
Abstract
Amniotic fluid represents an abundant source of multipotent stem cells, referred as broadly multipotent given their differentiation potential and expression of pluripotency-related genes. However, the origin of this broadly multipotent cellular fraction is not fully understood. Several sources have been proposed so far, including embryonic and extraembryonic tissues. In this regard, the ovine developmental model uniquely allows for direct comparison of fetal fluid-derived cells from two separate fetal fluid cavities, the allantois and the amnion, over the entire duration of gestation. As allantoic fluid mainly collects fetal urine, cells originating from the efferent urinary tract can directly be compared with cells deriving from the extraembryonic amniotic tissues and the fetus. This study shows isolation of cells from the amniotic [ovine amniotic fluid cells (oAFCs)] and allantoic fluid [ovine allantoic fluid cells (oALCs)] in a strictly paired fashion with oAFCs and oALCs derived from the same fetus. Both cell types showed cellular phenotypes comparable to standard mesenchymal stem cells (MSCs), with trilineage differentiation potential, and expression of common ovine MSC markers. However, the expression of MSC markers per single cell was higher in oAFCs as measured by flow cytometry. oAFCs exhibited higher proliferative capacities and showed significantly higher expression of pluripotency-related genes OCT4, STAT3, NANOG, and REX1 by quantitative real-time polymerase chain reaction compared with paired oALCs. No significant decrease of pluripotency-related gene expression was noted over gestation, implying that cells with high differentiation potential may be isolated at the end of pregnancy. In conclusion, this study suggests that cells with highest stem cell characteristics may originate from the fetus itself or the amniotic fetal adnexa rather than from the efferent urinary tract or the allantoic fetal adnexa.
Introduction
A
So far, the origin of human AFSCs during intrauterine development in general as well as the origin of this broadly multipotent cellular fraction in particular is not fully understood yet. AFSCs represent a heterogeneous population of cells and ultimately several sources of origin have been proposed, including embryonic (such as the urinary tract, the skin, the urogenital, respiratory, or gastrointestinal tract) as well as extraembryonic tissues (such as the placental tissue or the amniotic fetal adnexa) [9]. Also the origin of the amniotic fluid itself varies with increasing gestational age, whereas at the beginning, the production is mainly due to active water and electrolyte transport across the amniochorionic membrane and fetal skin. In the second half of gestation most of the fluid is produced by the fetus itself, either by secretion from the respiratory tract or by production of urine [10,11]. This further suggests that the origin of cells may also vary with different stages of gestation. However, anatomical and ethical limitations prevent further studies concerning the origin of AFSCs in humans.
On the contrary, the ovine developmental model uniquely enables investigation of prenatal extraembryonic (stem) cell compartments [12]. Although being structurally and ontogenetically similar to the human fetal anatomy [13] and thus, serving as a standard in vivo animal model for several fetal therapeutic interventions involving AFSCs [12,14 –21], it maintains two separate extraembryonic fluid compartments over the entire duration of pregnancy, the allantoic and the amniotic fluid cavities (Fig. 1).
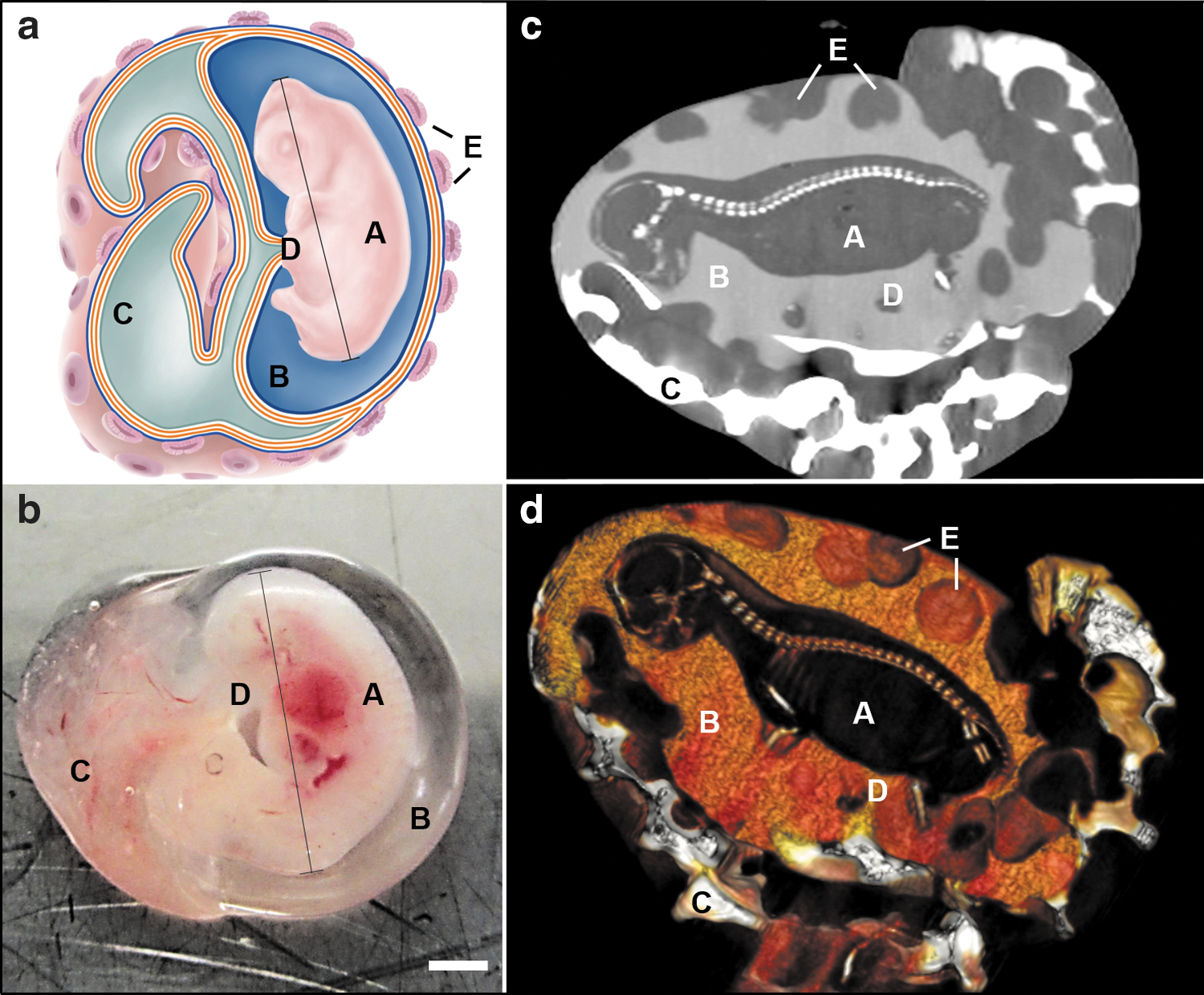
The ovine developmental model and its corresponding fetal fluid cavities.
In human embryogenesis, the allantois is only present in the first 3–5 weeks after conception and afterward it involutes to form the intraembryonic urachus [22]. Interestingly, in sheep the allantoic fluid compartment mainly collects fetal urine, which is drained from the urinary bladder through the urachus [8]. It therefore represents a model that allows for a direct comparison of fetal fluid-derived cells originating from the efferent urinary tract and the allantoic fetal adnexa with cells derived from the fetus itself and amniotic fetal adnexa directly surrounding the fetus. A comparison of the pluripotency-related gene expression of fetal fluid-derived cells from different compartments would shed some first light on the actual origin of the (broadly) multipotent cellular component of human amniotic fluid cells, in particular on whether these cells seem to originate from efferent urinary tissues or not.
Therefore, the present study compares the expression of pluripotency-related genes between cells isolated from the amniotic versus the allantoic fluid cavity in the ovine developmental model over gestation in a strictly paired fashion.
Materials and Methods
Postmortem harvest of ovine fetal fluids
Ovine amniotic and allantoic fluids were harvested from gravid uteri postmortem from the local slaughterhouse Zurich/Hinwil, Switzerland (n = 9). The fetuses were selected according to crown rump length (CRL) and allocated to three corresponding gestational age groups—group 1: CRL ≤15 cm; group 2: >15 cm, ≤30 cm CRL; group 3: >30 cm CRL [23].
For illustration purposes of the ovine fluid cavities, a computer tomography (Brilliance 16; Philips AG, Zurich, Switzerland) examination was performed with different concentrations of contrast medium injected into each fluid cavity (Ioversol; Optiray 300, Guerbet, Switzerland).
For the harvest of ovine fetal fluid, the uterus and the fetal fluid cavities were opened surgically and the fetal fluids were aspirated with a 50-mL syringe and a 10G needle under visual guidance as previously published [12]. In case of inaccurate surgical puncture or blood contamination, isolated samples were excluded from the study. Following centrifugation for 10 min at 300 g, residual cells were separated from their corresponding fetal fluid. For each animal, one aliquot of harvested supernatant was stored overnight at 4°C to verify the accuracy of fetal fluid harvest (and consequently accurate cell isolation) according to previous studies [12].
Therefore, values of total protein, albumin, sodium, chloride, and magnesium were compared with reference values from an existing database previously published [12]. These parameters revealed a high discriminative power to distinguish between the two fetal fluids. If selected values deviate from the reference values, isolated cells were excluded from subsequent analysis, as their pure origin could not be proven.
Isolation and expansion of cells
Ovine amniotic fluid cells and ovine allantoic fluid cells
Ovine amniotic fluid cells (oAFCs; n = 9) and ovine allantoic fluid cells (oALCs; n = 9) were isolated by centrifugation of the fetal fluids at 300 g. The obtained cell pellet was seeded into standard culture flasks and incubated in a humidified incubator at 5% CO2 at 37°C. The proliferation medium consisted of endothelial basal medium (EBM™; Lonza, Switzerland) supplemented with 10% fetal calf serum (FCS; Sigma-Aldrich, Switzerland), growth factors (recombinant long insulin-like growth factor-1, human fibroblast growth factor, human epidermal growth factor), and ascorbic acid, as well as an antibiotic–antimycotic solution containing 100 U/mL penicillin, 100 μg/mL streptomycin, and 25 ng/mL amphotericin B.
This medium has been used in previous studies [5,12,24,25] and demonstrated to prevent predifferentiation of isolated fetal cells and maintain their stem cell phenotype over in vitro passaging. Nonadherent cells were removed by replacing the medium 72–96 h following the seeding procedure. At 80% confluency, cells were split using Accutase (Gibco™; Thermo Fisher Scientific) or trypsin–ethylenediaminetetraacetic acid (EDTA) solution (Sigma-Aldrich). Cellular viability was assessed using a 0.4% Trypan Blue exclusion staining (Gibco™; Thermo Fisher Scientific). A stock of frozen cells at every passage was stored in liquid nitrogen [10% dimethyl sulfoxide (DMSO) in FCS; Sigma-Aldrich) until further analysis.
Control ovine bone marrow-derived mesenchymal stem cells
The isolation of ovine bone marrow-derived mesenchymal stem cells (oBMSCs; n = 3) as control cells was performed according to standard procedures. In brief, bone marrow aspirate was obtained from the sternum or iliac crest of an adult sheep in oblique supine position under general anesthesia according to the cantonal ethical permission No. 9/14. Int. 25040. Bone marrow aspirate was diluted 1:2 with phosphate-buffered saline (PBS) for a histopaque (Histopaque-1077; Sigma-Aldrich) density gradient centrifugation at 400 g for 30 min. The obtained mononuclear cell layer was washed with PBS and seeded in the abovementioned proliferation medium for further expansion. A stock of frozen cells at every passage was stored in liquid nitrogen (10% DMSO in FCS) for further analysis.
Phenotypic characterization of oAFCs, oALCs, and control oBMSCs
Antibodies
Primary antibodies used were specific against CD11b (clone CC126; AbD Serotec, United Kingdom), CD29 (A. Zannettino, Adelaide, Australia) [26], CD44 (clone MAC329; LifeSpan BioSciences; sc-59758; Santa Cruz Biotech), CD166 (clone 3A6; BioLegend), STRO-4 (A. Zannettino) [27], Desmin (clone D33; Dako, Denmark), Vimentin (clone Vim3B4; Dako), α-smooth muscle actin (αSMA) (clone 1A4; Dako), Nanog (hNanog.2; eBioscience), Oct3/4 (H-134; Santa Cruz), and Stat3 (BD Transduction Laboratories). All stainings were accompanied by isotype controls for IgG1 (clone MOPC-21; BioLegend) and IgG2a (clone MOPC-173; BioLegend or AbD Serotec), IgG2b (clone C.SW IgG2b, k; BD Pharmingen™), as well as secondary antibody-only control stainings.
For immunofluorescence stainings, primary antibodies were detected with secondary goat anti-mouse Cyanine-2-conjugated antibodies (Alexa Fluor 488; Jackson ImmunoResearch or Life Technologies), donkey anti-rat Cyanine-2-conjugated antibodies (Alexa Fluor 488; Jackson ImmunoResearch) or goat anti-rabbit Cyanine-2-conjugated antibodies (Alexa Fluor 488; Life Technologies), phalloidin (Alexa Fluor 546; Invitrogen, Thermo Fisher Scientific), and 4′,6-diamidino-2-phenylindole (Sigma-Aldrich). For flow cytometry, primary antibodies were detected with either a fluorescein isothiocyanate (FITC)-conjugated goat anti-mouse antibody (BD Bioscience) or a FITC-conjugated goat anti-rat antibody (BioLegend).
Immunofluorescence staining
For immunofluorescence stainings, oAFCs (n = 3), oALC (n = 3) and oBMSC (n = 3) were washed with PBS and fixed with 4% paraformaldehyde/PBS for 10 min. Following a second washing step with PBS, cells were permeabilized with 0.2% Triton X-100/PBS (Sigma-Aldrich) for 10 min at room temperature. After blocking with 5% goat serum in 1% bovine serum albumin (BSA)/PBS for 30 min at room temperature, primary antibodies (diluted in 5% goat serum/1% BSA/PBS) were added and incubated for 1 h or overnight at room temperature. Cells were washed three times for 5 min with PBS and incubated with the secondary antibodies for 45 min at room temperature. Again, three washing steps with PBS were performed (3 × 5 min) and samples were mounted with Aqua-Poly/Mount (Polysciences, Inc.). Stained cells were visualized using an inverted fluorescence microscope equipped with a CCD camera (Zeiss Axiovert II; Zeiss, Germany and Leica DM6000B).
Flow cytometric analysis
For flow cytometry analysis, 2 × 105 oAFCs (n = 3), oALC (n = 3) and oBMSC (n = 3) were used per primary antibody staining, each diluted in fluorescence-activated cell sorting (FACS) buffer (2% FCS/PBS, 5 mM EDTA) and incubated for 30 min at 4°C. After two washing steps with FACS buffer, cells were incubated for another 30 min at 4°C with secondary FITC-conjugated antibodies. Again, two washing steps with FACS buffer were performed. Between 50,000 and 100,000 events were acquired using a FACSCanto II (BD Bioscience) and the datasets were analyzed with FlowJo software (Tree Star, Inc.). Median fluorescence intensity ratio (MFIR) was calculated relative to the unstained controls. Specificity of the antibodies was verified using the corresponding isotype controls.
Cell proliferation kinetics
Proliferation kinetics were monitored over the complete expansion period for every isolated fetal fluid. Only paired cell lines were integrated into the analysis, meaning that oAFCs (n = 9) and oALCs (n = 9) from the same fetus were expanded and compared. The population doubling (PD) rate was determined by integrating the harvested viable cell number (following a Trypan Blue exclusion staining) into the formula mentioned below [28]. N
1 is defined as the plated and N
H the harvested cell number.
To define the absolute number of times each cell has doubled since their first isolation in vitro, the cumulative population doublings (CPD) were calculated up to passage 3. Therefore, the PD of each passage was summed up to the PD of the previous one. The generation time (GT, average time between two cell doublings) was calculated at all three passages using the following formula [28]:
Trilineage differentiation potential
Multilineage differentiation was assessed by inducing differentiation of oAFCs (n = 3), oALCs (n = 3), and oBMSCs (n = 3) to osteogenic, adipogenic, and chondrogenic lineages according to standard protocols. Briefly, ovine cells were cultured in either (i) osteogenic medium: 10 nM glycerol 2-phosphate (Sigma, Switzerland), 50 μM
Pluripotency-related gene expression
Total RNA was extracted using the RNeasy Mini Kit (Qiagen, Switzerland) and reverse transcription was carried out using Superscript III RT (Invitrogen, Switzerland) according to the manufacturer's instructions. Quantitative real-time PCR (qRT-PCR) was performed using the Rotor-Gene SYBR Green PCR Kit (Qiagen) and ovine pluripotent stem cell primers (Microsynth, Switzerland) for OCT4, STAT3, NANOG, SOX2, and REX1 (Table 1). Primers have been established and evaluated by the use of a previously published ovine induced pluripotent stem cell line kindly provided by Paul Verma and Jun Liu of the Stem Cell and Genetic Engineering Group, Monash University, Clayton, Australia [29]. The expression levels were analyzed twice in triplicates using standard conditions on a Rotor-Gene Q (Qiagen) for n = 9 oAFCs, n = 9 oALCs, and n = 3 oBMSCs. GAPDH served as housekeeping gene to quantify relative stemness expression levels using the ΔCT threshold cycle method. Expression levels were calculated with 2−dCT and a multiplication factor of 100.
Statistical analysis
Quantitative data are presented as mean ± standard deviation (GraphPad Prism; GraphPad Software, Inc.). For statistical comparison of the paired results on oAFCs and oALCs paired students t-test was performed and P-values <0.05 were considered statistically significant. Shapiro–Wilk normality test proved that the datasets were normally distributed (P > 0.05). Grubbs' test determined whether one of the data points represented a significant outlier from the rest (P < 0.05).
Results
Cell isolation, proliferation, and phenotypic cell characterization
After harvest of fetal fluids, the origin and purity of the fetal fluids were verified using a biochemical differential fluid analysis according to previously published protocols [12] before inclusion of the samples into the present study. Cellular yield, cell attachment, and phenotypes were analyzed using light microscopy and compared with oAFCs (n = 9), oALCs (n = 9), and control oBMSCs (n = 3) (Fig. 2). Bacterial contamination and premature senescence, defined by stagnation of proliferation, high granularity of the cells, and cellular detachment, were equally distributed between oAFCs and oALCs. Compared with oBMSCs, the oAFCs and oALCs showed a more heterogeneous intra- and interpatient phenotypic variability, including cell size and phenotype.
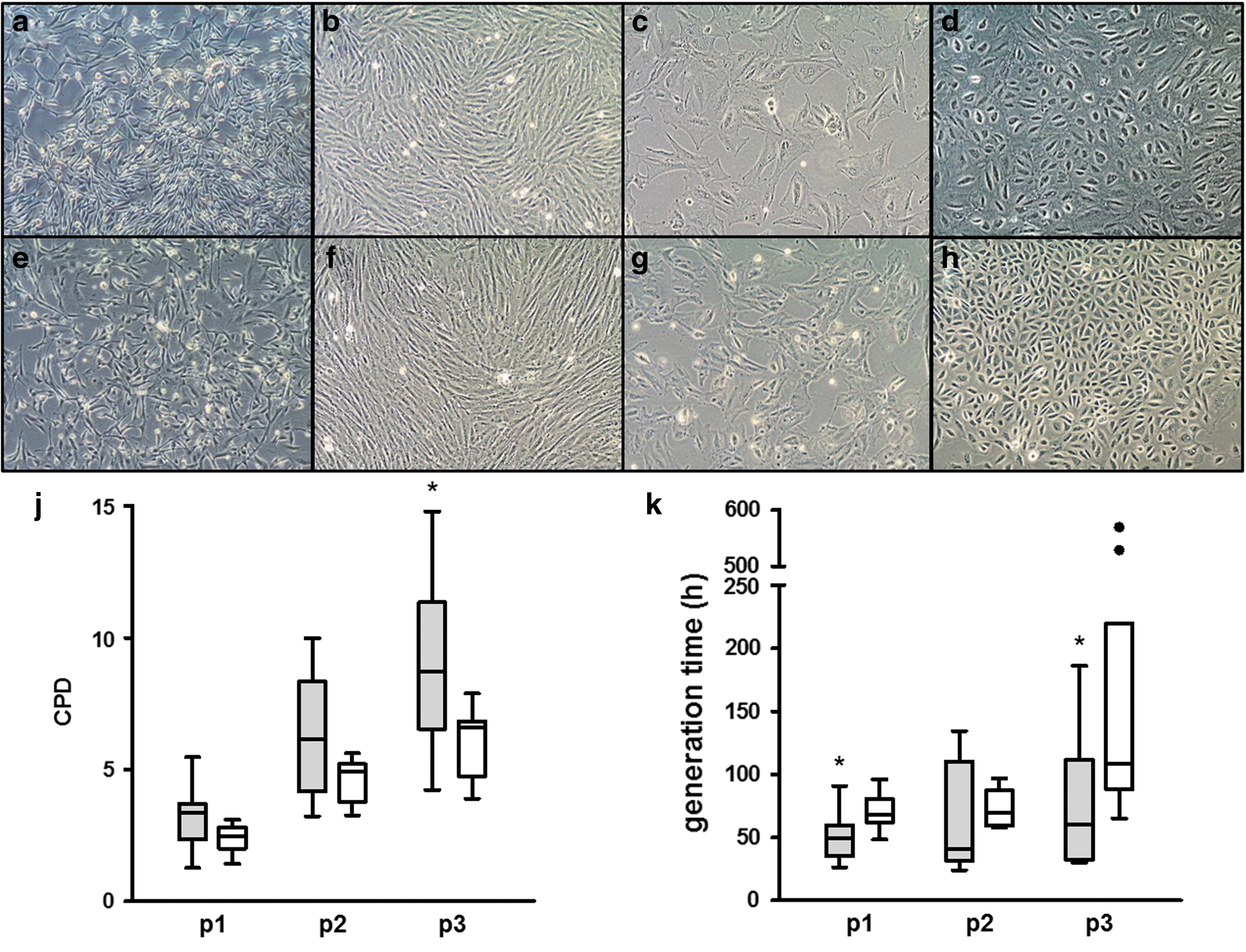
Morphology and proliferation capacity of isolated oAFCs and oALCs. Isolated oAFCs (n = 9)
Three morphological cell types (Fig. 2a–h) were predominantly evident in both oAFCs (Fig. 2a–d) and oALCs (Fig. 2e–h): (I) small spindle-shaped cells (Fig. 2a, b, e, and f), (II) cobblestone-like cells, resembling epithelioid cells (Fig. 2d, h), and (III) larger flattened fibroblastic cells (Fig. 2c, g). After direct plating, a highly heterogeneous population of cells was observed, whereas with passaging, the cell population became more homogeneous with either phenotype I or III. At higher passages, a higher senescence rate was found with morphological changes of the cells and stagnated proliferation. Concerning proliferation (Fig. 2j, k), oAFCs showed a tendency for lower GTs and higher CPD when compared with oALCs derived from the same mother animal and fetus (n = 9 per source). Significant differences were predominantly found at passage 3 (P < 0.05). No differences in proliferation parameters were detected when cells derived from different gestational ages were compared with each other.
Mesenchymal stem cell surface marker expression and trilineage differentiation of oAFCs versus oALCs
Immunofluorescence stainings (passage 3–5) of representative paired oAFCs (n = 3) and oALCs (n = 3) from the same fetus revealed a positive expression of common ovine mesenchymal stem cell (MSC) markers CD29, CD44, STRO-4, as well as Vimentin and α-SMA (Fig. 3). A myogenic, mesodermal phenotype is supported by the positive expression of the intermediate filament Vimentin and α-SMA. Partial positivity was detected in CD166 and α-SMA in both oAFCs and oALCs. Isotype control staining did not produce any signals. Overall, no differences between oAFCs, oALCs, and oBMSCs (n = 3) could be detected by immunohistochemical analyses on a qualitative level.
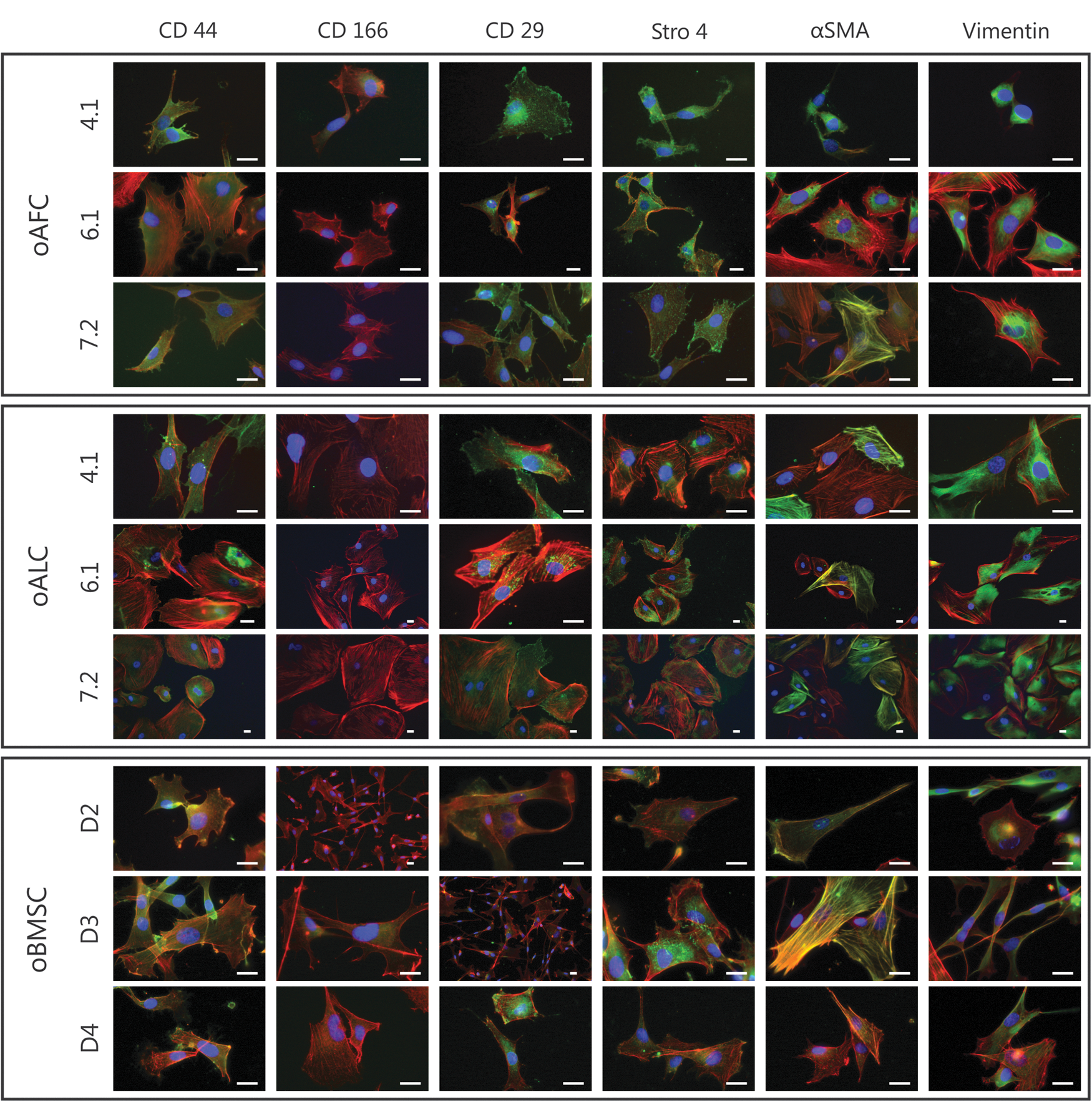
Phenotypic characterization of isolated oAFCs and oALCs. Immunohistological analyses of representative paired oAFCs (n = 3; animal 4.1, 6.1, and 7.2) and oALCs (n = 3; animal 4.1, 6.1, and 7.2) revealed positive expression of common MSC markers CD29, CD44, Stro4, and partial expression of CD166 (Phalloidin in red, DAPI in blue and respective antibody in green). Positive expression of Vimentin and αSMA show a myogenic, mesodermal phenotype of the cells. No phenotypic differences to standard oBMSCs (n = 3; animal D2, D3, and D4) were detected. Scale bar = 10 μm. MSC, mesenchymal stem cell; oBMSC, ovine bone marrow-derived mesenchymal stem cell; αSMA, α-smooth muscle actin; DAPI, 4′,6-diamidino-2-phenylindole. Color images available online at
Therefore, to further quantify MSC stem cell marker expression levels and to also detect differences between the three cell sources quantitatively, flow cytometry analyses for the same set of markers were conducted at passage 3–4 (Fig. 4). Again positive expression of CD29, CD44, CD166, and STRO-4 was found in oAFCs (n = 3), oALCs (n = 3), and control oBMSCs (n = 3) (with no expression of CD11b) (Fig. 4a). Notably, oAFCs and oALCs were paired and derived from the same fetus for flow cytometry analyses. No statistically significant differences in the percentage of positive cells concerning the expression of CD11b, CD29, CD44, and STRO-4 could be detected between the three cell sources (Fig. 4b, all P's > 0.05). CD166-positive cells were more abundant in the fetal cell sources (oAFCs and oALCs) than in adult oBMSC control cells. In contrast, MFIRs of the common MSC markers showed high heterogeneity between the three paired animal samples analyzed (Fig. 4c). However, a reduced MFIR of common MSC markers was observed in oALCs when compared with oAFCs from the same mother animal and fetus (Fig. 4d). This could be better appreciated when calculating the ratio of MFIR of oAFC/oALC samples for the following ovine MSC markers CD29 (mean ratio = 2.4), CD44 (mean ratio = 1.4), and STRO-4 (mean ratio = 3) (Fig. 4e).

MSC marker expression of isolated oAFCs and oALCs. Expression of common ovine MSC markers was confirmed by flow cytometry with positive expression of CD29, CD44, and STRO-4, partial expression of CD166 and no expression of CD11b in paired oAFCs (n = 3; animal 4.1, 6.1, and 7.2) and oALCs (n = 3; animal 4.1, 6.1, and 7.2). oBMSCs served as a control cell source (n = 3; animal D2, D3, and D4).
These results demonstrate a higher expression of MSC surface markers per single cell in oAFC compared with paired oALC samples (even if the overall percentages of cell populations with MSC marker expression were equal in oAFCs and oALCs). Contrary to human cells, positive expression of the following established set of markers—CD29, CD44, CD166, and STRO-4—defines MSC origin in the ovine animal model [26,27].
In addition, trilineage differentiation into the adipogenic, osteogenic, and chondrogenic lineages was proven for oAFCs (n = 3), oALCs (n = 3), and oBMSCs (n = 3). However, high interindividual variances were evident and successful differentiation had to be assessed by direct comparison to the control setup (Fig. 5). In particular, osteogenic differentiation resulted in a weak Alizarin Red staining even if clearly different to the control set-up.
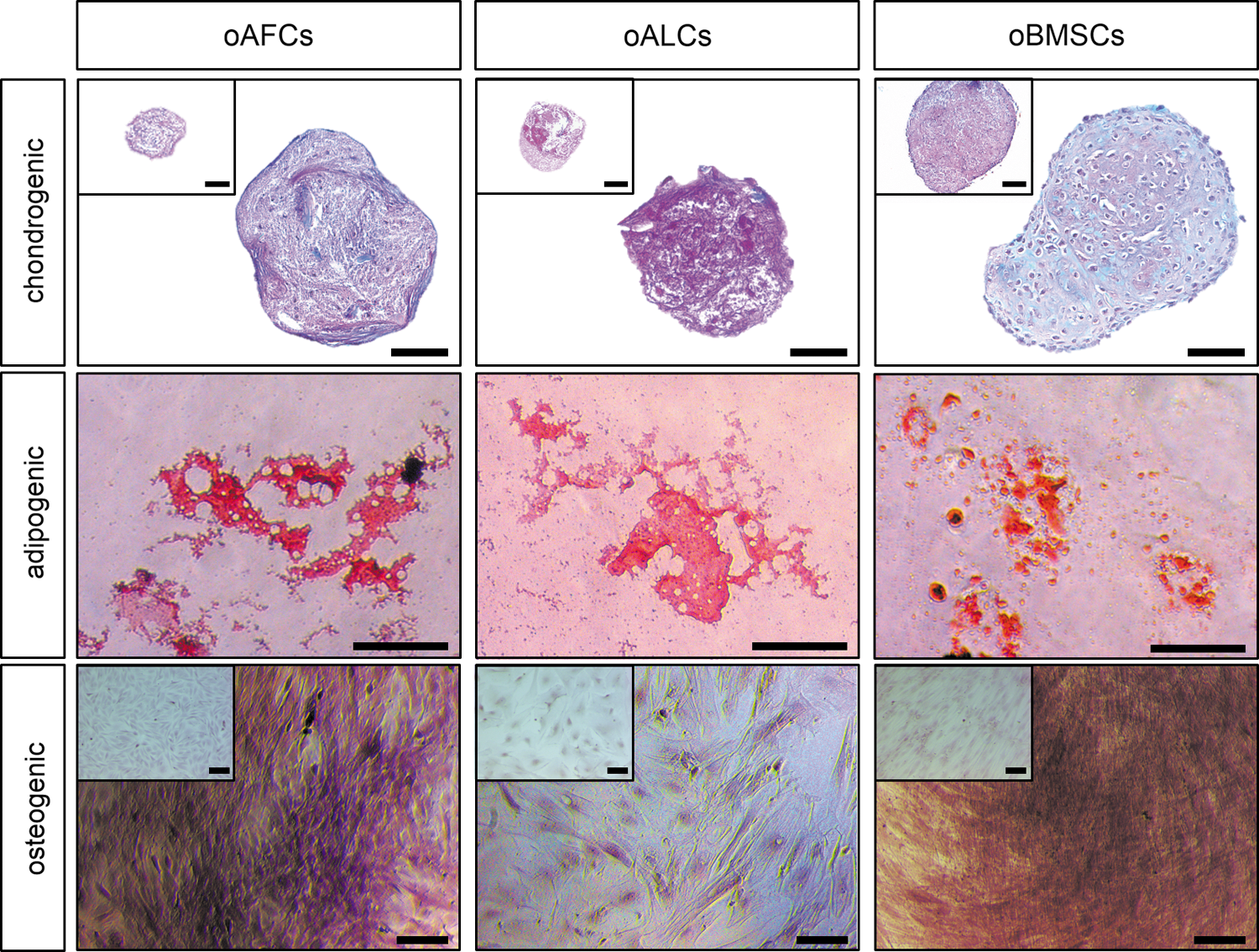
Multilineage differentiation potential of isolated oAFCs and oALCs. The capacity of oAFCs (n = 3), oALCs (n = 3), and oBMSCs (n = 3) to differentiate into adipogenic, osteogenic, and chondrogenic lineages was demonstrated by positive staining with Oil Red O, Alizarin Red S, and Alcian Blue PAS. The upper squares represent stained controls. Scale bar = 100 μm. Color images available online at
Pluripotency-related gene expression of oAFCs versus oALCs
The expression of OCT4, STAT3, and NANOG was qualitatively shown with immunofluorescence staining of representative cell lines of each cell group (Fig. 6). Higher expression of all pluripotency-related markers has been detected for fetal oAFCs (n = 3) and oALCs (n = 3) in direct comparison to control oBMSCs (n = 3). Ovine endothelial cells served as control cells to verify the specificity of the particular staining and did not express any of the analyzed pluripotency-related markers (data not shown).
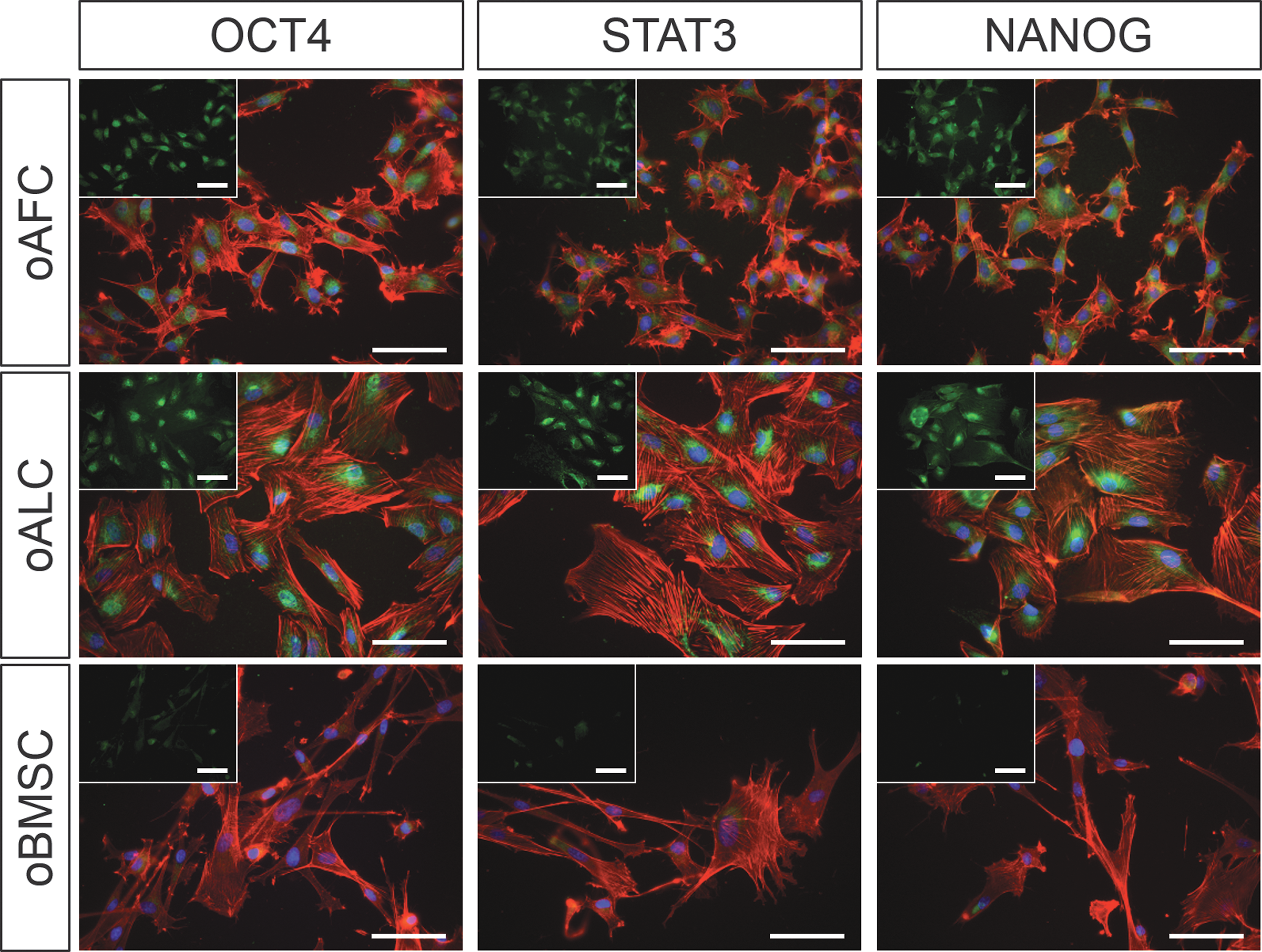
Phenotypic characterization of pluripotency-related stem cell markers of isolated oAFCs and oALCs. Immunohistological analyses of representative paired oAFCs (n = 3) and oALCs (n = 3) (Phalloidin in red, DAPI in blue, and respective antibody in green) revealed positive expression of the pluripotency-related markers OCT4, STAT3, and NANOG. In direct comparison to oBMSCs (n = 3) higher expression has been detected in fetal cells. Scale bar = 100 μm. Color images available online at
To quantify the differences in expression of genes involved in the maintenance of pluripotency, qRT-PCR analyses for OCT4, STAT3, NANOG, SOX2, and REX1, at early (p1 = passage 1) and late (p4 = passage 4) passages, were performed (Fig. 7). Significantly higher expression (P < 0.05) of OCT4, STAT3, NANOG, and REX1 was found in oAFCs (n = 9) compared with oALCs (n = 9) from the same offspring at p4 (Fig. 7a, d, g, j, and m). No significant difference was detected for SOX2 (P = 0.09). At p1, only a tendency of higher expression in oAFCs could be observed for OCT4, STAT3, NANOG, and SOX2, but with no statistically significant differences (Fig. 7a, d, g, j, and m). Given these results with no significant differences at p1, the observation of a decreasing heterogeneity of cell populations following further expansion might be of particular interest. REX1 revealed a tendency of higher expression in oALCs in p1 (P = 0.07), which could reflect a substantially higher expression of REX1 at early gestational ages (gestational age group 1). Unpaired standard oBMSCs (n = 3) served as a baseline MSC source for direct comparison.
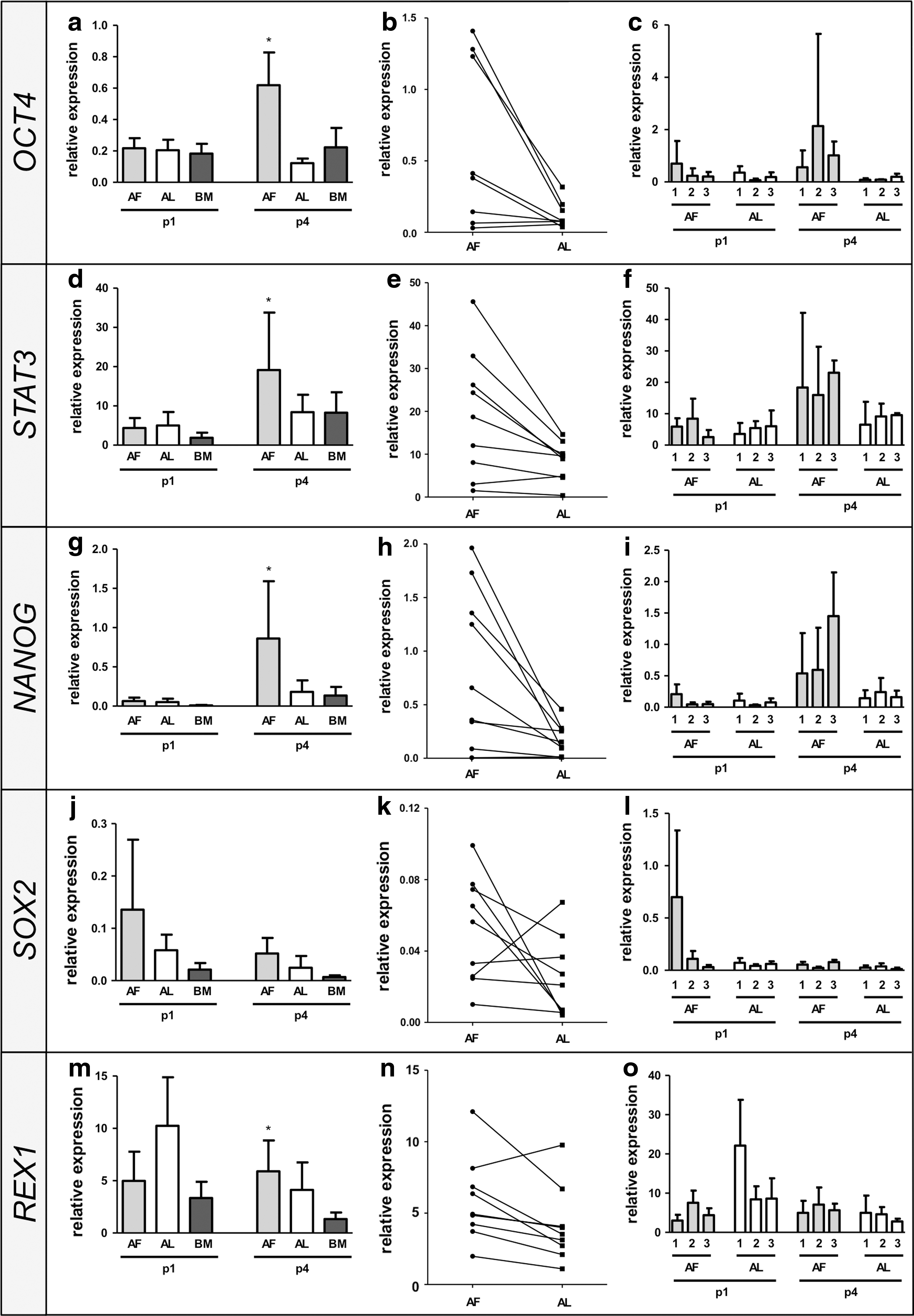
Quantitative real-time polymerase chain reaction of pluripotency-related stem cell markers of isolated oAFCs and oALCs. Genes involved in the maintenance of pluripotency, including OCT4
By pairing each oAFC cell line with the corresponding oALC cell line from the same offspring, a significantly reduced expression of pluripotency-related genes in oALCs becomes evident at p4 (Fig. 7b, e, h, k, and n). To further analyze the gestational age dependency of pluripotency-related gene expression, three gestational age groups have been established based on the CRL (n = 3 per group). However, qRT-PCR revealed no gestational age dependency in the expression of OCT4, STAT3, NANOG, and SOX2 (Fig. 7c, f, i, l, and o). Interestingly, higher Rex1 expression levels were observed in oALCs at gestational age group 1 compared with gestational age group 2 and 3.
Discussion
The present study aimed at addressing the origin of the broadly multipotent stem cell fraction in amniotic fluid by the use of the ovine developmental model, which is frequently used as an in vivo model for studying prenatal disease and fetal therapeutic applications before human clinical use.
However, one major difference to the human anatomy is the retention of an allantoic fluid cavity, which enabled us to derive samples of fetal fluid and cells from two separate extraembryonic fluid compartments from the same animal. In this manner, cells derived from the efferent urinary tract and the allantoic fetal adnexa (released into the allantoic fluid compartment) can directly be compared with those derived from the fetal skin, gastrointestinal, respiratory tracts, and amniotic fetal adnexa (released into the amniotic fluid compartment). In light of several preclinical studies also investigating amniotic fluid stem cell-based therapies in this animal model [15 –21], these differences may have to be taken into account in future trials focusing on a pure harvest of amniotic fluid only. However, in the presented study, this embryological difference enabled us to derive samples of fetal fluids and cells from both extraembryonic fluid cavities from the same animal and compare the expression of pluripotency-related markers between both cell types, including oAFCs and oALCs, over the entire duration of gestation.
In this regard, the present strictly paired analyses of ovine fetal fluid-derived cells, revealed significantly higher pluripotency-related gene expression in oAFCs than in oALCs, although showing similar cellular phenotypes. Significantly higher expression has been detected by qRT-PCR for OCT4, STAT3, NANOG, and REX1, whereas only a tendency for higher SOX2 was found. Interestingly, SOX2 expression has been associated with neural induction and enhanced neural differentiation [30], even in preselected SOX2 expressing AFSC [31], implying that in the present study only a small portion of neural progenitor cells have been detected in both fetal fluids analyzed. In the ovine preclinical model, the expression of several pluripotency-related genes has been reported in amniotic fluid-derived cells, including NANOG [12,15,16,32 –34], SOX2 [15,16,32 –34], OCT4 [15,16,33 –35], TERT [16,32 –35], and STAT3 [12], and further underlines that the ovine broadly multipotent cell compartment shows a similar expression pattern to the human counterpart [2,3,31,36 –42].
In humans, AFSC genes being involved in the undifferentiated state of cells have received major attention given that AFSCs are thought to represent an intermediate stage between pluripotent stem cells and lineage-restricted adult stem cells and ultimately can reacquire pluripotency through reprogramming more easily [5 –8]. AFSCs can form three-dimensional embryoid bodies, which were positive for alkaline phosphatase and expressed specific genes of ESCs [37]. Therefore, the presented results suggest that the cells with higher expression of selected markers representing hallmark genes of pluripotency, and thus broad(er) differentiation potential, originate rather from the fetus itself (including skin, respiratory, and gastrointestinal tract) or the extraembryonic amniotic tissues. The allantoic fluid cavity, being mainly composed of fetal urine, seems to contain a cell compartment with significantly lower pluripotency-related gene expression suggesting that the urogenital tract may not be the primary source of broadly multipotent cells, contrary to the speculation of previous reports [43 –45].
In humans, urine becomes evident in the amniotic fluid at week 8 of gestation, given that the urethra is fully developed and the fetal kidneys became functional [11]. In the second half of gestation (∼25 weeks), the production of amniotic fluid is predominately based on fetal urine (∼300 mL/kg fetal weight/day or 600–1,200 mL/day near term) and secretion of oral, nasal, tracheal, as well as pulmonary fluids (∼60–100 mL/kg fetal weight/day) [11]. Consequently, urine represents a possible abundant source of stem cells. A population of stem cell-like cells has also been recently described in human adult urine displaying MSC markers [46] and feasibility of using these cells for tissue engineering applications has been demonstrated [47 –49].
Previous studies described a heterogeneous cell population in amniotic fluid, where ultimately a wide spectrum of different cellular origins has been proposed, in particular, the fetal skin, urinary, gastrointestinal, and respiratory tracts as well as extraembryonic membranes. Few studies have described subpopulations of progenitor cells committed to specific lineages or prone to differentiate easily under appropriate culture conditions, including renal [43 –45], pancreatic [50], and neuronal [51] progenitors. In particular, renal/podocyte precursors are suggesting the urogenital tract as the possible source of amniotic fluid (stem) cells [43 –45]. On the other hand, human AFSCs derived from early gestational periods have shown to express ubiquitously Keratin-8, suggesting an epithelial origin of the cells [31]. However, conclusive evidence is missing so far and further research is necessary to investigate the origin of broadly multipotent stem cell component in particular.
Besides, the higher expression of pluripotency-related genes, oAFCs also exhibit a higher proliferative capacity in a direct, paired comparison to oALCs, which would be in line with their higher stem cell characteristics reported above. Particularly at higher passages, increased proliferation capacities of oAFCs were observed, where ultimately a more morphologically homogeneous cell population became evident in both cell types analyzed, in agreement with previous studies [52]. The authors suggest that stem cell-like cells are enriched over progressive passaging through selected proliferation medium toward a more homogeneous population, which further underlines the higher pluripotency-related gene expression pattern at later passages (p4) compared with early passages (p1) reported here. Immunofluorescence and flow cytometry stainings verified a mesodermal, myogenic phenotype of isolated fetal cells, with higher expression of common MSC markers per single cell (measured with MFIR) in oAFCs compared with oALCs. Selected MSC markers differ from the ones used with human MSCs given interspecies differences. They have previously been described to uniquely characterize ovine BMSCs for future translational studies involving this animal model [26,27]. Besides, one needs to emphasize the limited amount of well-established markers in the ovine model and the necessity for better standardized protocols for future clinical studies implying the ovine developmental model. This interspecies difficulty was also observed in the multilineage differentiation of ovine fetal cells, even if their multilineage differentiation potential could be positively confirmed.
Nevertheless, this study for the first time uniquely provides (i) a systematic comparative analysis of fetal fluid cells from both cavities, the amnion and the allantois, (ii) paired analysis of AFCs and ALCs from the same offspring, and (iii) quantitative information on the pluripotency-related gene expression over gestation. Initial studies on the isolation of fetal fluid cells have been performed in canine [53] and feline [54] models, proving the principal feasibility of ALC isolation with a differentiation potential and surface marker expression equivalent to multipotent MSCs from the amniotic fluid. However, oALCs have not been broadly characterized so far, and solid evidence for their stem cell nature is missing. In particular, the expression of pluripotency-related genes has not been analyzed. Furthermore, strictly paired quantitative analyses between AFCs and ALCs were missing and nonpaired analyses may neglect the substantial interindividual variances also detected in the present study.
In spite of the differences between oAFCs and oALCs, the present study did not find any gestational age-dependent differences of cellular phenotype, proliferation, and the expression of pluripotency-related markers. So far, most studies on human AFSCs using cells from the second trimester of gestation, as routine amniocentesis in humans, are usually performed between 14 and 26 weeks of pregnancy [55]. Nevertheless, several studies have demonstrated that human AFSCs can also be successfully isolated at the first [6,38] and third [39,56] trimesters of gestation. In agreement with our results, comparable cellular phenotypes have been observed in human AFSCs isolated in the second or third trimester of gestation, including expression of Oct4 and Nanog [39,56].
However, Moschidou et al. illustrated that cells isolated from the first trimester of gestation express a larger number of organ-specific genes, suggesting a more undifferentiated state of cells [38]. As development proceeds, cells from the second trimester do not express the complete pattern of pluripotent factors anymore and become more committed with a limited differentiation potential [6,38,39]. Also when isolating circulating CD34+ hematopoietic progenitors cells and multipotent stem cells from the human umbilical cord blood, the frequency and immature pool of cells showed a decrease toward the later stages of pregnancy [57,58]. However, in the present study, the complete pattern of pluripotency-related genes, including OCT4, NANOG, STAT3, SOX2, and REX1, showed a consistent expression independent of the developmental stage at which cells were harvested. In addition, also no morphological phenotype tendency could be assigned to a certain gestational age, all three morphological phenotypes were equally distributed, and abundant at every stage of fetal development.
These results suggest that cells with high pluripotency-related gene expression seem to be equally abundant within the amniotic fluid over the entire duration of gestation. Consequently, amniotic fluid harvest for regenerative applications may also be performed shortly before or directly at birth, to minimize any risk of amniocentesis for the fetus in cases where no diagnostic amniocentesis is necessary. A single study focusing on the analysis of human amniotic fluid obtained at the time of elective cesarean delivery also reported expression of Oct-4; however, the analysis was limited to this single pluripotency-related factor only [59]. Further research on human amniotic fluid samples pre- and perinatally seems mandatory to confirm these findings and to establish amniotic fluid as a potential perinatal cell source.
In conclusion, the presented data suggest that the expression of pluripotency-related genes of cells derived from the fluid directly surrounding the fetus is substantially higher than the one of cells derived from the efferent urinary tract. These findings suggest that the cells expressing the highest stem cell characteristics may originate from the fetus itself (eg, the fetal skin, the respiratory, or the gastrointestinal tract). Interestingly, contrary to previous reports on human cord blood [57,58], no significant decrease of the pluripotency-related gene expression was noted over pregnancy, implying that even at the end of pregnancy, AFSCs with high differentiation potential may be isolated. These findings will have to be confirmed by future studies in humans, at least as far as the major ethical and medical restrictions allow for it. However, if confirmed, these findings may have major implications for harvesting, banking, and usage of AFSCs for future therapeutic applications.
Footnotes
Acknowledgments
The authors would like to thank Paolo Cinelli, Ursula Steckholzer, Petra Wolint, Laura Frese, Agnieszka Książek, and Burkhardt Seifert (IFSPM) for their technical and statistical help and assistance. Imaging and flow cytometry analyses were performed with equipment maintained by the Center for Microscopy and Image Analysis, University of Zurich, and the Flow Cytometry Facility, University of Zurich, respectively. Differential fluid analyses were performed in collaboration with the Institute of Clinical Chemistry, University Hospital Zurich, Switzerland. The authors would like to further acknowledge Paul Verma and Jun Liu of the Stem Cell and Genetic Engineering Group, Monash University, Clayton, Australia, for providing iPSC-cDNA for the establishment of all primers. This work was supported by the Swiss National Science Foundation (project 310030_143992), the Alfred and Anneliese Sutter-Stöttner-Foundation (doctoral dissertation Debora Kehl, Münchwilen, Switzerland), and the Foundation for Research in Science and the Humanities at the University of Zurich, Switzerland.
Author Disclosure Statement
No competing financial interests exist.