
Abstract
The periodontal ligament (PDL) plays an important role in anchoring teeth in the bone socket. Damage to the PDL, such as after severe inflammation, can be treated with a therapeutic strategy that uses stem cells derived from PDL tissue (PDLSCs), a strategy that has received intense scrutiny over the past decade. However, there is an insufficient number of PDLSCs within the PDL for treating such damage. Therefore, we sought to induce the differentiation of induced pluripotent stem (iPS) cells into PDLSCs as an initial step toward PDL therapy. To this end, we first induced iPS cells into neural crest (NC)-like cells. We then captured the p75 neurotrophic receptor-positive cells (iPS-NC cells) and cultured them on an extracellular matrix (ECM) produced by human PDL cells (iPS-NC-PDL cells). These iPS-NC-PDL cells showed reduced expression of embryonic stem cell and NC cell markers as compared with iPS and iPS-NC cells, and enrichment of mesenchymal stem cell markers. The cells also had a higher proliferative capacity, multipotency, and elevated expression of PDL-related markers than iPS-NC cells cultured on fibronectin and laminin (iPS-NC-FL cells) or ECM produced by human skin fibroblast cells (iPS-NC-SF cells). Overall, we present a culture method to produce high number of PDLSC-like cells from iPS cells as a first step toward a strategy for PDL regeneration.
Introduction
P
Stem cells in PDL tissue (PDLSCs), which are derived from cranial neural crest (NC) cells [3] and involved in maintenance and regeneration of periodontium, were previously identified and characterized [4], and shown to possess self-renewal properties [5,6] and express surface markers typical of mesenchymal stem cells (MSCs), such as CD90 and CD105 [6,7]. PDLSCs also display multipotentiality, capable of differentiating into osteoblasts and adipocytes in vitro [4,8], and forming PDL-like fibers with cementum-like tissues when transplanted into the dorsal aspect of immunodeficient mice [4]. It has been considered that PDLSCs were the most suitable cells for periodontal tissue regeneration therapy among other dental stem cells [9,10]. Transplantation of autologous PDLSCs into human and swine periodontal defect portions was reported to regenerate PDL tissue [11,12]. However, since the percentage of stem cells resident in PDL tissue is very low [13], and isolation of PDLSCs involves tooth extraction, it has been difficult to stably obtain a mass of PDLSCs for research and clinical application.
In this context, so far we have had to use SV40- and hTERT-immortalized human undifferentiated PDL cell lines [14] to study the characteristics of PDLSCs. These cell lines display the typical multipotent properties of PDLSCs; for example, cell line 1–17 can differentiate into osteoblasts, adipocytes, chondrocytes, and even neurocytes, and expresses PDL-related markers, such as COL1, FBN1, and POSTN [15,16]. However, because these cell lines are immortalized, they cannot be used in clinical applications.
We, therefore, shifted our focus onto using induced pluripotent stem (iPS) cells as a potential source of PDLSCs. Yamanaka and colleagues first established iPS cells from skin fibroblasts (SFs) and showed that they expressed markers of embryonic stem cells (ESCs), NANOG, and OCT3/4 [17]. Since then, several groups have been able to derive iPS cells from other somatic cell types, including umbilical cord blood cells and blood cells [18 –20]. iPS cells can be induced to differentiate into various cell types, such as retinal pigmented epithelium cells, dopaminergic neurons, cardiomyocytes, and oocytes [21 –25]. In addition, organs, such as kidney, liver, and pancreas, have been, in part, generated from iPS cell technology [26 –28]. However, there is no method as yet of inducing PDLSCs from iPS cells.
The biological rejection of autologous or allogeneic transplantation materials derived from iPS cells is considered to be low [29]; however, concerns have been raised about the potential pathogenesis of teratomas from iPS cells [30]. Teratoma formation occurs in residual, undifferentiated pluripotent stem cells; therefore, in clinical applications, iPS cells must be differentiated into their target progenitor cells, such as NC cells, before transplantation or grafting [31]. Embryologically, PDL cells are derived from the NC cells [3,32], which express nestin and p75 neurotrophic receptor (p75NTR) [33 –35].
The extracellular matrix (ECM) has key roles in determining stem cell fate [36]. PDL tissues abundantly express COL1 and POSTN [37,38], two ECM proteins that are also involved in PDL tissue development in mice [39,40]. Therefore, in this study, we also examined the potential of the ECM from primary human PDL cells (HPDLCs) to guide the development of PDLSC-like cells from iPS cells after their induction into NC cells.
Materials and Methods
Cell culture
Human iPS cells from SFs were purchased from RIKEN (Saitama, Japan; HPS No. 0063). Human iPS cells were cultured on mouse embryonic fibroblasts (MEFs; ReproCELL, Inc., Kanagawa, Japan) and maintained in primate ESC medium (ReproCELL, Inc.) containing 5 ng/mL human recombinant basic fibroblast growth factor (b-FGF; ReproCELL, Inc.) at 37°C in a humidified atmosphere of 5% CO2 and 95% air. HPDLCs were isolated from the third molar of a healthy 25-year-old female patient, who visited Kyushu University for extraction, as described previously [14]. SFs and HPDLCs were maintained in alpha-minimum essential medium (α-MEM; Gibco-BRL, Grand Island, NY) containing 10% fetal bovine serum (FBS; Biowest, Nuaille, France), 50 U/mL penicillin, and 50 μg/mL streptomycin (10% FBS/α-MEM) at 37°C in a humidified atmosphere of 5% CO2 and 95% air. All procedures were performed in compliance with the Research Ethics Committee, Faculty of Dentistry, Kyushu University.
Isolation and culture of human PDLSCs
HPDLCs were isolated from a healthy patient, a third molar from a 22-year-old female, who visited Kyushu University Hospital for extraction, as described previously [41]. In brief, PDL tissues were from the root surface and digested with 0.2% collagenase and 0.25% trypsin (Wako Pure Chemical Industries Ltd.) for 20 min at 37°C to obtain single-cell suspension. Explant culture was maintained in α-MEM containing 10% FBS, 50 μg/mL of streptomycin, and 50 U/mL of penicillin at 37°C in a humidified atmosphere with 5% CO2 and 95% air. Some cell populations were isolated from colonies through colony rings (Asahi Techno Glass) and expanded for further cultivation to establish HPDLC clones. A HPDLC clone, which showed potential for osteoblastic and adipogenic differentiation, and expressed cell surface markers (CD90 and CD105), were used as human PDLSCs in this study. All procedures were performed in compliance with the Research Ethics Committee, Kyushu University.
Development of iPS-NC-PDL cells
Human iPS cells were first differentiated into NC-like cells, according to a previous report [42]. In brief, colonies of iPS cells were separated into single cells and cultured on gelatin-coated dishes for 30 min. Unattached cells were collected and seeded onto Matrigel-coated dishes at a density of 1 × 105 cells/dish. These cells were cultured with MEF-conditioned medium supplemented with 10 μM Rho-associated protein kinase inhibitor (Enzo Life Sciences, Inc., Farmingdale, NY) and 10 ng/mL b-FGF (ReproCELL, Inc.). The medium was changed every day.
When the cells reached 50%–70% confluence, they were cultured in the differentiation medium according to culture periods as shown in Table 1 to differentiate into NC cells. In brief, the culture medium was changed to knockout serum replacement (KSR) medium to initiate differentiation into NC cells. KSR medium was composed of 15% KSR/knockout DMEM (Gibco-BRL), 1%
KSR, knockout serum replacement; NB, neurobasal.
Thereafter, the medium was switched to neurobasal (NB), composed of NB medium (Gibco-BRL), 5% B-27 supplement (Gibco-BRL), 1% N-2 supplement (Gibco-BRL), 1%
iPS-NC cells were further cultured for 2 weeks on dishes coated with various substrates: (1) fibronectin and laminin (designated as iPS-NC-FL cells), (2) COL1, (3) POSTN, (4) ECM produced by cultured HPDLCs (designated as iPS-NC-PDL cells), or (5) ECM produced by cultured human SF cells (designated as iPS-NC-SF cells).
Coating of culture plates
HPDLCs and human SFs were cultured until reaching confluency in a 24-well plate, and then detached with 2% EDTA (nacalai tesque, Kyoto, Japan) treatment to obtain the plate coated with ECM produced by each cell population. These plates were used as ECM-coated plates in this study.
In addition, a fibronectin (10 μg/mL) and laminin (1 μg/mL)-coated plate, a COL1 (300 μg/mL)-coated plate, and a POSTN (10 μg/mL)-coated plate were also prepared. In brief, after each protein solution was put into a 24-well plate and incubated for 1 h at room temperature, each plate was used as a protein-coated plate.
Immunofluorescence staining
iPS, iPS-NC, iPS-NC-PDL, iPS-NC-FL, and iPS-NC-SF cells were fixed with 4% paraformaldehyde and 0.5% dimethyl sulfoxide in phosphate-buffered saline (PBS) for 30 min. Cells were blocked in 2% bovine serum albumin in PBS for 1 h, and then incubated with rabbit polyclonal antihuman COL1 (1:500 dilution; Abcam Systems, Inc., Minneapolis, MN), rabbit polyclonal antihuman OPG (5 ng/mL; Abcam), goat polyclonal antihuman NANOG (1:40 dilution; R&D Systems, Inc., Minneapolis, MN), mouse monoclonal antihuman p75NTR (1:200 dilution; Advanced Targeting Systems, San Diego, CA), or goat polyclonal antihuman POSTN antibody (1:50 dilution; Santa Cruz Biotechnology, Dallas, TX) overnight at 4°C. Cells were then washed three times with PBS and incubated for 30 min with an Alexa Fluor 488-conjugated chicken antigoat antibody, an Alexa Fluor 488-conjugated chicken antimouse antibody, Alexa Fluor 488-conjugated chicken antirabbit antibody, Alexa Fluor 568-conjugated goat antirabbit antibody, or Alexa Fluor 568-conjugated rabbit antigoat antibody (1:200 dilution; Invitrogen), as appropriate. Nuclei were stained using Fluoro-KEEPER antifade reagent, nonhardening type with DAPI (Nacalai Tesque). Stained cells were imaged and analyzed using a Biozero digital microscope (Keyence Corporation, Osaka, Japan).
Proliferation assay
iPS-NC-FL, iPS-NC-PDL, and iPS-NC-SF cells were seeded at 3 × 103 cells/well into the wells of 48-well plates for up to 3 days. On days 0, 1, 2, and 3, proliferation was examined using a cell proliferation assay kit (Takara Bio, Inc.). In brief, 25 μL of the WST-1 kit reagent was added to the culture medium of each well. After 1 h, 100 μL of supernatant was collected from each well, and the density was measured by an immunoMini NJ-2300 (System Instruments Co., Ltd., Tokyo, Japan) at an absorbance of 450 nm.
Flow cytometric analysis
Cells (iPS-NC, iPS-NC-FL, iPS-NC-PDL, iPS-NC-SF, and PDLSC) were detached with accutase (ReproCELL, Inc.) and the number of cells was adjusted to 5 × 105 cells/tube. Cells were then incubated with R-phycoerythrin-conjugated mouse antihuman p75NTR, CD90, or CD105 antibodies for 1 h at 4°C. Cells were then washed with flow cytometry buffer (R&D Systems) and the percentage of positive cells was measured by flow cytometry (EC800 Cell Analyzer; Sony, Tokyo, Japan). Data were analyzed using Eclipse software (Sony).
Quantitative reverse transcription polymerase chain reaction
First-strand cDNA synthesis was performed as described for semiquantitative reverse transcription polymerase chain reaction (RT-PCR). Quantitative RT-PCR assays were performed with a KAPA SYBR Fast qPCR kit (Nippon Genetics Co., Ltd, Tokyo, Japan) using a Thermal Cycler Dice Real-Time System (Takara Bio, Inc.) under the following conditions: 95°C for 10 s (initial denaturation), 40 cycles of 95°C for 5 s, and 60°C for 30 s, followed by a dissociation program at 95°C for 15 s, 60°C for 30 s, and 95°C for 15 s. The expression of the target genes was calculated from ΔΔCt values. Primer sequences, annealing temperatures, and product sizes for human bone morphogenetic protein 2 (BMP2), bone sialoprotein (BSP), CCAAT/enhancer-binding protein α (CEBPα), COL1, FBN1, lipoprotein lipase (LPL), OCT3/4, OPG, NANOG, NESTIN, POSTN, p75NTR, and β-actin are listed in Table 2. The expression levels of these genes were normalized against β-actin and the results are shown as the fold increase over the control. Primer sequences were designed using the GenBank database (NCBI), and a BLAST search of GenBank was performed on the primer sequences to ensure specificity.
Osteoblastic differentiation
iPS-NC-FL, iPS-NC-PDL, iPS-NC-SF cells, and PDLSC were seeded at 2 × 104 cells/well into the wells of a 24-well plate and cultured in 10% FBS/α-MEM. After reaching confluence, the culture medium was changed to an osteoblastic differentiation medium (OM) composed of 10% FBS/α-MEM supplemented with 2 mM β-glycerophosphate and 50 μg/mL ascorbic acid. After 4 weeks, the cells were fixed with 10% formalin, washed with distilled water, and stained with 0.3% Alizarin Red S for calcium detection.
Adipogenic differentiation
iPS-NC-FL, iPS-NC-PDL, iPS-NC-SF cells, and PDLSCs were seeded at 2 × 104 cells/well into the wells of a 24-well plate and cultured in 10% FBS/α-MEM. After reaching confluence, the culture medium was changed to an adipogenic differentiation medium (AM) composed of 10% FBS/α-MEM supplemented with 0.1 mM ascorbic acid, 0.5 mM methylisobutylmethylxanthine (Sigma-Aldrich, St Louis, MO), 0.5 mM hydrocortisone (Sigma-Aldrich), and 60 mM indomethacin (Sigma-Aldrich). After 4 weeks, the cells were fixed with 10% formalin, washed with distilled water, and stained with 0.3% Oil Red O for lipid detection.
Statistical analysis
All values are expressed as the mean ± standard deviation. Statistical analyses were performed using the appropriate tests (unpaired doubled tailed t-test or one-way analysis of variance). A P value of <0.05 was considered statistically significant.
Results
Expression of ES-, NC-, and PDL-related markers in iPS, iPS-NC, and iPS-NC-PDL cells
iPS-NC-PDL cells were obtained according to the protocol described in Fig. 1A. iPS cells were first differentiated into NC-like cells, as previously described [42]. Since NC-like cells also included glial cells, we captured the NC cells expressing p75NTR using MACS (iPS-NC cells) for further cell culture. Additional flow cytometric analysis demonstrated that our final population of iPS-NC cells was 97% homogeneous for p75NTR-positive cells (Supplementary Fig. S1; Supplementary Data are available online at
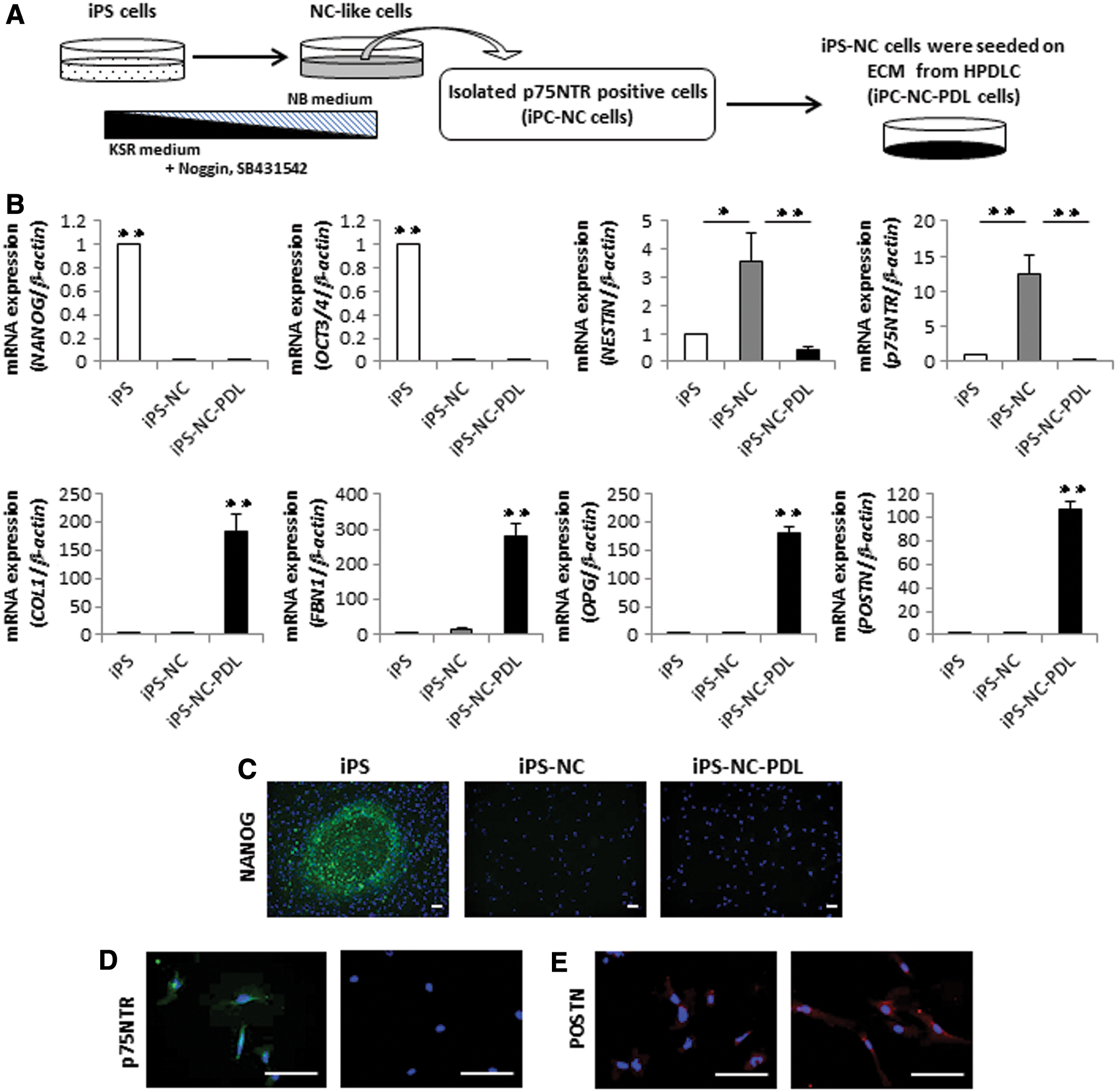
Expression of ES-, NC-, and PDL-related markers in iPS, iPS-NC, and iPS-NC-PDL cells.
We next grew the iPS-NC cells on an ECM substrate produced by HPDLCs to induce PDLSC formation, and measured changes in the expression of ES-, NC-, and PDL-related genes among iPS, iPS-NC, and iPS-NC-PDL cells by quantitative RT-PCR. iPS cells predictably expressed ES-related genes, NANOG and OCT3/4, and iPS-NC cells notably expressed NC-related genes, NESTIN and p75NTR. iPS-NC-PDL cells, by comparison, had a decreased expression of ES- and NC-related genes and a higher expression of PDL-related genes, COL1, FBN1, OPG, and POSTN (Fig. 1B). We further used immunofluorescence staining to examine the expression of ES-, NC-, and PDL-related markers in iPS, iPS-NC, and iPS-NC-PDL cells. We show that NANOG was highly expressed in iPS cells, with little expression noted in iPS-NC and iPS-NC-PDL cells. p75NTR was highly expressed in iPS-NC cells but not in iPS-NC-PDL cells. Expression of the PDL-related marker, POSTN, was high in iPS-NC-PDL cells (Fig. 1C–E). These results indicated that iPS-NC-PDL cells lost their ES and NC features but acquired PDL-related properties.
Expression of NC-, PDL-, and MSC-related markers and proliferation assay of iPS-NC-FL, iPS-NC-PDL, and iPS-NC-SF cells
To evaluate the efficiency of ECM from PDL cells, iPS-NC cells were cultured on wells coated with ECMs from HPDLCs or SFs, or FL-coated wells (Fig. 2A). To test the potential of other substrates to induce PDLSC production, we grew iPS-NC cells on laminin/fibronectin (iPS-NC-FL) or an ECM produced by human SFs (iPS-NC-SF cells), and measured changes in the expression of certain genes through quantitative RT-PCR analysis. We found that the expression of NC-related genes was almost completely reduced in iPS-NC-FL, iPS-NC-PDL, and iPS-NC-SF cells than in iPS-NC cells (Fig. 2B). We further used flow cytometric analysis to measure the expression of MSC markers, CD90 and CD105, in these cell types. We found that iPS-NC-PDL cells expressed elevated levels of MSC markers than iPS-NC-FL or iPS-NC-SF cells (Fig. 2C). Finally, we measured the proliferative activity of the various cell types using WST-1 assay, and found that, overall, iPS-NC-PDL cells exhibited the highest proliferation (Fig. 2D).

Expression of NC-, PDL-, and MSC-related markers and proliferation of iPS-NC-FL, iPS-NC-PDL, and iPS-NC-SF cells.
Osteoblastic and adipogenic differentiation of iPS-NC-FL, iPS-NC-PDL, and iPS-NC-SF cells
Next, we sought to assess the multipotentiality of the cells by longer term culture in osteogenic or adipogenic conditions. First, we cultured iPS-NC-FL, iSP-NC-PDL, and iPS-NC-SF cells in control medium (CM) or osteoblastic differentiation medium (OM) for 4 weeks to test mineralization rates using Alizarin Red S staining (Fig. 3A). iPS-NC-FL and iPS-NC-SF cells grown in OM showed some Alizarin Red-positive staining, whereas iPS-NC-PDL cells grown in OM had a much higher Alizarin Red-positive reaction (Fig. 3A). Likewise, the expression of osteoblast-related genes BMP2 and BSP was higher in iPS-NC-PDL cells than in the other cell types (Fig. 3B), as determined by quantitative RT-PCR analysis.
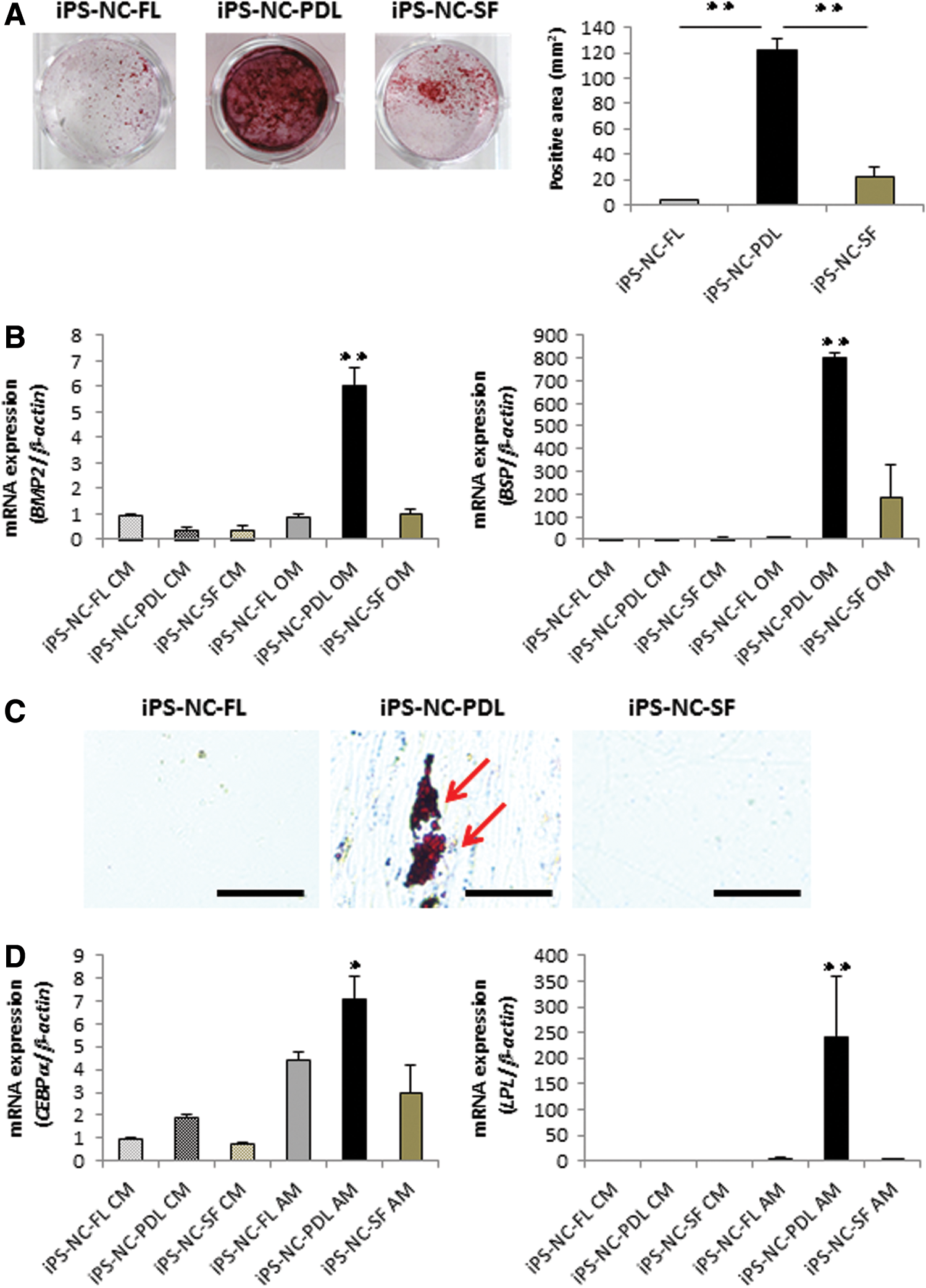
Osteoblastic and adipogenic differentiation of iPS-NC-FL, iPS-NC-PDL, and iPS-NC-SF cells.
Cells were also cultured in CM or AM for 4 weeks to test for the production of fatty lipids by Oil Red O staining (Fig. 3C). We found that iPS-NC-FL and iPS-NC-SF cells grown in AM produced no fatty lipids, whereas there was a clear production of Oil Red O-positive fat lipids in iPS-NC-PDL cells grown in AM. As expected, the expression of adipocyte-related genes CEBPα and LPL was higher in iPS-NC-PDL cells than in the other cells (Fig. 3D), as determined by quantitative RT-PCR analysis.
Expression of PDL-related markers in iPS-NC-FL, iPS-NC-PDL, and iPS-NC-SF cells
Quantitative RT-PCR was used to analyze the expression of PDL-related genes, COL1, FBN1, OPG, and POSTN, in iPS-NC-FL, iPS-NC-PDL, and iPS-NC-SF cells. We found an increase in the expression of each of these PDL-related genes in iPS-NC-PDL cells as compared with iPS-NC-FL and iPS-NC-SF cells (Fig. 4A). The findings for COL1, OPG, and POSTN were further confirmed by immunofluorescence staining, which showed a much higher expression of COL1, OPG, and POSTN in iPS-NC-PDL cells than in the two other cell types (Fig. 4B).
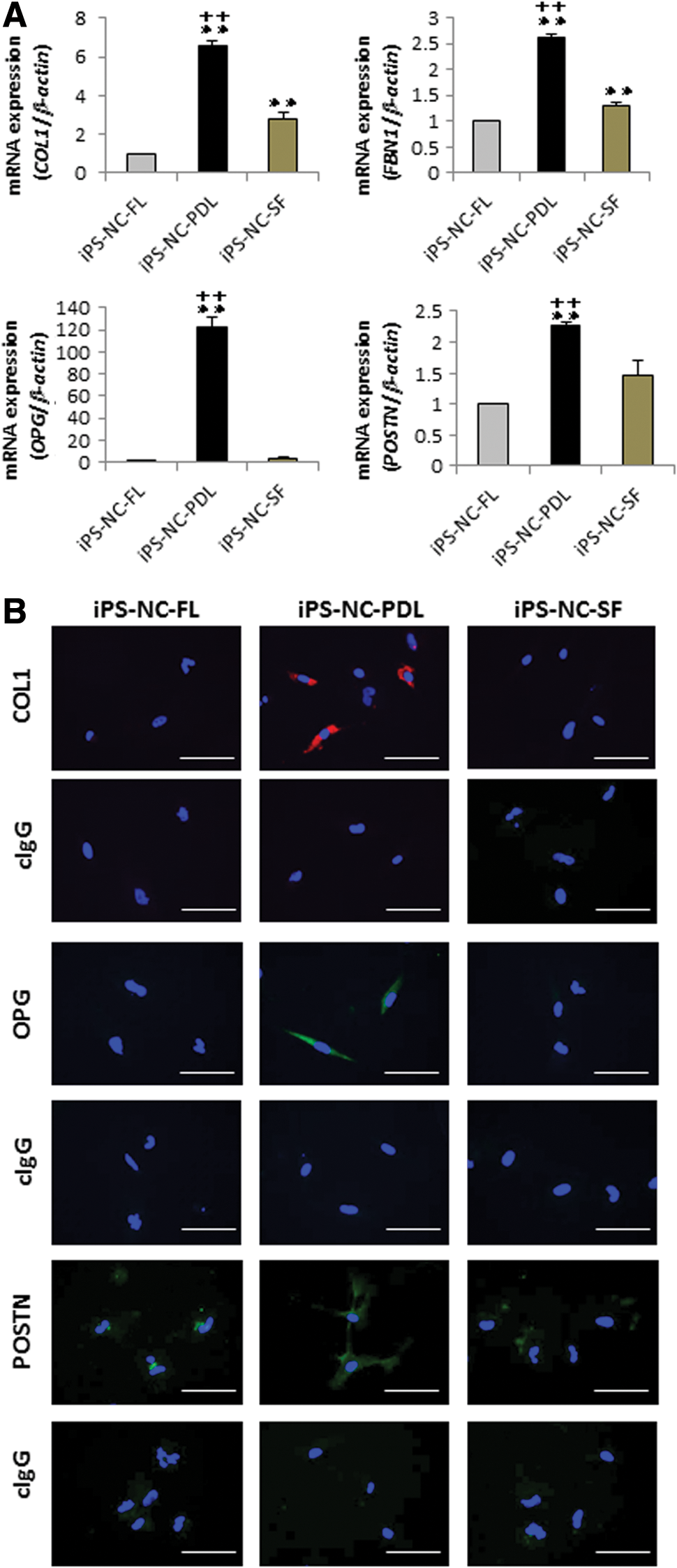
Expression of PDL-related markers in iPS-NC-FL, iPS-NC-PDL, and iPS-NC-SF cells.
We also compared the expression of PDL-related genes between our iPS-NC-PDL cells and human PDLSC line, using quantitative RT-PCR analysis. Before analysis, stem cell features of this PDLSC were examined (Supplementary Fig. S2A, B). Human PDLSCs expressed CD90 and CD 105 with high frequency and showed osteoblastic and adipocytic differentiation capacity. We then found that COL1, FBN1, and OPG were expressed in iPS-NC-PDL cells similarly to PDLSCs. Interestingly, the expression of POSTN in iPS-NC-PDL cells was higher than that in PDLSCs (Supplementary Fig. S2C). Overall, we observed similar expression patterns of PDL-related genes in the two cell types. When compared with expression of HPDLCs, the expression levels of COL1 and OPG in iPS-NC-PDL cells were lower, and the expression level of POSTN was also higher (Supplementary Fig. S3), indicating that iPS-NC-PDL cells acquired PDL-related features.
Expression of PDL-related genes in iPS-PDL and iPS-NC-PDL cells
We next examined the possibility of directly inducting PDLSC formation from iPS cells without going through the iPS-NC stage as shown in Fig. 5A. However, we found that iPS cells cultured on the ECM from HPDLCs showed no increase in the expression of PDL-related genes (Fig. 5B), retaining a similar phenotype as that of ESCs, with a high expression of ES-related markers, NANOG and OCT3/4 (Fig. 5C).

Expression of PDL-related markers in iPS-PDL cells with iPS-NC-PDL cells.
Expression of PDL-related markers in iPS-NC-PDL and iPS-NC cells cultured on COL1 or POSTN
Finally, we sought to examine which ECM proteins from HPDLCs trigger the induction of PDLSC-like features in iPS-NC cells (Fig. 6A). We found that the expression of PDL-related genes in iPS-NC cells cultured on COL1 or POSTN was significantly lower than that in iPS-NC-PDL cells (Fig. 6B). Overall, neither COL nor POSTN could reproduce the effects of the ECM from the cultured HPDLCs.
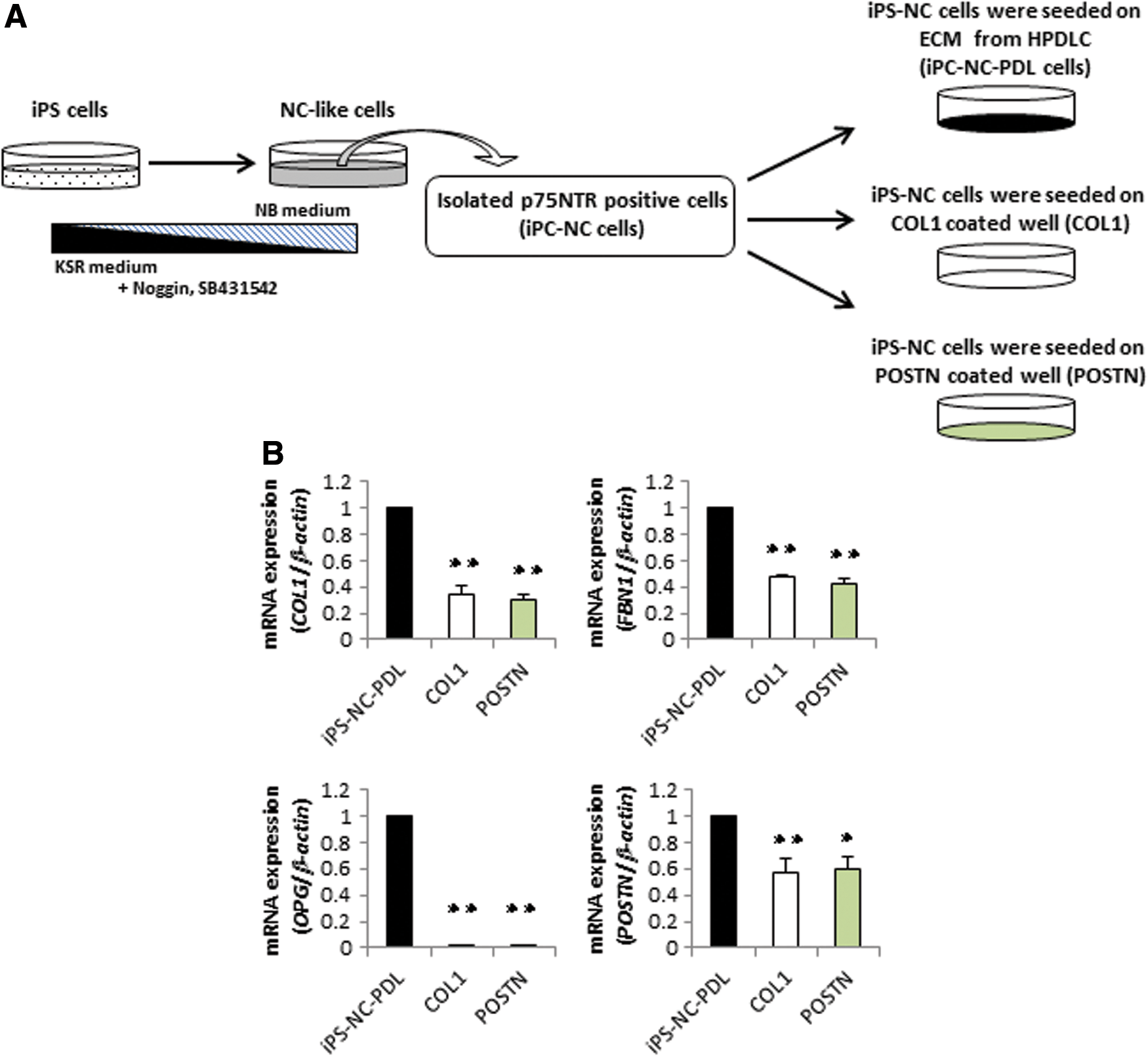
Expression of PDL-related markers in iPS-NC-PDL and iPS-NC cells cultured on COL1 or POSTN.
Discussion
The aim of this study was to elucidate an effective method for inducing the production of PDLSCs from iPS cells for the clinical treatment and regeneration of PDL tissue. Overall, we found that the following three steps were important for such induction: (1) differentiation of iPS cells into NC-like cells, (2) isolation of p75NTR-positive cells from the NC-like cells (iPS-NC cells), and (3) culture iPS-NC cells on an ECM produced by HPDLCs (iPS-NC-PDL cells). Our analysis showed that iPS-NC-PDL cells acquired the proliferative and differentiative properties of PDL cells and expressed markers typical of MSCs. This is the first report to outline the production of PDL stem-like cells from iPS cells.
In tissue regeneration, it is important to follow the sequential developmental process of embryogenesis. Therefore, as per a previously reported method [42], we first induced the differentiation of iPS cells to NC cells. The resultant cells exhibited NC features, similar to that shown by others [33,35]. Then, we reasoned that culturing iPS-NC cells on an ECM derived from PDL cells could help their further differentiation into PDLSCs, as, in vivo, PDLSCs reside in a niche environment composed of ECM components produced by PDL cells. We have examined the inductive effects of some HPDLCs from different patients (data not shown), but found that the HPDLCs isolated from the 25-year-old patient used in this study were the most effective, compared with other HPDLCs that also included the inductive effects basically, but at lower level.
iPS cells can be induced to differentiate into mesodermal progenitor cells using specific culture media or substrates. For example, others have shown that laminin-coated dishes can efficiently induce the production of vascular endothelial cells [43]. Therefore, to identify an effective ECM for inducing iPS-NC cells into PDLSCs, we cultured iPS-NC cells on fibronectin and laminin, COL1, or POSTN, or an ECM produced by HPDLCs or SFs. Among these, we found that ECM from PDL cells was the most efficient at promoting the proliferation, multipotency, and expression of PDL and MSC markers of iPS-NC-PDL cells, with a phenotype similar to that of primary PDLSCs. Although COL1 and POSTN are the major ECM proteins in PDL tissue [37,38], these proteins failed to increase the expression of PDL-related markers. Therefore, we suggest that COL1 and POSTN alone are insufficient to drive PDLSC-like differentiation of iPS-NC cells. Although fibronectin and laminin showed some increase, it was not as efficient as the PDL ECM, suggesting that a combination of proteins is likely required to drive differentiation. Further work will be required to identify the key proteins in the ECM that are necessary for inducing PDL cells from iPS cells.
Of note, we were unable to produce the characteristics of iPS-NC-PDL cells without first inducing iPS cells into NC cells. We found that iPS-PDL cells retained high expression levels of ES-related genes, suggestive of an undifferentiated state. Therefore, we conclude that iPS cells must transit through an NC cell stage before becoming PDLSCs. From our data, we described the schema explaining the gene expression pattern of iPS, iPS-NC, and iPS-NC-PDL cells according to the differentiation stages in Supplementary Fig. S4.
Overall, our findings demonstrate a new method for inducing sufficient number of PDLSC-like cells for clinical treatment, repair, and regeneration of the lost PDL tissue. The results of this study provide a first step toward the efficient regeneration of periodontal tissue for therapeutic purposes.
Footnotes
Acknowledgments
We thank Drs. Serita, Mizumachi, Sonoda, Nozu, Itoyama, Ono, and Taketomi-Saito for their great support in the preparation of this work. This work was supported by JSPS KAKENHI grant numbers JP15H05023, JP16K20457, JP16K20458, JP17H01598, and JP17H04385.
Author Disclosure Statement
No competing financial interests exist.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.