
Abstract
Very small embryonic-like cells (VSELs) are a population of very rare pluripotent stem cells isolated in adult murine bone marrow and many other tissues and organs, including umbilical cord blood (UCB). VSEL existence is still not universally accepted by the scientific community, so for this purpose, we sought to investigate whether presumptive VSELs (pVSELs) could be isolated from human UCB with an improved protocol based on the isolation of enriched progenitor cells by depletion of nonprogenitor cells with magnetic separation. Progenitor cells, likely including VSELs, cultured with retinoic acid were able to form dense colonies and cystic embryoid bodies and to differentiate toward the ecto-meso-endoderm lineages as shown by the positivity to specific markers. VSEL differentiative potential toward mesodermal lineage was further demonstrated in vitro upon exposure to an established inductive protocol, which induced the acquisition of renal progenitor cell phenotype. VSEL-derived renal progenitors showed regenerative potential in a cisplatin model of acute kidney injury by restoring renal function and tubular structure through induction of proliferation of endogenous renal cells. The data presented here foster the great debate that surrounds VSELs and, more in general, the existence of cells endowed with pluripotent features in adult tissues. In fact, the possibility to find and isolate subpopulations of cells that fully fit all the criteria utilized to define pluripotency remains, nowadays, almost unproven. Thus, efforts to better characterize the phenotype of these intriguing cells are crucial to understand their possible applications for regenerative and precision medicine purposes.
Introduction
U
To our knowledge, these cord blood-derived pluripotent stem cells share embryonic-like characteristics with embryonic stem (ES) cells and for this reason, based on our final results, we agreed to call them VSELs [8]. They are epiblast-derived cells and have been isolated and described in different tissues such as bone marrow [9], ovary [10], and heart [11]. They have a very primitive morphology with small size and large nuclei mostly comprising euchromatin; they are negative for CD45 and lineage, but they do express CD133 and many pluripotent genes and meet almost all the in vitro criteria established to define pluripotency [5].
Although their existence is still highly debated [12] and several articles reported different isolation and classification protocols, we developed an improved method to isolate presumptive VSELs (pVSELs) from UCB based on the depletion of nonprogenitor cells with magnetic separation. In this way, we were able to grow purified pVSELs in medium containing all-trans retinoic acid (RA), to form embryoid bodies (EBs) and cystic embryoid bodies (CEBs), and to differentiate them toward the extraembryonic lineages, thus providing evidences on the bona fide nature of VSELs.
RA plays a key role for differentiation of both mouse and human ES cells toward the extraembryonic lineages [13 –15] and can thus be considered one of the key factors, inducing VSELs, to undertake the differentiation process together with deacetylase inhibitors, valporic acid, or nicotinamide, all able to induce proliferation by remethylating the differentially methylated regions of parentally imprinted genes (Igf2, H19, Rasgrf1) erased in VSELs, as recently shown by Ratajczak et al. [16].
For future applications in the regenerative field, it would be relevant to test, once the potential to differentiate toward the ecto-meso-endoderm lineages is proved, the VSEL capability to be further differentiated into more specialized cells. Recently, it has been shown that induced pluripotent stem cells (iPSCs) can be differentiated into renal progenitor cells (RPCs) through exposure to an efficient inductive protocol and that iPSC-derived RPCs were able to be engrafted into damaged tubuli, restoring both renal function and structure in cisplatin mice with acute kidney injury (AKI) [17]. Since VSELs seem to share many characteristics with both ES cells and iPS cells, we sought to investigate their ability to differentiate in vitro into renal precursors and to test in vivo their potential to promote kidney regeneration in a mouse model of AKI.
Based on our preliminary data, we believe that VSELs represent the future of the embryonic-like stem cells as wisely suggested by Kassmer and Krause [18] and we also believe that they represent a valuable treasure that should be exploited for regenerative medicine purposes assigning an additional value to UCB.
Materials and Methods
Umbilical cord blood sample
pVSELs were isolated from fresh UCB units (<24 h after birth) collected for banking, but discarded either for volume or cell content, according to international protocols adopted for UCB banking (FACT-NETCORD; mother's written consent for research use No. 11601399). In total, 54 UCB units (21 males and 22 females) were used and, among them, 34 have been in vitro cultured to get colonies and CEBs, 9 have been differentiated toward extraembryonic lineages, and 11 have been used for renal differentiation.
Briefly, 10% of sterile sodium citrate was added to UCB diluted then with 1:1 sterile phosphate-buffered saline (PBS; Gibco). The solution was carefully overlaid with Lympholyte Cell Separation media (Cederlane, Ontario, Canada), and the mononuclear cell fraction containing pVSELs was recovered after centrifugation at 800g for 20 min from the layer at Lympholyte–plasma interface and the cells were then centrifuged at 300g for 7 min. The supernatant containing the smallest cells (including pVSELs) was centrifuged at 1,000g for 10 min and the pellet resuspended in PBS +2% fetal bovine serum (FBS) and diluted at 5 × 107 cells/mL before negative immunoselection.
A human progenitor cell enrichment kit was used to isolate pVSELs according to the manufacturer's instructions (EasySep, Human Progenitor Cell Enrichment Kit; Stemcell Technology, Vancouver, Canada). Introduction of this step allowed us to collect pluripotent progenitor cells (containing or totally represented by pVSELs) residing in the mononuclear fraction of UCB with immunomagnetic selection excluding all the mature blood cells expressing CD2, CD3, CD11b, CD11c, CD14, CD16, CD19, CD24, CD56, CD66b, and glycophorin A. The cellular fraction obtained from this procedure was then centrifuged at 1,000g for 10 min and the pellet containing pVSELs (∼105 cells/mL) was resuspended in culture media [15% FBS, 1% Pen/Strep, 0.1 mM NEAA, 0.1 mM 2-mercaptoethanol, Dulbecco's modified Eagle's medium (DMEM)+
A total of 2 × 106 pVSELs/mL were then seeded in Petri dishes containing culture media with different concentrations of RA (RA+; 1, 5, 10 μM; Sigma, St. Louis, MO) dissolved in dimethyl sulfoxide (DMSO; Euroclone, Milan, Italy) and in Petri dishes without RA (RA−). Media were changed every week. Colonies and EBs formed only in the RA+ dishes after 2 weeks in culture. No EB formation was assessed in RA− culture conditions.
All the colonies, EBs (collected after 7–15 days) and CEBs (collected after 15 days onward) in the presence of different concentrations of RA, were collected and immediately treated for further analyses.
pVSEL characterization by flow cytometry
Flow cytometry analysis was performed before pVSEL differentiation and immediately after isolation (day 0) and after 7, 12, and 30 days to evaluate the positivity/negativity of different antigens (CD45, CXCR4, CD34, and CD133). Flow cytofluorimetric events with very small dimensions were included and the complete blood count analysis validated the absence of erythrocytes and white blood cells. In particular, the CD45-negative fraction was first selected to later sort the CD45-negative, CD34-negative, and CD133-positive cells using a Boolean gating strategy. Last, we identified the CXCR4 (CD184+) positivity of this little cluster of cells.
The Beckman Coulter Navios instrument was employed with a no-wash technique, using the following fluorochrome-conjugated monoclonal antibodies (Beckman Coulter, Brea, CA): anti-CD45 allophycocyanin (APC)-Alexa Fluor 750, anti-CXCR4 (CD184) phycoerythrin (PE), anti-CD34 PE-cyanin 7 (PE-Cy7) or fluorescein isothiocyanate (FITC), and anti-CD133 APC (AC133 clone; Miltenyi Biotec, Bergisch Gladbach, Germany) following the manufacturer's instructions (Beckman Coulter). Samples were incubated in the dark for 10 min before analyses. Stained cells were analyzed with Kaluza software (Beckman Coulter).
Immunofluorescence of pVSELs, colonies, and EBs/CEBs
Undifferentiated pVSELs, isolated colonies, and EBs/CEBs were analyzed by immunofluorescence with antibodies against human OCT4, SOX2, and NANOG (triple immunostaining), and Nestin, Brachyury (T), and α-fetoprotein (AFP; triple immunostaining; Table 1). Briefly, after fixation in 4% paraformaldehyde (PFA; Thermo Scientific, Roskilde, Denmark) for 20 min and permeabilization in 1 × PBS, 10% FBS, and 0.5% Triton X-100 for 15 min at room temperature, they were blocked in 1 × PBS, 0.1% Tween 20, and 10% FBS for 15 min at room temperature. All the antibodies were properly diluted in blocking solution.
All the antibodies are produced by Abcam (Cambridge, UK) except FITC-conjugated donkey anti-goat (Santa Cruz Biotechnology, Dallas, TX).
FITC, fluorescein isothiocyanate.
Colonies and EBs/CEBs were mounted on slides using Vectashield Mounting Medium with DAPI (Vector Laboratories), while undifferentiated pVSELs fixed and immunostained directly in Petri dishes were counterstained with NucBlue Live Cell Stain Ready Probes (Life Technologies, Grand Island, NY) following the manufacturer's instructions.
Images were collected using an all-in-one confocal laser scanning microscope Olympus Fluoview Fv10i and processed using Fluoview software. All the images obtained are the Z projections of optical sections. Negative controls (no primary antibodies) were always added to each immunofluorescence experiment.
Ectoderm, mesoderm, and endoderm differentiation
The Human Pluripotent Stem Cell Functional Identification Kit (R&D Systems, Minneapolis, MN) was used to differentiate pVSELs toward ectoderm, mesoderm, and endoderm lineages. The differentiation procedure was applied on cells cultured in normal media and induced to differentiate immediately after isolation (day 0) and after 1 week, 2 weeks, and 1 month (days 7, 12, and 30) of in vitro culture, following the manufacturer's instructions. Media and specific differentiation cocktails were replaced each day and the cells were fixed for mesoderm differentiation after 48 h and for ectoderm and endoderm differentiation after 72 h. Controls (ie, cells cultured at the same conditions without adding any differentiating factor in the culture media) have been added to the analyses to verify that cells did not start the differentiation process spontaneously.
Ectoderm, mesoderm, and endoderm analysis by immunofluorescence
Differentiated cells were washed in PBS and fixed in 4% PFA for 20 min at room temperature. Cells were permeabilized for 45 min at room temperature, blocked for 30 min at room temperature, and incubated with specific antibodies diluted in blocking solution at 4°C overnight (Human Pluripotent Stem Cell Functional Identification Kit; R&D Systems: OTX2 for ectoderm, Brachyury for mesoderm, and SOX17 for endoderm at the concentration of 1 μg/100 μL). Cells have then been incubated with secondary antibody NL557 at the concentration of 2 μg/mL for 60 min at room temperature in the dark (R&D Systems). Nuclei were counterstained with NuncBlue. Three controls, specific for each differentiation, were analyzed simultaneously together with cross-check controls to test the specificity of the differentiation that occurred.
Images were taken with the confocal microscope Fluoview FV10i and then analyzed with the FV10i viewer software.
pVSEL differentiation into RPCs
To induce pVSEL differentiation toward the RPC phenotype, a differentiative protocol initially set up for differentiating human iPSC into RPCs was followed [17]. Briefly, freshly isolated pVSELs were incubated with DMEM/F12 medium containing 5% FBS (Hyclone; ThermoFisher, Milan, Italy), 0.1 mM nonessential amino acids, 0.1 mM 2-mercaptoethanol supplemented with 0.1 μM all-trans RA (Sigma-Aldrich, Milan, Italy), 1 μM CCG1423 (RhoA Inhibitor; Vinci Biochem, Florence, Italy), and 5 μM LY294002 (PI3K inhibitor) for 6 days. Ten nanograms per milliliter activin A (Peprotech) was added for 2 days starting on day 2. Then, the medium was replaced with fresh basal medium containing nephrogenic factors: 50 ng/mL BMP7 (Peprotech), 10 ng/mL FGF2, and 15 ng/mL GDNF (Abcam, Cambridge, MA). The medium was changed every 2 days.
Immunofluorescence of renal differentiated VSELs
Cells were fixed in 4% PFA in PBS for 30 min at room temperature and permeabilized in 0.5% Triton X-100 for 30 min at room temperature, followed by incubation with 5% bovine serum albumin diluted in PBS. The primary antibodies diluted according to the manufacturer's recommendations were incubated overnight at 4°C, followed by incubation with the appropriated secondary antibody for 1 h at room temperature. Primary antibodies included anti-NANOG (Santa Cruz Biotechnology, Santa Crus, CA); anti-WT1 (R&D Systems), anti-SIX2 (Proteintech, Manchester, UK), and anti-PAX2 (Invitrogen), followed by the secondary antibody Alexa Fluor 546 (Jackson ImmunoResearch). Images (representative of n = 3 experiments) were acquired by Apotome Axio Imager Z2 (Zeiss, Jena, Germany).
Cisplatin-induced AKI mouse model and VSEL-derived RPC injection
Animal care and treatment were in accordance with institutional guidelines in compliance with national (D.L. n.116, G.U., suppl 40, February 18, 1992, Circolare No. 8, G.U., July 14, 1994) and international laws and policies (EEC Council Directive 86/609, OJL 358, Dec 1987; NIH Guide for the Care and Use of Laboratory Animals, U.S. National Research Council, 1996). Animal studies were submitted to and approved by the Institutional Animal Care and Use Committee of “Mario Negri” Institute (Milan, Italy). Animals were housed in a constant temperature room with a 12-h dark:12-h light cycle and fed a standard diet.
Cisplatin (13.9 mg/kg; Ebewe Italia Srl) was subcutaneously injected in 2-month-old female NOD-SCID mice (Charles River Italia S.p.a., Lecco, Italy), and after 24 h, mice were divided into two groups receiving injection in the tail vein of saline or VSEL-derived RPCs (5 × 105 cells/mouse). In selected experiments, VSEL-derived RPCs were prelabeled with cell tracker PKH26 (Sigma-Aldrich) following the manufacturer's instructions. Mice were sacrificed 4 days after cisplatin and kidneys were used for histology and immunohistochemistry. Renal function was assessed as blood urea nitrogen (BUN) by the Reflotron test (Roche Diagnostics Corporation, Milan, Italy). BUN levels exceeding 30 mg/dL were considered abnormal. Untreated mice were used as controls.
Immunohistochemical analysis of renal tissues
Kidney samples were fixed in Duboscq-Brazil and paraffin sections were stained with hematoxylin and eosin or periodic acid-Schiff reagent (PAS). Luminal hyaline casts and tubular necrosis (denudation of tubular basement membrane) were assessed in nonoverlapping fields (up to 28 for each section) (40 × , high-power field, HPF).
PKH26 prelabeled VSELs were identified in PLP-fixed renal sections costained with FITC-WGA lectin (Vector Laboratories) and DAPI.
Cell proliferation was investigated in renal tissues 4 days after cisplatin treatment, on PLP-fixed cryosections incubated with anti-Ki67 (Abcam), followed by the appropriate Cy3-conjugated secondary antibody, FITC-WGA lectin, and DAPI. Cell proliferation has been quantified as the number of Ki-67-positive cells/HPF (15 fields/section, n = 10 animals/group).
Statistical analysis
Data are expressed as mean ± standard error of the mean (SEM) of biological replicates. Data analysis was performed using the computer software Prism (GraphPad Software, Inc.). Comparisons were made using analysis of variance (ANOVA) with the Tukey post hoc test. Statistical significance was defined as P < 0.05.
Results
Immunomagnetic cell selection
The recovered fraction of pluripotent, small, round progenitor cells collected after immunomagnetic cell selection was able to grow and expand rapidly during the first 7 days and slow down to a more stationary phase around day 10 in culture. From day 20 onward, cells started to detach and die, and by day 30, only few cells were vital.
pVSELs appeared round shaped with a variable diameter from 2 to 5 μm up to 10 μm (Fig. 1a–c). Interestingly, they did not show any change in morphology during culture.
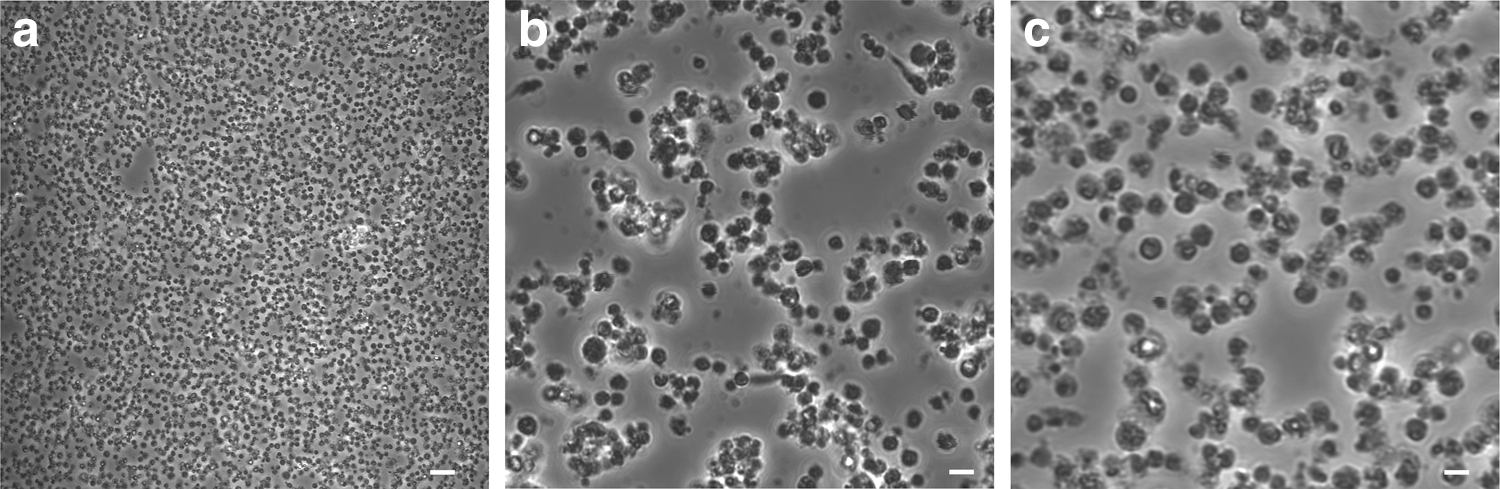
Images of isolated pVSELs. Magnification 120 ×
Cytofluorimetric analyses
The cytofluorimetric analyses on freshly isolated samples confirmed that our improved protocol is able to isolate and purify cells with very small dimensions (2–10 μm), very low cellular complexity, and well clustered in the CD45− region (>98% in all the analyses performed; Fig. 2a). Flow cytofluorimetric events matching with larger cells were present, but always in a very limited amount. The recovered CD45−/CD133+ fraction was extremely reduced (Fig. 2b), confirming what is already published in literature [19,20], and the CD34 signal indicates a small cluster of positive cells (Fig. 2c).
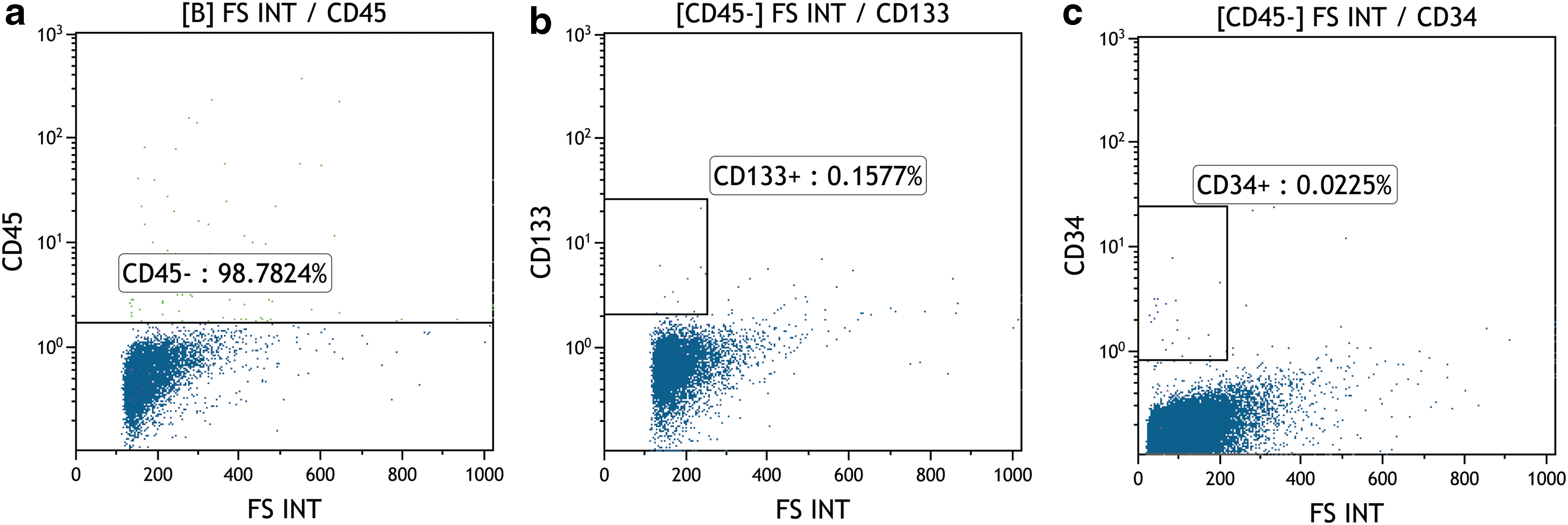
Cytofluorimetric analyses showing the smallest cells clustered in
The analysis on pVSELs confirmed our observations about rapid expansion during the first week, followed by a stationary phase and a subsequent decrease in number. During days 12–30 of culture, the number of contaminants (ie, larger cells, likely not pluripotent and with a different phenotype) decreased compared with cells plated at days 1–7, showing that our media were somewhat specific for undifferentiated cells and not suitable for more differentiated cells.
Interestingly, the CD45−/CD133+ percentage increases from day 0 (ie, right after isolation from UCB) as a further indication of the optimal culture conditions used. In fact, at day 7, the recovered CD133+ fraction was higher than at day 0, while from day 12 onward, we found a significant subfraction of the CD133+ cells with a CD184+ configuration (including a very rare fraction of CD34−) representing the more undifferentiated cells (Fig. 3).
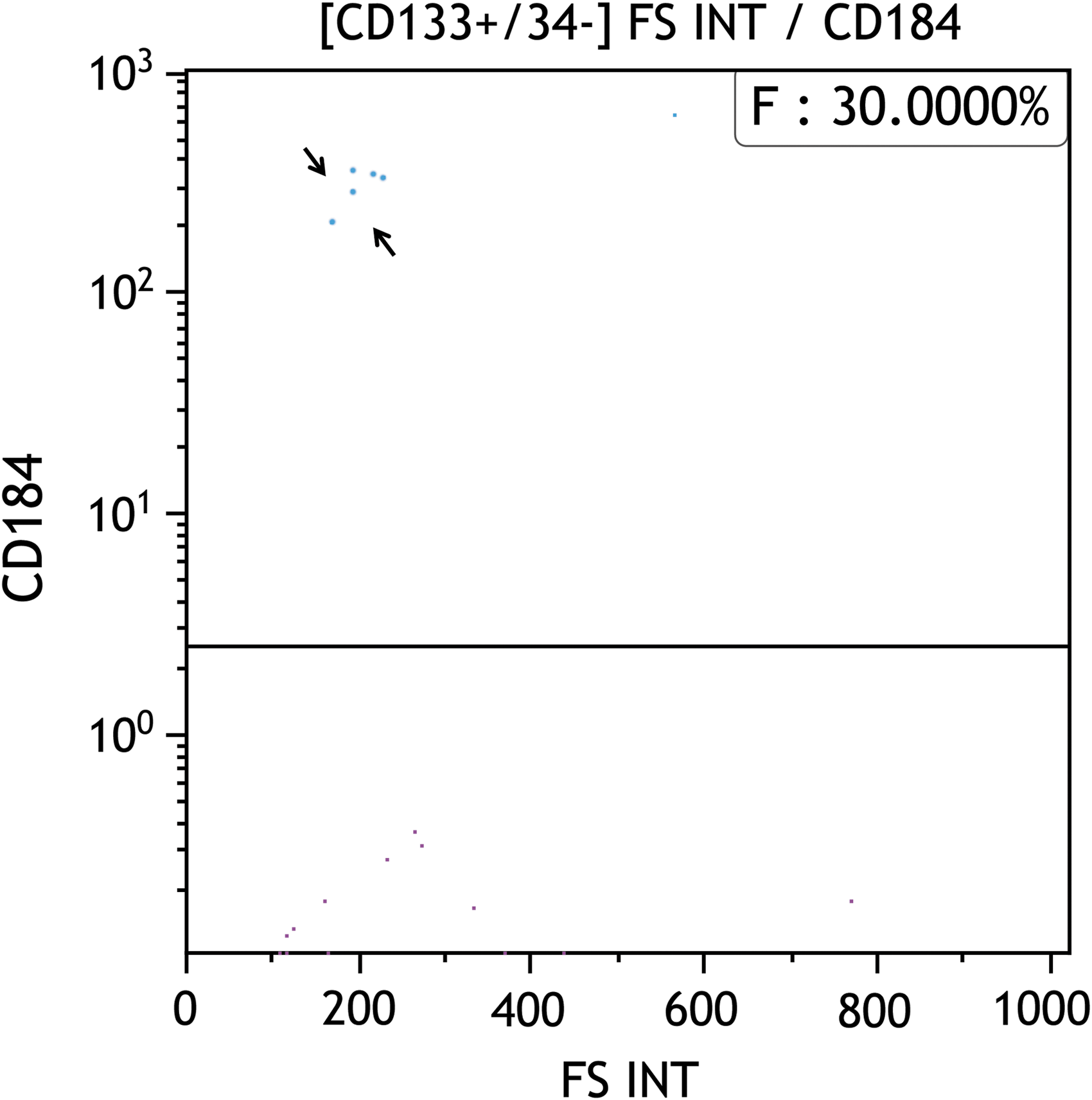
Representative image of cytofluorimetric analysis of day 12 undifferentiated pVSELs showing very limited, but significant, fraction of CD45−/CD133+/CD34−/CD184+ cells (arrows). In this figure, 30% of CD45−/CD133+/CD34− cells have a CD184+ phenotype.
Immunofluorescence of pVSELs, colonies, and EBs/CEBs
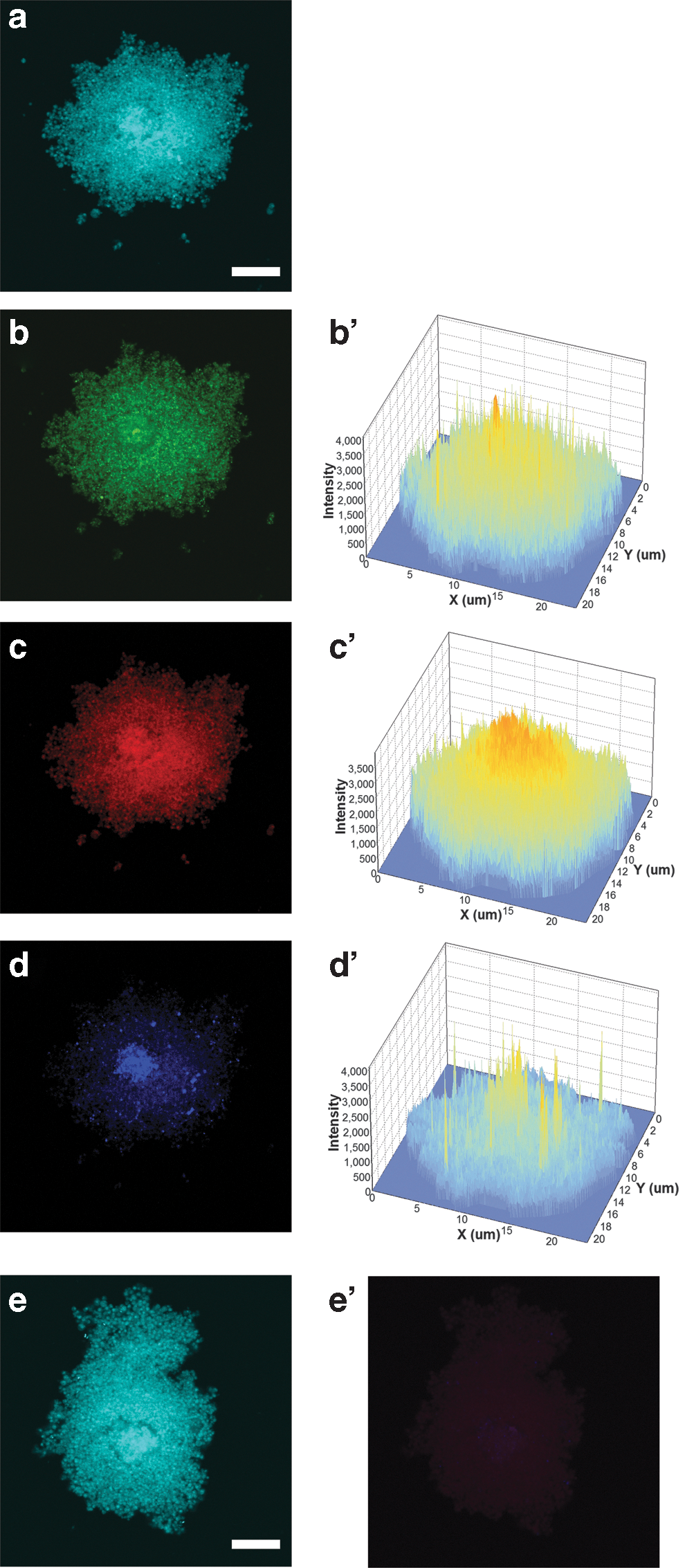
Immunofluorescence of undifferentiated pVSELs (Fig. 4) with antibodies specific for pluripotent proteins showed positivity for OCT4 and NANOG (Fig. 4b, d) and a negative signal for SOX2 (Fig. 4c).

Z-stack section of plated pVSELs triple stained with pluripotent proteins. DAPI
Since pVSELs show characteristics resembling ES cells, we analyzed the effects of different RA concentrations in culture media showing that some cells started to detach and dye at the highest concentrations of RA (10 μM), while many other healthier and probably selected pVSELs were able to form two different types of aggregates: dense colonies with heterogeneous morphology and dimensions (Fig. 5a–c) and spherical floating aggregates resembling EBs (Fig. 5d) and CEBs (Fig. 5e, f), as suggested by Kim et al. [21].

Images of different colonies cultured in the presence of 1 μM
Colony immunofluorescence shows an intense and diffused OCT4 signal (Fig. 6b) and well-defined SOX2 and NANOG positivity in the central region (Fig. 6c, d), supported by further analyses of three-dimensional (3D) localization (Fig. 6b’–d’), probably indicating the presence of different cell types forming or joining this central area. We investigated whether the cells forming the colonies (Fig. 7) spontaneously began (or because they were induced by RA) to differentiate toward precursor cells of the ecto-meso-endoderm lineages by analyzing the localization of Nestin (ectoderm), Brachyury, and AFP (meso-endoderm), showing a central positive staining for the ectoderm-specific marker (Fig. 7b), no signal for the mesoderm marker (Fig. 7c), and a brilliant spotted-like signal for AFP (Fig. 7d), for all the RA concentrations used (1 and 5 μM).
Representative Z-stack section of a colony stained with pluripotent proteins. DAPI

Representative Z-stack section of a colony (RA +5 μM) stained with proteins specific for ecto-meso-endoderm markers. DAPI
Fixed EBs/CEBs have been analyzed by immunofluorescence and the signals appeared very faint for OCT4 (Fig. 8b) and mostly diffused with some positive cells mainly located on the outer part of the aggregates for SOX2 and NANOG (Fig. 8c, d), as verified by the analysis of 3D localization. Actually, an accurate analysis of the 3D images gives us interesting hints: for example, the number of NANOG-positive peaks corresponding to NANOG-positive cells seems higher compared with SOX2, although their intensity is totally comparable (Fig. 8c’, d’).
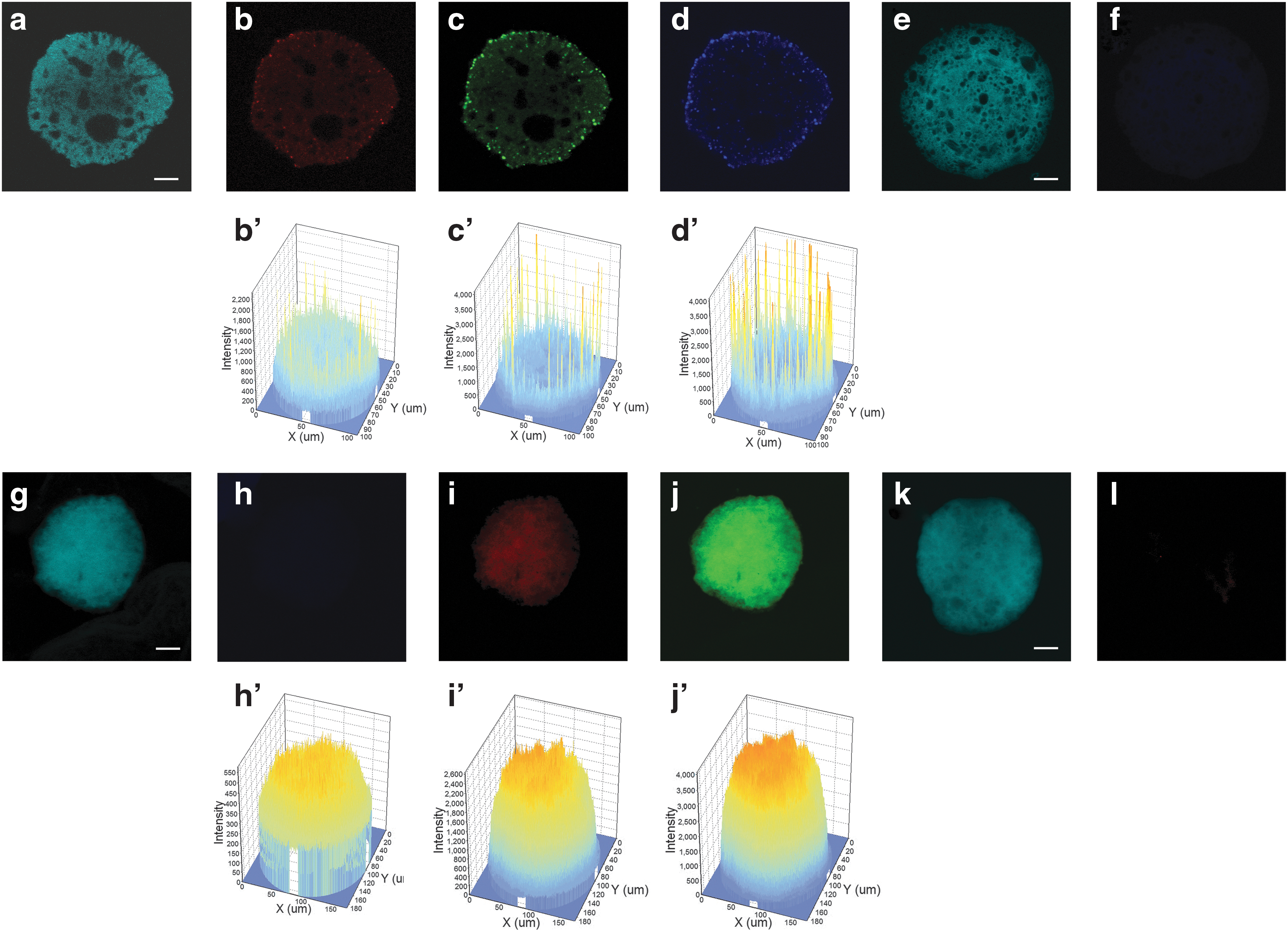
Representative Z-stack section of CEB (RA +5 μM) triple stained with pluripotent proteins. DAPI
EBs seem to share the same cellular heterogeneity with the colonies as shown by our further analyses on Nestin, Brachyury, and AFP. Interestingly and unexpectedly, Nestin signal is totally absent (Fig. 8h), Brachyury is mostly diffused in the central region (Fig. 8i), and only AFP is highly diffused (Fig. 8j). 3D protein localization shows several intense orange-colored peaks for AFP mostly concentrated in the center of the CEB (Fig. 8j’), a lower signal for T (Fig. 8i’) and only background signal as shown by a thick blue layer, and a very low-intensity signal for Nestin (Fig. 8h’).
As for the colonies, no appreciable differences in protein signals have been detected in EBs/CEBs obtained with different RA concentrations in culture media (1 and 5 μM).
Three germ layer differentiation
To finally assess the pluripotency features of pVSELs, from now on called VSELs, once the positivity to specific markers and the ability to form EBs and CEBs were proved, we induced the ectoderm, mesoderm, and endoderm differentiation using nine different UCB samples to better prove the VSEL commitment to differentiation, as shown in Fig. 9 and Supplementary Figs. S1 and S2 (Supplementary Data are available online at
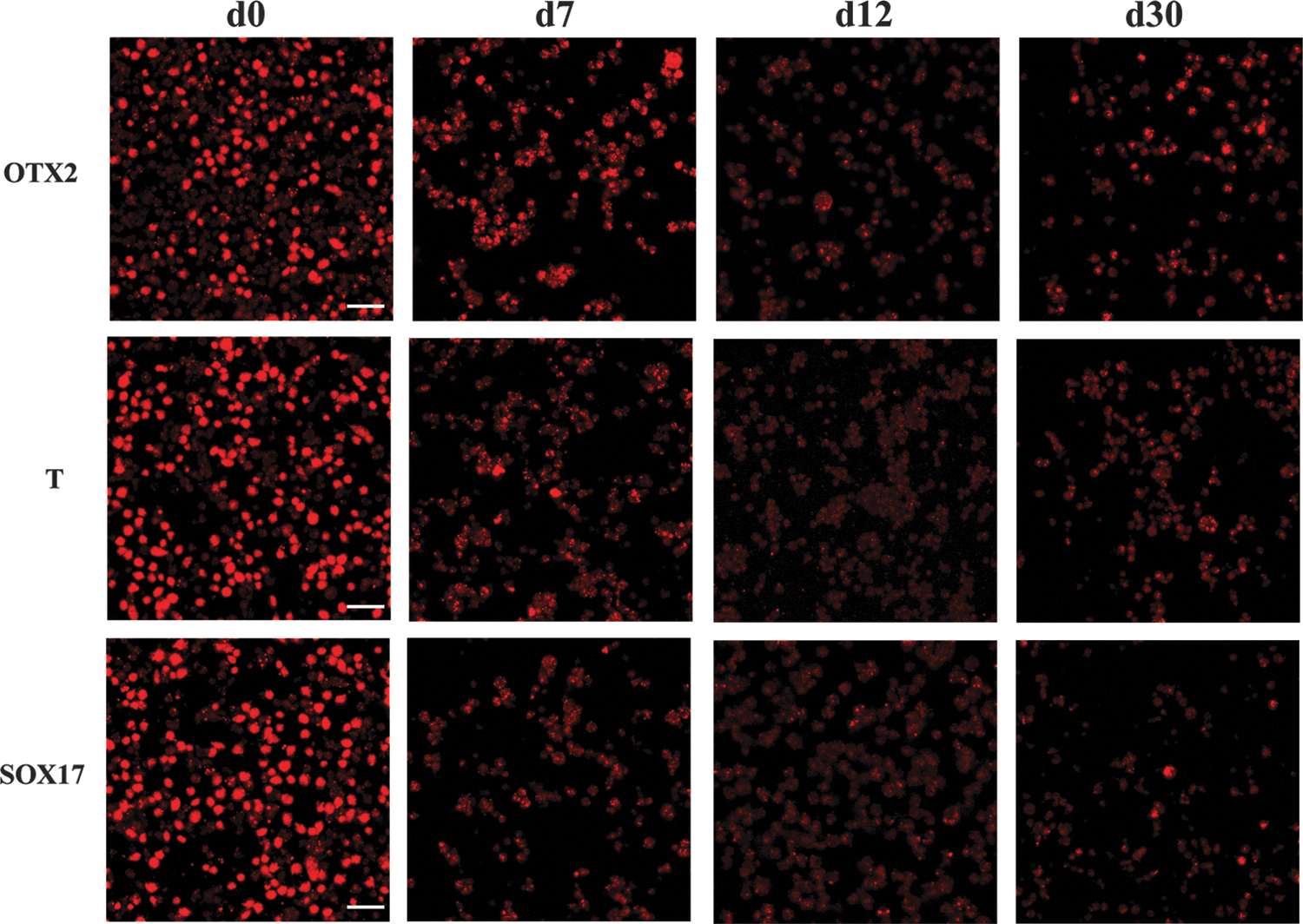
Representative panel of VSEL differentiation in ectoderm (OTX2), mesoderm (T, Brachyury), and endoderm (SOX17) from d0 to d30. Magnification 180 × . Bar = 20 μm. d0, day 0; d7, day 7; d12, day 12; d30, day 30.
As detailed in the Materials and Methods section, VSELs have been induced to differentiate at different times of in vitro culture in normal media to understand if their behavior (ie, ability to respond to differentiation supplements) changed in some way during these time points. Immunofluorescence with antibodies specific for ectoderm (OTX2), mesoderm (Brachyury), and endoderm (SOX17) lineages and flow cytometry analyses were then performed.
Our data showed that VSELs are more committed to differentiate when freshly isolated (day 0) than after weeks in culture (days 7–30) as shown by the very brilliant signals shown in the first panel of Fig. 9. At days 7 and 12, the number of positive cells seems to be highly reduced, probably indicating that a prolonged VSEL culture induces a strong selection: many of the cells died and detached from the dish, but few healthy cells properly differentiate showing a good signal for all the markers used. Negative controls and cross-check controls have been added to differentiated cells at day 0 (ie, differentiated cells toward ectoderm have been stained with Brachyury and SOX17; mesoderm differentiated cells have been stained with OTX2 and SOX17; and endoderm differentiated cells have been stained with OTX2 and Brachyury; Supplementary Figs. S1 and S2).
pVSEL in vitro differentiation toward the renal progenitor phenotype
The pVSEL ability to acquire the renal progenitor cell phenotype has been tested to both study their differentiative capacity and to obtain tissue-specific progenitors potentially useful for regenerative purposes. pVSELs have been exposed to a two-step inductive protocol that (as previously reported [17]) was able to specifically drive iPSCs toward renal commitment. Based on our previous experience with iPSC differentiation into RPCs, we decided to expose pVSELs to inductive media for 12 days. At this time point, immunofluorescence analysis showed that VSELs had lost the pluripotency marker NANOG, while they had acquired markers expressed by RPCs typically expressed in the metanephric mesenchyme such as SIX2, PAX2, and WT1 (Fig. 10). These results demonstrate the potential of VSELs to differentiate in RPCs.
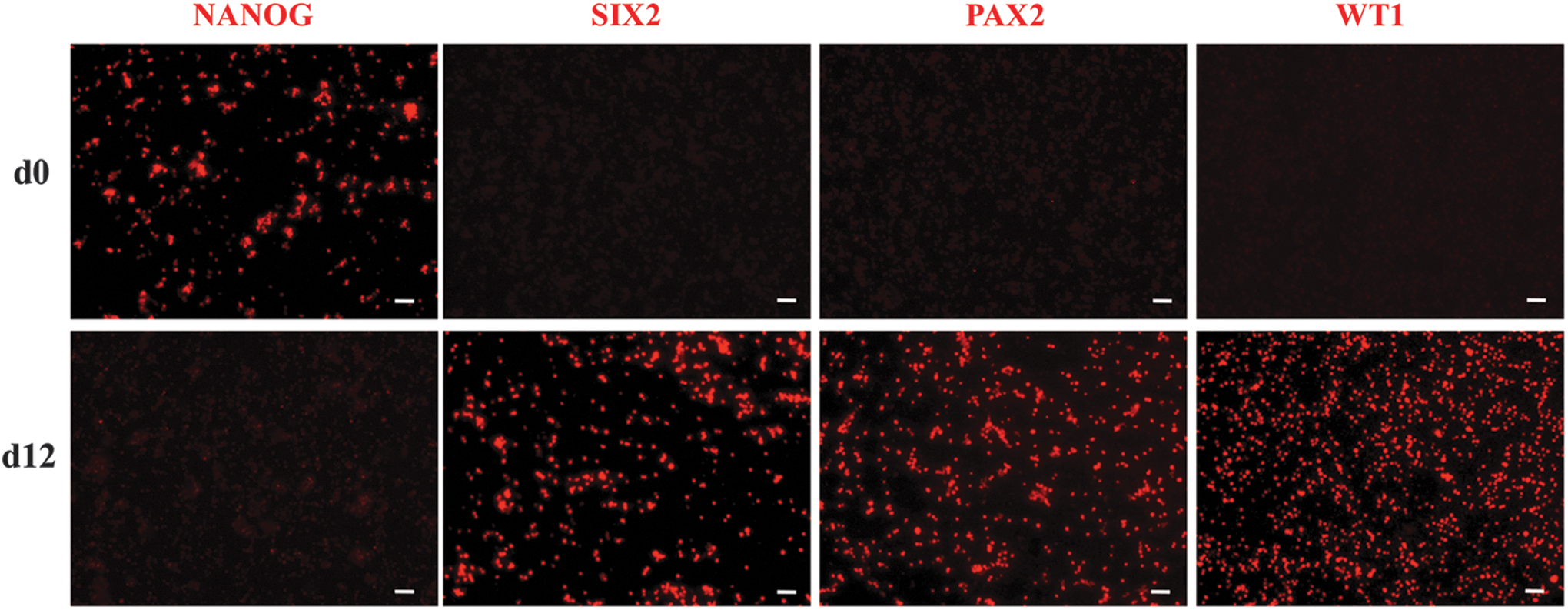
Differentiation of VSELs toward renal commitment. Representative immunofluorescence images of VSELs, incubated for 12 days with renal inductive media, stained for the pluripotency marker (NANOG) and renal progenitor markers (SIX2, PAX2, and WT1). Bar = 20 μm.
VSEL-derived RPCs as cell therapy for AKI
The renoprotective effect of VSELs differentiated into RPCs was tested in a murine model of AKI induced in NOD-SCID mice by cisplatin administration [22]. Renal progenitor-derived VSELs, intravenously injected 1 day after cisplatin (Fig. 11a), showed protection of renal function as indicated by reduction of BUN compared with mice receiving saline (Fig. 11b). As well, renal structure ameliorated in mice treated with VSEL-derived RPCs compared with saline-treated mice, as shown by reduction of cast deposition and tubular necrosis (Fig. 11c).
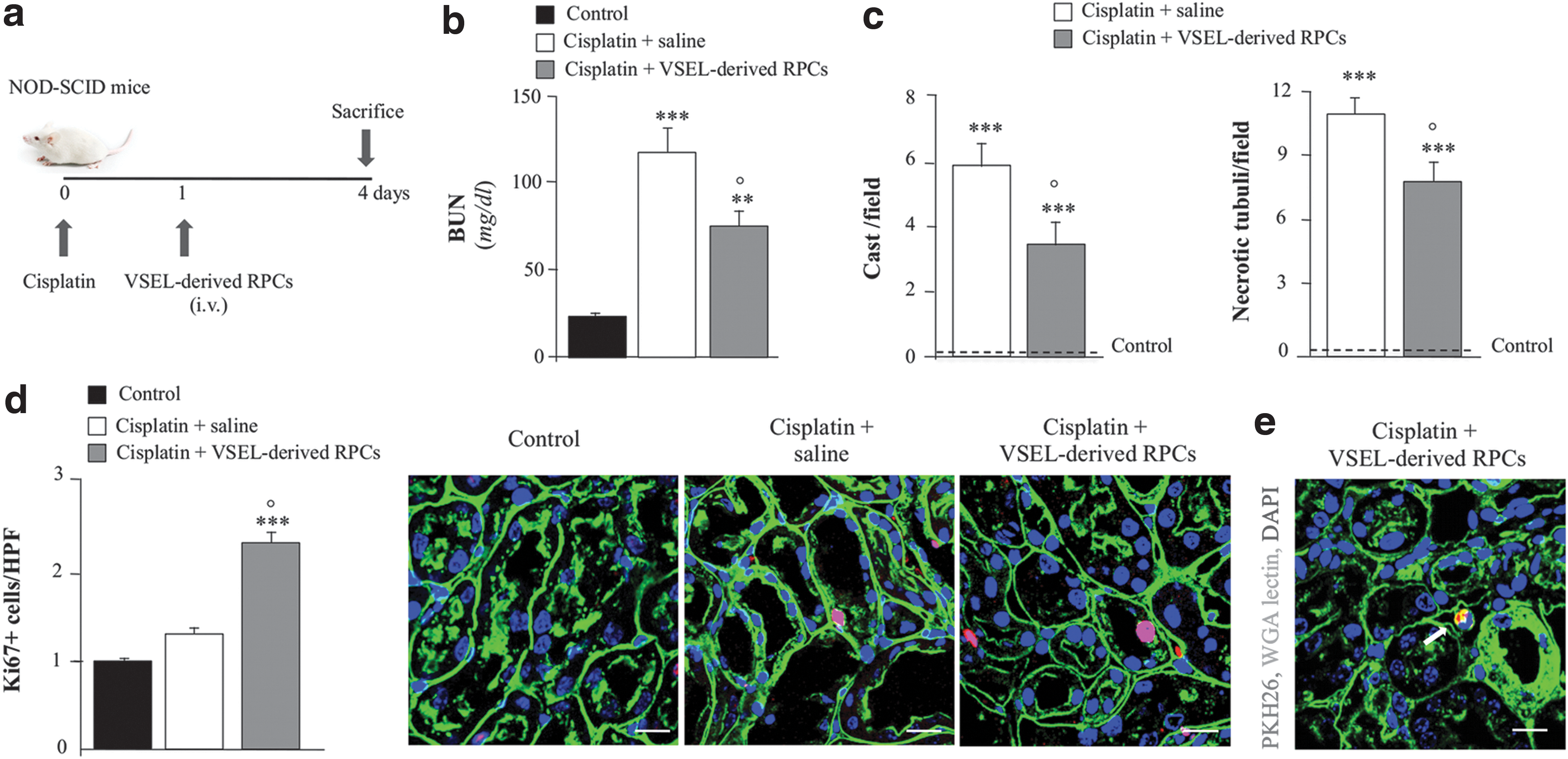
VSEL regenerative potential in experimental AKI.
Renal sections from mice receiving VSEL-derived RPCs showed increased proliferation of endogenous tubular cells, suggesting an ongoing renal regenerative process (Fig. 11d). To assess cell engraftment in renal tissues, VSEL-derived RPCs have been labeled prior injection with cell tracking PKH-26. VSEL-derived RPCs at 4 days averaged 0.7 ± 0.3 cells/105 renal cells and were localized in peritubular areas (Fig. 11e).
Discussion
In this article, we presented a method for isolating small and rare (105 cells/100 mL of UCB) pluripotent stem cells from UCB. The majority of UCB-VSELs isolated are CD45− (>98%) with a variable number of CD133+ cells. This variability can be explained by high heterogeneity of the UCB manipulation, thus affecting the number of undifferentiated stem cells that we were eventually able to isolate together with the intrinsic ability of these cells to form colonies and CEBs in the presence of RA, an important morphogen and effective player of ES cell differentiation [23,24]. There are probably many more issues responsible for the abovementioned heterogeneity (eg, the time window of UCB manipulation maintained around 24 h after birth, the sex of the newborn, birth conditions, and mother's health,) that open the way for further investigations.
Although more analyses are required to understand the correct behavior and differentiation potentials of these debated cells, we obtained interesting results that may contribute as a stone thrown in the big pond of knowledge pertaining to VSELs: (1) a small number of VSELs can be isolated from human UCB and form EBs, peculiar structures of ES cells endowed with a pluripotent phenotype. Thus, VSELs resemble the ES cells of embryos at the blastocyst stage. In fact, it is known that during early embryonic development, a population of primitive Oct4 + VSELs is deposited in developing organs serving as precursors for tissue-committed stem cells [25].
Besides, VSELs akin to primordial germ cells of epiblast origins are thought to be quiescent in adult tissues to prevent teratoma formation due to changes in the epigenetic status [26]; (2) VSELs cultured with RA form colonies showing a central area rich with cells positive for NANOG and SOX2. This peculiar region is round or elliptical shaped and covers about 30% of the entire colony area and it probably comprises more undifferentiated cells required to sustain pluripotency [27,28]; (3) EBs/CEBs showed, interestingly, a very high signal for AFP, an early endoderm marker and a well-known protein produced by the fetus.
In our opinion, this is a very intriguing result considering the endoderm as the innermost of the three germ layers formed at the beginning of the third week of gestation for invagination of epiblast cells.
This is in line with previous researches demonstrating that this protein localizes both in the inner and peripheral layers during CEB differentiation [29]; (4) the high heterogeneity found in all the analyzed UCB units may also explain our data on VSEL differentiation during the first 10 days in culture. We believe that between 7 and 14 days of in vitro culture, VSELs undergo a selecting process to let the more undifferentiated cells (ie, those cells that do not change shape or dimensions) survive, divide, and possibly differentiate. If VSELs are cultured in the presence of RA, they are able to form both colonies and EBs that conversely did not form in our control plates without RA. This sustains the abovementioned idea suggesting that 2 weeks in culture are indispensable for a good selection of these small, rare, and pluripotent embryonic-like stem cells: our data support their bona fide nature of VSELs; (5) as proof of concept that VSELs can be committed toward more specialized cell phenotype, we demonstrated their capability to give rise to RPCs in vitro as it has been reported for iPS [17].
These data represent a novel and exploitable therapeutic possibility for VSELs as indicated by the capacity of renal-committed VSELs to protect from renal functional impairment and acute tubular injury in a mouse model of AKI. The observed renoprotective effect of VSEL-derived RPCs does not seem to be related to their incorporation and differentiation into host renal structures, as evidenced by their exclusive peritubular localization. The low number of cells engrafted in renal tissues may indicate a higher susceptibility of VSEL-derived RPCs to the hypoxic environment of AKI kidneys that may hinder cell integration and survival. Moreover, we cannot exclude that VSEL-derived RPCs express a pattern of adhesive molecules with low efficiency to engraft the kidney. Nevertheless, it is conceivable that the renoprotective effect of VSEL-derived RPCs passed through a paracrine mechanism as indicated by enhanced tubular cell proliferation.
Notably, the presence of VESLs identified in adult kidneys [30] may suggest a possible intrinsic role of VSELs in tissue maintenance and regeneration, thus further supporting the use of VSELs as cell therapy for kidney disease.
In conclusion, our results foster the great debate surrounding VSELs and, more in general, the presence of cells with pluripotent features in adult tissues. This concept is widely questioned by the consistent data about the specific rare population's ability to differentiate in cells derived from all the three germ layers. VSELs represent until now the greater attack to the theories on cell differentiation and restriction of stemness potency during the first phase of development. Those theories attribute the roles to renew and repair adult tissues only to tissue-committed stem cells, excluding the presence of a pool with pluripotent potentials after gastrulation. We think that the discovery of these cells, proven by many laboratories, although questioned by others [31 –33], breaks, definitively, this idea highlighting the real differentiation capability of a particular population of cells. The difficulty to isolate and differentiate VSELs does really exist, confirming the contrasts that appear even in very recent literature [34], thus further considerations are required on the tissues/organs where VSELs can be isolated. After their discovery, many articles were published confirming the presence of these cells in several organs and tissues, but lacking, perhaps, appropriate controls and analyses, thus lighting a strong and still active debate. We choose UCB to investigate the presence of pluripotent stem cells such as VSELs due to its richness in other stem cell populations (eg, MSCs) and its prompt availability to eventually widen the UCB employment for regenerative medicine purposes.
In our opinion, it is important to make efforts to better characterize these young cells to understand their real potentials to exploit possible clinical applications. Anyway, even if critical thinking is absolutely required, we believe that this field of investigation can give precious help in understanding the molecular basis of pluripotency to fulfill unmet therapeutic needs.
Footnotes
Acknowledgments
The authors wish to thank Marianna Longo (Research Center for Regenerative Medicine, Fondazione IRCCS Policlinico San Matteo) for help in data analysis and Gianluca Viarengo and Bina Romano (Immunohaematology and Transfusion Service, Fondazione IRCCS Policlinico San Matteo) for help in cytofluorimetric analyses. This study was funded by Istituto di Ricovero e Cura a Carattere Scientifico, Policlinico San Matteo (to M.M. and C.P.), and by Ministero della Salute, grant “Bando progetti di ricerca, Giovani ricercatori——Ricerca finalizzata 2009” cod. GR-2009-1547415.
Author Disclosure Statement
No competing financial interests exist.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.