment from plates was performed every day using enzymatic digestion by trypsin (Life Technology Corporation), followed by neutralization with two volumes of growth medium. MSCs were counted by loading into a TC20 automated cell counter (Bio-Rad) using the capillary-filled disposable loading chambers. Data were collected from three replicates.
Detection of cell cycle by flow cytometry
Cells were seeded into T25 flasks at a density of 106/mL and exposed to LLLs. Cells were washed in phosphate-buffered saline (PBS) and harvested by centrifugation. They were fixed in 1 mL 70% pre-cold ethanol for 24 h at 4°C. Cells were resuspended in 200 μl of PBS and incubated with 50 μg/mL propidium iodide (PI) in the dark for 30 min at 4°C, following which flow cytometric analysis was performed using BD Accuri™ C6 flow cytometer (BD Biosciences). Data were analyzed with CFlow plus 1.0.
Mitochondrial fission and fusion state
Chemotherapy drugs paclitaxel and 5-fluorouracil were used to restrain each cycle of the cells in the corresponding period; 5-fluorouracil can make cells mainly stay in S phase (88.38%, data not shown); paclitaxel can make cells mainly stay in G2/M phase (63.89%, data not shown). Mitochondrial measurement by staining with MitoTracker Red (Invitrogen) was performed. After stimulation, cells were incubated in serum-free medium with 150 nM MitoTracker Red FM for 30 min in the dark. After staining, cells were washed twice with cold PBS and suspended in 200 μL PBS. Subsequently, cells were analyzed on Olympus FV1000 confocal microscope (Olympus, Japan) and analyzed using FV10-ASW 4.0 Viewer software (Olympus). Fragmented mitochondria (fragmented, 0–2 μm), intermediate state of mitochondria (intermediate, 2–5 μm), and tubular mitochondria (tubular, 5–10 μm) were randomly selected in 100 cultured cells, the proportion of each type of mitochondria was statistically analyzed.
Transmission electron microscope
For transmission electron microscope (TEM), MSCs with or without stimulation were fixed in a 2% glutaraldehyde solution in 0.1 M cacodylate buffer, then MSCs were postfixed in 1% osmium tetroxide (OsO4) for 1 h and dehydrated in sequential steps of acetone (25%, 50%, 75%, and 100% twice) before impregnation in increasing concentrations (25%, 50%, 75%, and 100% three times) of resin in acetone over a 24-h period. To analyze MSC mitochondrial morphology, cells were photographed at × 8,000 and × 20,000 magnifications. Regions containing numerous MSC mitochondria were selected for analysis. Mitochondrial length was measured by tracing the mitochondria using ImageJ software.
Measurement of intracellular ATP content
Intracellular ATP content was detected using an ATP Assay Kit (Beyotime, China). After centrifugation at 12,000 g for 10 min at 4°C, the supernatant was removed and mixed with dilution buffer. The relative light unit was measured by a luminometer according to the manufacturer's instructions. A fresh standard curve was prepared each time and ATP content was estimated using the curve.
Measurement of intracellular reactive oxygen species
Mitochondrial reactive oxygen species (ROS) formation was detected with 2,7-dichlorofluorescein diacetate (DCFH2-DA) according to the instructions of ROS assay kit (Beyotime, China). The irradiated (or not) MSCs were incubated with 10 μM DCFH2-DA dissolved in none-serum DMEM at 37°C for 30 min. The fluorescence was then measured at 488 nm excitation and 525 nm emission by BD Accuri C6 (BD Biosciences).
DAF-FM diacetate for nitric oxide indication
Prepare viable cells in suspension. Dilute the DMSO stock solution into a suitable buffer with concentration about 5 μM. Incubate the cells with the diluted DAF-FM diacetate for 30 min at 4°C. Wash the cells to remove excess probe. Replace with fresh buffer, then incubate for an additional 30 min to allow complete de-esterification of the intracellular diacetates. Fluorescence excitation and emission maxima are 495 and 515 nm, respectively.
Oxygen consumption
The Seahorse XF24 Extracellular Flux Analyzer (Seahorse Biosciences) was used to measure oxygen consumption in MSCs. LLL-treated MSCs and control group were adhered onto XF24-well microplates at 40,000 cells/well in 250 μL culture media on the day before analysis. XF assay medium was supplemented with additional 250 mM glucose and 2 mM sodium pyruvate. The next day, cells were equilibrated in XF assay medium in a non-CO2 incubator for 1 h, then for simultaneous analysis of oxygen consumption rate (OCR). Following basal respiration measurements, cells were treated sequentially with oligomycin (1 μM) to measure the nonphosphorylating OCR, carbonyl cyanide-4-(trifluoromethoxy)phenylhydrazone (FCCP) (0.5 μM) to get the maximal OCR, and rotenone and antimycin A mixture (0.5 μM) to measure the extra mitochondrial OCR; the changes in respiration were recorded.
Induction of the scrape wound
Scrape wounds were performed in confluent monolayer cultures of the MSCs. A linear scrape was introduced over the entire diameter of the six wells. The wound area was marked with blue ink string on one side of the wound for reference. Cultures were rinsed with culture medium to remove floating cellular debris, and fresh culture medium was added. After 12 h, the respective traveling distances were measured with MSCs under LLL treatment or normal MSCs.
Migration and invasion transwell study
MSCs at a density of 2 × 105 cells/mL in 0.2 mL of L-DMEM without fetal bovine serum (FBS) were added into the upper chamber of a 6.5-mm diameter transwell insert. The lower chamber contained 0.5 mL of L-DMEM with 10% FBS. For invasion assay using a sterile syringe, 0.1 mL of the diluted BD Matrigel matrix-coating solution was carefully added to each insert. After 24 h of incubation, the upper surface of the membrane was scraped gently to remove nonmigrating cells. The membrane was then fixed in 4% paraformaldehyde for 10 min and stained in hematoxylin for 30 min. The number of migrating cells was counted in five random fields per well under the microscope, and their proportion in total MSCs was considered as the migration ratio.
Cytoskeleton staining
A total of 105 cells were plated in a six-well chamber and grown for 24 h. They were fixed in 4% formaldehyde for 10 min and permeabilized with 0.1% Triton™ X-100. F-actin was stained with rhodamine-phalloidin (Molecular Probes) and Tubulin Tracker Green (Beyotime, China) and the nucleus with DAPI (Beyotime, China). Cells were visualized under an Olympus FV1000 confocal microscope (Olympus, Japan) and analyzed using FV10-ASW 4.0 Viewer software (Olympus).
Enzyme-linked immunosorbent assay
MSCs were plated at 106 cells per well of a six-well plate and allowed to attach for 12 h. Cells were serum-deprived for 24 h following treatment with LLLs for 1 h. Levels of secreted HGF, PDGF, EGF, and bFGF from cell supernatants were quantified using ELISA. Data are representative of the average concentration in pg/mL from three individual wells.
Fluorescence recovery after photobleaching analysis of membrane fluidity
Fluorescence recovery after photobleaching (FRAP) was carried out by using the Leica TCS SP5 confocal microscope. The confocal plane was centered in the middle of the cell membrane, about 1 μm2 area. Before bleaching, a stack of 10 images was scanned to obtain a baseline. The region of interest (ROI) was then bleached for 3 s by the argon and He-Ne laser and allowed to recover for 30 s. Calculation of the half-time of recovery of fluorescence (t1/2) was performed with the Leica LAS AF Lite software (Leica Microsystems).
Apoptotic staining
In this study, we established a model by using 5-fluorouracil as in vitro stimulation to induce MSC apoptosis. With drug concentration gradient (1, 5, and 10 μg/mL), 24 and 48 h after treatment, respectively, cells were collected for Annexin V/PI dyeing. The quantitative analysis of apoptotic cells was performed using the Annexin V–FITC/PI Apoptosis Detection Kit (BD Biosciences). After treatment, the cells were harvested and resuspended in 200 μL of binding buffer, then incubated with 5 μL of Annexin V–FITC/binding buffer mixture for 30 min at 37°C in the dark, subsequently the cells were incubated with 10 μL of PI for 5 min. The cells were then analyzed by BD Accuri C6 flow cytometry (BD Biosciences).
Analysis of caspase-3/7 activity
Apoptosis is mediated by a cascade of aspartate-specific cysteine proteases (caspase). The Cell Event™ Caspase-3/7 Green Detection Reagent (Invitrogen) was applied to distinguish viable and apoptotic cells. After activation of caspase-3 or caspase-7 in apoptotic cells, the substrate bound to DNA to exhibit a green fluorescence signal. Cell Event Caspase-3/7 Green Detection Reagent was added at a final concentration of 1 μM and incubated for 1 h at 37°C. Cells were analyzed by BD Accuri C6 (BD Biosciences).
Mitochondrial membrane potential assay
Cells were exposed to LLLs for 1 h and then mitochondrial membrane potential was measured using a mitochondrial membrane potential (▵Ψm) assay kit with 5,50,6,60-tetrachloro-1,10,3,30-tetraethyl benzimidazolyl carbocyanine iodide (JC-1) (Beyotime, China) according to the manufacturer's protocol. After LLL exposure, cells were incubated with JC-1 for 20 min in the dark at 37°C and then fluorescence levels were quantified by BD Accuri C6 (BD Biosciences) with 514 nm excitation and 529 nm emission wavelength for JC-1 monomer; 585 nm excitation and 590 nm emission wavelength for JC-1 aggregates.
Evaluation of cell death
We established a coculture system of apoptosis-induced MSCs with LLL-treated or untreated MSCs. Cell membrane were labeled with Cell Tracker CM-DiI (Invitrogen), then 10 μM 5-Fu induced apoptosis for 24 h. Labeled and 5-Fu-treated MSCs were cultured alone; or cultured with LLL-treated MSCs, while unlabeled and 5-Fu-untreated MSCs were in 1:1 ratio; or cocultured with normal MSCs in 1:1 ratio. MSCs grown in above experimental conditions were stained with Annexin V-AF488 (1:500; Invitrogen) at 37°C for 45 min and then imaged at 400 nm (for Cell Tracker CM-DiI) and at 488 nm (for Annexin V-AF488) with the Olympus FV1000 confocal microscope (Olympus, Japan). The numbers of Annexin V-positive cells in the Cell Tracker CM-DiI-labeled and unlabeled populations were counted. Evaluation of 100 cells per condition and cell death was expressed as a percentage of Annexin V-positive cells in the Cell Tracker CM-DiI-labeled (finally yellow) or unlabeled (finally green) population.
Quantification of tunneling nanotubes
To test whether tunneling nanotubes (TNTs) have a role in the rescue of apoptotic cells, MitoTracker® Red CMXRos (Invitrogen)-labeled and 5-Fu-treated MSCs were cocultured with MitoTracker® Green FM (Invitrogen)-labeled and LLL-treated or normal MSCs. MitoTracker Green FM (Invitrogen)-labeled and LLL-treated MSCs were then treated with or without cytochalasin B. The number of TNTs between MitoTracker Red CMXRos-labeled MSCs, between MitoTracker Green FM-labeled MSCs, and between both types of populations were counted and expressed as the number of TNTs per 20 pairs of cells.
Quantitative polymerase chain reaction
Total RNA of cultured cells was extracted with TRI reagent (Sigma-Aldrich), and 1 μg RNA was reverse transcribed to cDNA. The primers were designed based on the NCBI website. Gene expression levels in the cells were normalized by GAPDH. Relative expression of mRNA was evaluated by the 2−ΔΔCt method and normalized to GAPDH.
Western blot analysis
Cells were harvested in RIPA lysis buffer (Beyotime) with 1 mM phenylmethanesulfonyl fluoride (PMSF), quantified by a BCA Protein Assay Kit (Beyotime), and separated by 10% SDS-PAGE and transferred onto polyvinylidene fluoride membranes (Millipore). The membranes were blocked in 5% nonfat milk in Tris-buffered saline Tween-20 (TBST) for 1 h, and incubated in primary antibodies overnight at 4°C, followed by horseradish peroxidase (HRP)-conjugated secondary antibodies for 1 h at room temperature. Signals were visualized with HRP substrate (Proteintech) and detected by an Image Quant LAS 4000 mini imaging system (GE Healthcare). β-Actin was used as an internal control. Primary antibodies used were as follows: ERK (1:1,000, rabbit IgG, Proteintech), p-ERK (1:1,000; rabbit IgG, Proteintech), AKT (1:1,000, rabbit IgG, Proteintech), p-AKT (1:1,000, rabbit IgG, Proteintech), PI3K (1:1,000, rabbit IgG, Proteintech), FAK (1:1,000, rabbit IgG, Proteintech), p-FAK (1:1,000, rabbit IgG, Abcam), caspase3 (1:1,000, rabbit IgG, Proteintech), Bcl-2 (1:1,000, rabbit IgG, Abcam), Bax (1:1,000, rabbit IgG, Abcam), β-actin (1:1,000, rabbit IgG, Proteintech), and HRP-conjugated anti-rabbit-IgG (1:2,000, Proteintech).
Statistics and data analysis
Results are expressed as mean ± SD of three or six independent experiments. All data were analyzed with SPSS 22.0. Differences between two groups were assessed using unpaired two-tailed t tests. Data involving more than two groups were assessed by analysis of variance (ANOVA). P values <0.05 were considered significant.
Results
Effects of LLLs on stem cell proliferation and cell cycle
We have demonstrated that LLL treatment increases the MSC proliferation rate and this occurs through cell cycle progression [11]. For cells under LLLs for 1, 2, and 3 h, respectively, MTS experiment shows that LLL processing can increase cell proliferation ability—1 h is of the strongest influence on cell proliferation (Fig. 1A)—and at the same time, cell doubling time from the control group of 19.64 ± 3.63 h to experimental group of 12.44 ± 4.48 h (Fig. 1B). The 1-h treated MSCs displayed a more rapid growth rate than the control group as revealed by cell counting (Fig. 1C). LLL stimulation also affects the cell cycle (Fig. 1D, E), S phase proportion in control group was 16.11% ± 0.74%, and with LLL treatment, S phase proportions in experimental group were 32.00% ± 4.54% (1 h), 29.53% ± 0.93% (2 h), and 26.17% ± 2.61% (3 h), respectively. In the following study, we chose 1 h of treatment as the process method. We collected cells 6 or 24 h after 1 h of irradiation; 6 h after irradiation compared with control group, cells in S phase increased significantly (32.47% ± 2.08% vs. 13.45% ± 1.32%, P < 0.05). After 24 h, the S phase proportion decreased (17.87% ± 6.99%). This indicates time window effect of the effects of LLLs, and also presents the strong ability that MSCs hold to maintain steady state.

Effects of the LLLs on stem cell proliferation and cell cycle.
Effects of LLLs on mitochondrial biogenesis and function
LLLs promoted mitochondrial biogenesis
Mitochondrial biogenesis and function are altered in response to external stimuli with changes in mitochondrial size and number [12,13]. Figure 2A shows mitochondria morphology at S or M cell cycle. Mitochondria in 100 cells were visually scored as tubular, fragmented, or intermediate, as in the diagram (Fig. 2B). Fusion (Mfn1, Mfn2, and Opa-1) and fission (Fis1, Drp-1, and MTP18) regulators [14] were evaluated by q-PCR during commitment to irradiation. The increased fission and fusion in respective cell cycle observed in Fig. 2A correlated with increased OPA1, Mfn1, and Mfn2 expression (Fig. 2C, P < 0.05) and upregulated fission effectors Fis1, Drp1, and MTP18 (Fig. 2C, P < 0.05). With the help of TEM, we observed increased nucleus–cytoplasm ratio and slightly more number of cytoplasmic organelles, especially mitochondria with elongated morphology (Fig. 2D). Black squares indicated where the ×20,000 magnification image (Fig. 2D, down) was magnified in the ×8,000 magnification image (Fig. 2D, up). The mitochondria length scale was counted by ImageJ, presenting 2.96 ± 0.36 μm long after LLL treatment compared with control group (0.8 ± 0.29 μm, P < 0.01) (Fig. 2E). We evaluated the expression of three mitochondrial biogenesis-associated genes (NRF1, Tfam, and PGC-1α) [15] by q-PCR after LLL treatment; the expression of PGC-1a was upregulated compared with control group (Fig. 2F). These results indicated that LLL exerts its biologic effects through modulating the physiologic status of mitochondria.

LLLs promoted mitochondrial biogenesis.
The impact of LLLs on mitochondria biochemical function
We evaluated the expression of 3 mitochondrial metabolism-associated genes by q-PCR and found that 6 h after LLL treatment, Cyt-c in mitochondrial respiratory chain, carnitine palmitoyl transferase-1 (CPT-1) in long chain fatty acid oxidation, and citrate synthase in tricarboxylic acid cycle were less expressed in LLL group compared with the control group. After 24 h, the expression of those genes was upregulated, while still less than control group (Fig. 3A). LLL processing did not cause significant changes of expression of heat shock proteins (HSP70/HSP90) [16], indicating nonthermal effect when LLL was applied to MSCs (Fig. 3B). The expression of mitochondria ferritin [17] increased and cytoplasmic ferritin decreased as assayed by q-PCR; we speculated the change of iron Fe3+ deposition was caused by LLLs (Fig. 3B). It demonstrated an increase in ROS [18], nitric oxide (NO) [19], and Ca2+ [20] generation in the LLL groups compared with the control (Fig. 3D, E). LLL-induced alteration in mitochondria function occurs through ATP production and respiration (Fig. 3C, F, and G).

The impact of LLL on mitochondria biochemical function.
The impact of LLLs on cell mobility and pertinent mechanism
In scrape wound experiment, the traveling distances on the last 12 h in the control group were 106 ± 10.09 μm, treatment with LLLs significantly accelerated the traveling distances to 223 ± 30.12 μm (Fig. 4A, P < 0.05). The invading assay showed that MSCs under two conditions were both with low invading ability (Fig. 4B). The migration assay results showed that LLLs produced a stimulated effect on MSC migration (Fig. 4B, P < 0.05). The influence of LLLs indicated a promoted expression and activation of cytoskeleton components, as shown in dyed F-actin, tubulin and the presence of TNTs (Fig. 4C). We investigated the membrane fluidity of MSCs by FRAP in variant situation (Fig. 4D, left). It showed FRAP curves derived from cells under different treatments (Fig. 4D, middle). The index for the speed of recovery is the time it takes for the curve to reach 50% of the plateau fluorescence intensity level and is called half-life (t1/2) (Fig. 4D, right), shorter half-life in LLL-treated group told us that the recovery was faster than in MSCs of control group, indicating that the mobility of the staining molecule was faster in the 6-h curve conditions than the control curve condition. The LLL effects of upregulated expression of growth factors (HGF, PDGF, EGF, basic fibroblast growth factor [b-FGF]) [21] on MSCs were measured by q-PCR (data not shown) and ELISA (Fig. 4E), which indicated that autocrine signaling was likely a response to LLL-induced activity of MSCs. MAPK/ERK, but not PI3K/AKT, [22] pathway was required for LLL-mediated migration of MSCs. The content and activity of FAK protein were markedly upregulated in LLL-treated group (Fig. 4F, G). In addition, pretreating MSCs with anti-ERK agent FR180204 significantly reduced the LLL-induced migration acceleration (data not shown). Our results indicated that HGF and PDGF can alter MSC migration through cytoskeletal rearrangements and the activation of ERK1/2 and FAK.

The impact of LLLs on cell mobility and pertinent mechanism.
LLLs interact with mitochondria to influence process of apoptosis
LLL-treated group presented antiapoptosis effect
After preparative experiment (data not shown), we selected the 10 μg/mL 5-Fu treating for 24 h as assay point and examined a list of apoptosis index in the following testing. Results showed that total level of Bcl-2 was upregulated around 50%, which was statistically significant, and the total level of Bax remained downregulated in MSCs treated with LLLs (Fig. 5A). Annexin V/PI staining showed that compared with the control group (6.3% ± 0.9%), the apoptosis ratio was significantly higher (11.23% ± 0.97% vs. 5.10% ± 0.72%, P < 0.05) after 5-Fu stimulated apoptosis. Apoptosis ratio of 5-Fu-treated group with LLL processing was decreased (Fig. 5B, 6.10% ± 0.70% vs. 11.23% ± 0.97%, P < 0.05) compared with 5-Fu-treated group. Western blot showed that the caspase 3 expression reduced after LLL processing compared with control group (Fig. 5C). Flow cytometer detected that after dealing with the LLLs, the activity of caspase-3/7 enzyme was significantly lower than that of 5-Fu-treated group (Fig. 5D, 6.30% ± 1.02% vs. 9.03% ± 0.86%, P < 0.05). FACS analysis was used to determine membrane potential changes by means of JC-1, a lipophilic cationic dye selectively entering mitochondria. JC-1 accumulation in mitochondria due to concentration-dependent formation of red fluorescent JC-1 aggregates was higher for MSCs treated with LLLs compared with MSCs treated with 5-Fu. Results are presented as JC-1 aggregates/JC-1 monomer with decreased JC-1 red/green fluorescence ratio, heralding a decline in mitochondrial membrane potential (Fig. 5E).
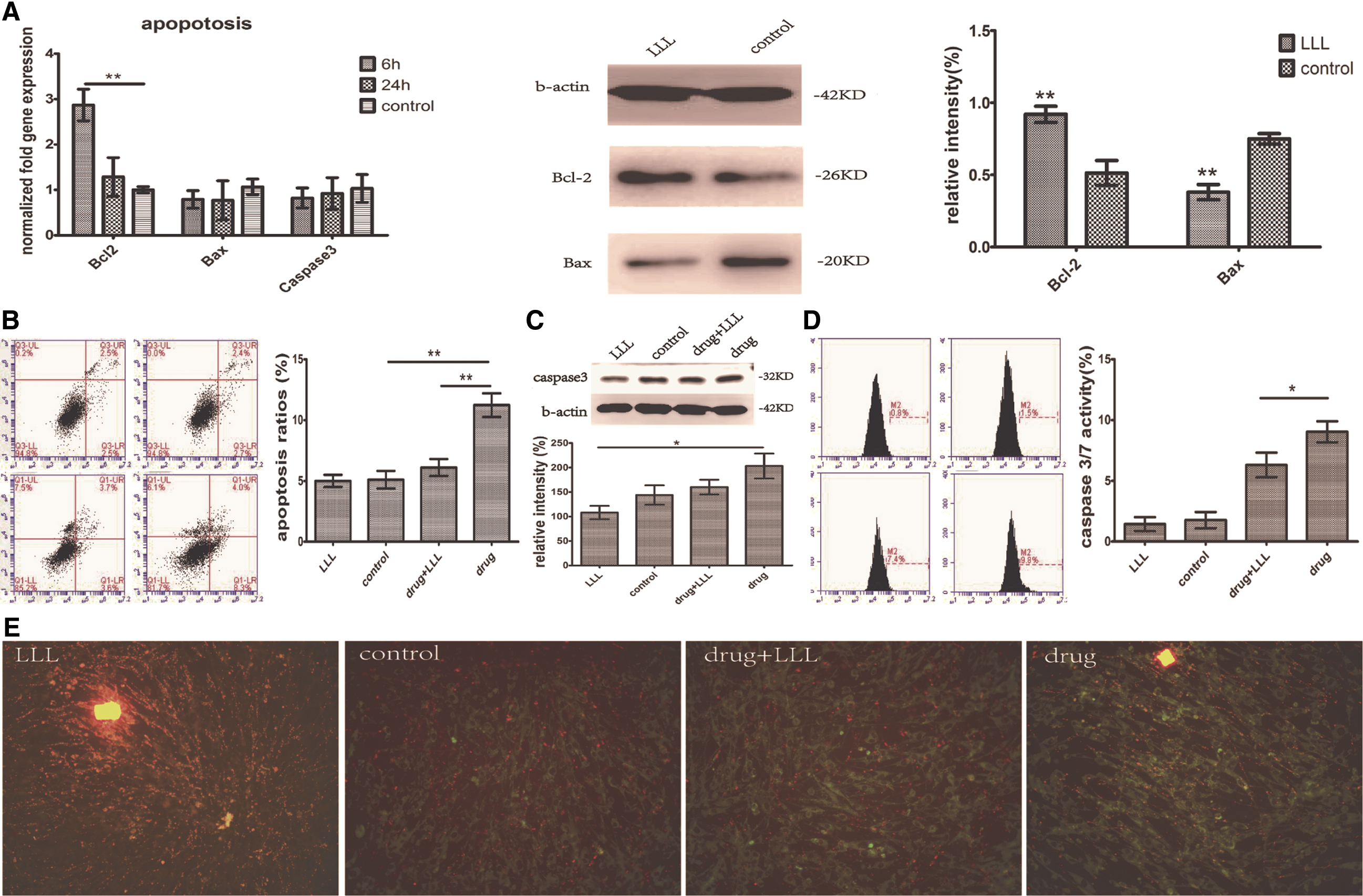
LLL-treated group presented antiapoptosis effect.
TNT-mediated transfer of functional mitochondria between LLL group and induced apoptotic group
Compared with alone cultured group, mortality of cells in coculture system was significantly lower (Fig. 6A, P < 0.05). It suggested that MSCs treated with 5-Fu were rescued, and the ability of saving was stronger in LLL-treated MSCs than normal cultured MSCs. The rescue effect did not appear in the 5-Fu-treated cells incubated with supernatant of LLL-treated MSCs, indicating a contact-dependent mechanism (Fig. 6A). The proportion of apoptotic cells in the 5-Fu-treated MSCs in coculture was significantly higher in the presence than in the absence of cytoB (Fig. 6B). Three types of TNTs [23,24] were calculated between 20 pairs in two types of cocultured systems. Elimination of TNTs with cytoB decreased the transfer of two types of MitoTracker-labeled mitochondria significantly (Fig. 6C). We concluded that TNT-mediated transfer of mitochondria reverse induced apoptotic trends of MSCs.
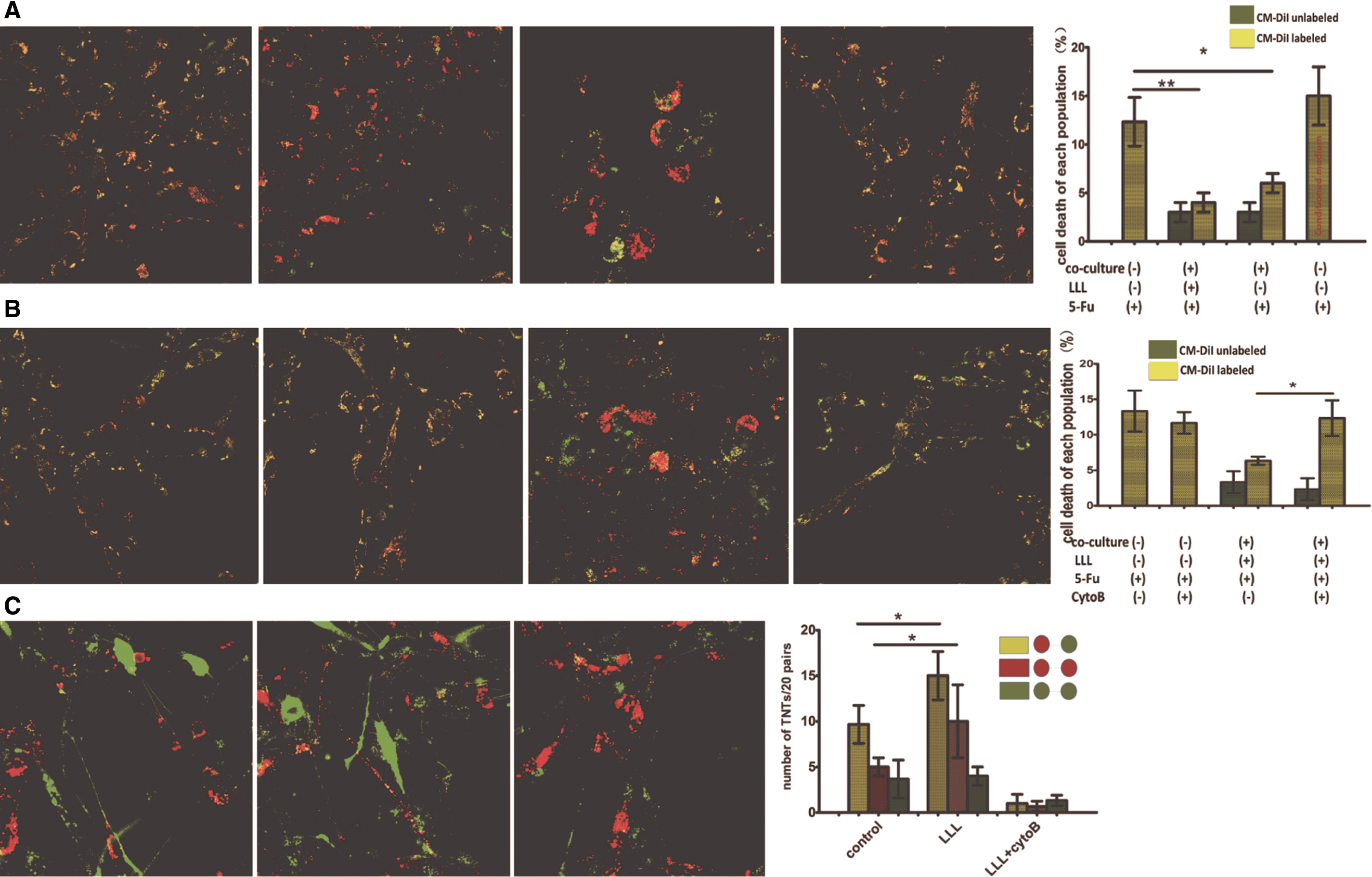
TNT-mediated transfer of functional mitochondria between LLL group and induced apoptotic group.
Discussion
Low-level lasers (LLLs) emit light in the visible red to near-infrared range [4], lack carcinogenic properties, and act on endogenous photoreceptors to initiate light-altered signaling pathways [25]. The conclusions referring to the effects of LLLs on stem cells were inconsistent [11,26,27]. One possible mechanism is photon absorption by cytochrome C oxidase in the mitochondrial respiratory chain [8,28]. We investigated that certain influence of LLLs on in vitro hAD-MSCs with discussion of the relationship between mitochondria and cellular function.
An increase in cellular viability and proliferation on MSCs was found using a diode laser at a wavelength of 636 nm and a fluence of 5 J/cm2 [29]. Proliferation of AD-MSCs significantly increased after exposure to a diode laser at 636 nm wavelength (5 J/cm2) was reported [30]. In our study, we used a fluence of 11–16 J/cm2 and the treatment was likely to be effective too.
In our study, LLL was found to promote cell proliferation through acceleration of G1 to S phase progression. Some mitochondrial biogenesis and function-related genes such as PGC-1α, NRF1/2, and Tfam [31] may be involved in the irradiation process. One possible reason for this increase might be that LLLs could be a stimulus to induce an activated state of the MSCs. Ferritin has effects on cellular proliferation and resistance to oxidative damage. A discovery of ferritin specific for the mitochondria, called mitochondria ferritin, indicated the relationships between iron and oxidative damage [17]. Interestingly, expression of mitochondria ferritin increased and cytoplasmic ferritin decreased during irradiation.
Wound healing is a highly coordinated process that reestablishes tissue integrity through various cell types and surrounding extracellular matrix with specific cytokines [32]. Clinical studies are using LLLs for skin wound repair [33]. The transplanted AD-MSC population can decline rapidly in the recipient tissue and is an obstacle to inhibit MSCs to be widely applied in skin tissue engineering. There were studies that suggested that LLL was an effective stimulator in wound healing that enhanced the survival of AD-MSCs and stimulated the secretion of vascular endothelial growth factor and b-FGF in the wound bed [34,35]. Studies have proven that MSC migration was regulated by numerous growth factors and their receptors [21,36], for example, bFGF was able to increase the migratory activity of MSCs significantly through activation of the PI3K/Akt pathway [37]. Our study indicated that the changed expression of HGF and PDGF was involved in irradiation. The LLL treatment would be good precondition of MSCs that were implicated in skin engineering.
Crosstalk within cytoskeleton promotes cell migration [38]. TNTs consisted of F-actin and formed between cells and mediate intercellular communication [39]. The mitochondria have essential roles in apoptosis and recovery of mitochondria injury through intercellular transference [40,41]. Our study revealed that LLLs regulate intercellular mitochondrial transport through TNTs and enhance MSC rescue efficacy in apoptotic situation. In addition, the promotion of secretion of Bcl-2 led to enhanced resistance to proapoptotic stresses.
Taken together, LLLs can be applied as stem cell preconditioning before transplantation toward the safe long-term efficacy of stem cell-based therapy.
Footnotes
Acknowledgments
This study was supported by grants from National Natural Science Foundation of China (Nos. 81370466, 81370879), Key Program for Beijing Municipal Natural Science Foundation (No. 7141006), National Key Research and Development Program (Nos. 2016YFA0101000, 2016YFA0101003), National Collaborative Innovation Program (for Biotherapy), and Beijing Key Laboratory of New Drug Development and Clinical Trial of Stem Cell Therapy.
Author Disclosure Statement
The authors declare no competing or financial interests.