
Abstract
Amniotic fluid is an alternative source of stem cells, and human amniotic fluid-derived stem cells (AFSCs) obtained from a small amount of amniotic fluid collected during the second trimester represent a novel source for use in regenerative medicine. These AFSCs are characterized by lower diversity, a higher proliferation rate, and a wider differentiation capability than adult mesenchymal stem cells. AFSCs are selected based on the cell surface marker c-kit receptor (CD117) using immunomagnetic sorting. Glial cell line-derived neurotrophic factor (GDNF) is expressed during early kidney development and regulates the proliferation and differentiation of stem cells in vitro. In this study, c-kit-sorted AFSCs were induced toward osteogenic or adipogenic differentiation. AFSCs engineered via the insertion of GDNF were cocultured with mouse renal tubular epithelial cells (mRTECs), which were preconditioned by hypoxia-reoxygenation in vitro. After coculture for 8 days, AFSCs differentiation into epithelial-like cells was evaluated by performing immunofluorescence, flow cytometry, and quantitative real-time polymerase chain reaction to identify cells expressing the renal epithelial markers, cytokeratin 18 (CK18), E-cadherin, aquaporin-1 (AQP1), and paired box 2 gene (Pax2). The GDNF gene enhanced AFSCs differentiation into RTECs. AFSCs possess self-renewal ability and multiple differentiation potential and thus represent a new source of stem cells.
Introduction
A
Glial cell line-derived neurotrophic factor (GDNF), an effective neurotrophic factor that protects nigral dopaminergic neurons, was isolated from a rat glial cell line by Lin in 1993 [12]. As an exogenous macromolecular protein, GDNF has difficulty passing through the blood-brain barrier and is easily degraded in the body; thus, GDNF gene therapy was considered the most promising route. Stem cells and genetic modifications further improved gene-based therapy [13 –15]. Rota et al., reported the enhanced regenerative potential of AFSCs and their survival, migration, and secretion of regeneration factors following preconditioning with GDNF [16].
In this study, we induced AFSCs to differentiate into RTECs by coculturing them with mouse RTECs in vitro. Additionally, we investigated whether the GDNF gene enhanced AFSCs differentiation capacity.
Methods
AFSCs extraction
Human amniotic fluid samples were collected from pregnant women at 15–20 weeks of gestation for prenatal diagnosis after obtaining written informed consent of subjects. The protocol was approved by Xuzhou Medical University Ethical Committee. After collection, 10 mL of amniotic fluid was centrifuged at 1,200 r/min for 5 min. The sedimented cells were cultured in Dulbecco's modified Eagle's medium/F12 (Gibco/BRL) supplemented with 15% fetal bovine serum (FBS) (Gibco/BRL), 4 ng/mL basic fibroblast growth factor (R&D Systems, Minneapolis, MN), 100 mg/mL glutamine (Sigma-Aldrich), and 1% antibiotics (pen-strep; Gibco/BRL) in 25-cm2 T-flasks and incubated at 37°C in a humidified atmosphere with 5% CO2. The medium was changed, and the nonadherent cells were removed on the fifth day, while adherent cells continued in culture. The cells were subjected to immunoselection when they reached 70%–80% confluence.
Immunomagnetic bead sorting
As described in previous reports [17], the primary passage of amniotic fluid cells was selected by performing immunomagnetic bead sorting using a CD117 MicroBead Kit (Miltenyi Biotec, Bergisch Gladbach, Germany). The sorted cells were cultured as described before. AFSCs were subcultured at a dilution of 1:3 at 80%–90% confluence. P1 to P3 AFSCs populations were detected by CD117 antibody (Abcam, Inc.) using flow cytometry (BD Biosciences).
Osteogenic and adipogenic differentiation of AFSCs
The third passage cells were cultured until they reached 70%–80% confluence. Then, osteogenic or adipogenic induction medium was applied, and the medium was replaced every 2 days. Osteogenic differentiation was detected by performing Alizarin Red S staining to observe calcium deposition after 3 weeks. Adipogenic differentiation was performed by performing Oil Red O staining to assess the presence of lipid vesicles, which were visible after 3 weeks in culture.
Lentivirus vector production
A green fluorescent protein (GFP) label for a lentivirus vector plasmid system carrying the GDNF gene was constructed by Shanghai Jikai Gene Technology Co., Ltd. Lv-GDNF-GFP vector was prepared using transient transfection of 293T cells and Lipofectamine 2000 (Invitrogen, Carlsbad, CA). In brief, 293T cells cultured in 15 cm2 tissue culture flask (Corning B.V.) were transfected with 100 μL of Lipofectamine 2000 and the mixed envelope and packaging plasmids, including 20 μg of pGC-LV recombinant vector, 15 μg of pHelper 1.0, and 10 μg of pHelper 1.0. Then, the 293T cells were transfected by the plasmids system. The viral supernatants were harvested 2d after 293T cells debris removal according to the manufacturer's instructions.
Lentivirus transfection
AFSCs were transfected with lentiviral vectors following the manufacturer's instructions at a multiplicity of infection (MOI) of 20. GFP expression was observed by performing fluorescence microscopy at 1, 3, and 5 days after lentiviral vector transfection. Untreated AFSCs were used as controls.
Assessment of GDNF by enzyme-linked immunosorbent assay and reverse transcription–polymerase chain reaction analysis
AFSCs were seeded to 96-well plates at a density of 1.0 × 104 cells/cm2, and the medium changed every 2 days. GDNF protein levels in AFSCs labeled with GDNF or unlabeled were detected with a GDNF ELISA kit (Abcam, Inc.). In brief, we added 100 μL of each standard and each sample into a 96-well enzyme-linked immunosorbent assay (ELISA) plate, followed by incubation for 2.5 h at room temperature. After discarding the solutions and washing four times with washing buffer, 100 μL of biotinylated GDNF detection antibody was added into each well with shaking for 1 h at room temperature. The wells were then incubated with 100 μL of horseradish peroxidase-streptavidin solution for 45 min at room temperature, followed by 100 μL of tetramethyl benzidine (TMB) one-step substrate reagent for 30 min at room temperature. Finally, we added 50 μL of stop solution to each well and immediately read the absorbance at 450 nm.
We performed reverse–transcription polymerase chain reaction (RT-PCR) to confirm GDNF gene incorporation into the nucleus. Total RNA was extracted from AFSCs using RnaEx (Shanghai Generay Biotech Co., Ltd.). Subsequently, cDNA was obtained using a TIANScript RT Kit (Tiangen Biotech Co., Ltd.). The following primers were used: forward primer for GDNF, ACTGACTTGGGTCTGGGCTATG; reverse primer for GDNF, TTTGTCACTCACCAGCCTTCTATTT; forward primer for β-actin, CGGGAAATCGTGCGTGACAT; reverse primer for β-actin, CGGACTCGTCATACTCCTGCTTG.
The PCR conditions were as follows: denaturation at 94°C for 5 min, followed by 35 cycles of denaturation at 94°C for 30 s, annealing at 55°C for 30 s and elongation at 72°C for 45 s, and a final elongation step at 72°C for 5 min.
Hypoxia-reoxygenation mouse RTECs
Mouse RTECs (mRTECs), obtained from the Chinese Academy of Sciences, Beijing, were cultured in Dulbecco's modified Eagle's medium/High Glucose supplemented with 10% FBS (Gibco/BRL) in 25-cm2 T-flasks and incubated at 37°C in a humidified atmosphere with 5% CO2.
mRTECs were seeded in six-well plates at a density of 4.0 × 105 cells/well and incubated at 37°C in a humidified atmosphere with 5% CO2 for 48 h. After discarding the medium and washing once with phosphate-buffered saline (PBS), amicrobic paraffin was added into each well and the cells were then incubated at 37°C in 5% CO2 for 60 min. We discarded the paraffin and added fresh medium, then cultured the cells for 0, 24, 48, or 72 h under the same conditions. Apoptosis was detected by flow cytometry (BD Biosciences) using an Annexin V-FITC/PI Apoptosis Detection Kit (Beyotime).
Renal tubular epithelial-like differentiation of AFSCs in vitro
A coculture system consisting of AFSCs and hypoxia-reoxygenation (H/R) mRTECs was established to induce AFSCs to differentiate into RTECs. Transwell experiments were performed in transwell tissue culture plates (0.4-mm pore-size polycarbonate Transwell filters; Corning B.V. Life Sciences, Schiphol-Rijk, The Netherlands). GFP-AFSCs and GDNF-AFSCs were cultured separately in the lower chamber of the transwell plate, whereas mRTECs were seeded on the semipermeable membrane of the transwell plate, thus physically separating both cell types. GFP-AFSCs and GDNF-AFSCs were assessed on day 8 of coculture with H/R mRTECs, and AFSCs were used as controls.
To perform immunofluorescent staining, cells were fixed in 4% paraformaldehyde for 30 min, washed in PBS three times, permeabilized with 0.2% Triton X-100 for 15 min, blocked with 10% goat serum (Peking, China) for 2 h, and incubated overnight at 4°C with a primary cytokeratin 18 (CK18) antibody (Abcam, Inc.) diluted 1:100 in PBS. A secondary antibody (TRITC-labeled goat anti-rabbit IgG) was added at a 1:50 dilution for 2 h at room temperature. DAPI (Beyotime) was used to stain the cell nuclei. The cells were observed by performing confocal laser scanning microscopy (OLS-3100; Olympus, Japan).
The cells were digested with trypsin, washed once with blocking buffer, fixed with 2% paraformaldehyde, and incubated at room temperature for 10 min. Then, the cells were washed once with blocking buffer, 0.5 mL of 1 × FACS permeabilization solution was added, and the cells were incubated at room temperature for 10 min. After a single wash step, the cells were incubated in blocking buffer for 30 min at room temperature. Then, the primary CK18 antibody (Abcam, Inc.) was added (3 μL) and incubated for 30 min. Afterward, the cells were washed twice with blocking buffer and incubated with goat anti-rabbit IgG-PerCP-Cy5.5 (Santa Cruz, Inc.) for 30 min at room temperature. Finally, the cells were washed twice with blocking buffer and resuspended in PBS followed by flow cytometry analysis.
Total RNA was extracted from AFSCs using RnaEx (Shanghai Generay Biotech Co., Ltd.) according to the manufacturer's instructions. Subsequently, cDNA was obtained via reverse transcription of total RNA with random hexamers and SuperScript III reverse transcriptase (Tiangen Biotech Co., Ltd.). Quantitative RT-PCR was performed in triplicate for each sample and each gene using a 7500 Real-Time PCR System and Power SYBR Green PCR Master Mix. The primer pairs spanning one intron were as follows: E-cadherin, CATCGCTTACACCATCCTCA, GGGAAACTCTCTCGGTCCA; Ksp-cadherin, GGAAGCTCCTGGAGTTGATGT, CCACGGTGACTGTGCTAGTG; AQP1, TGTGGGATTAACCCTGCTCG, GGAAGCTCCTGGAGTTGATGT; Pax2, GATTCCTCGCTCCAATGGTGAGAAG, GGTGAAGGTGTCAGCTCGCAAG; glyceraldehyde-3-phosphate dehydrogenase (GAPDH), ACAACTTTGGTATCGTGGAAGG, GCCATCACGCCACAGTTTC. All pairs were written five to three with the top and bottom strands in order.
Statistical analysis
All data are expressed as the mean values with standard deviations (mean ± SD). Differences between groups were assessed by performing one-way analysis of variance using SPSS 16.0 software. P < 0.05 was considered statistically significant.
Results
Characterization of AFSCs
Human amniotic fluid contains three major categories of cells: epithelial, fibroblast, and amniotic fluid cells. CD117-positive cells comprise ∼1% of total amniotic fluid cells. AFSCs adhered in T-25-cm2 tissue culture flasks on the third day of culture, and selected positive cells exhibited fibroblast-like properties, with no visible directionality when cultured to 90% confluence (Fig. 1A). P1 to P3 populations were positive for expression of CD117 using flow cytometry detecting. The results revealed the positive cell ratios were 96.8% ± 3.7%, 90.6% ± 3.2%, and 83% ± 4.0% respectively (Fig. 1E–G).
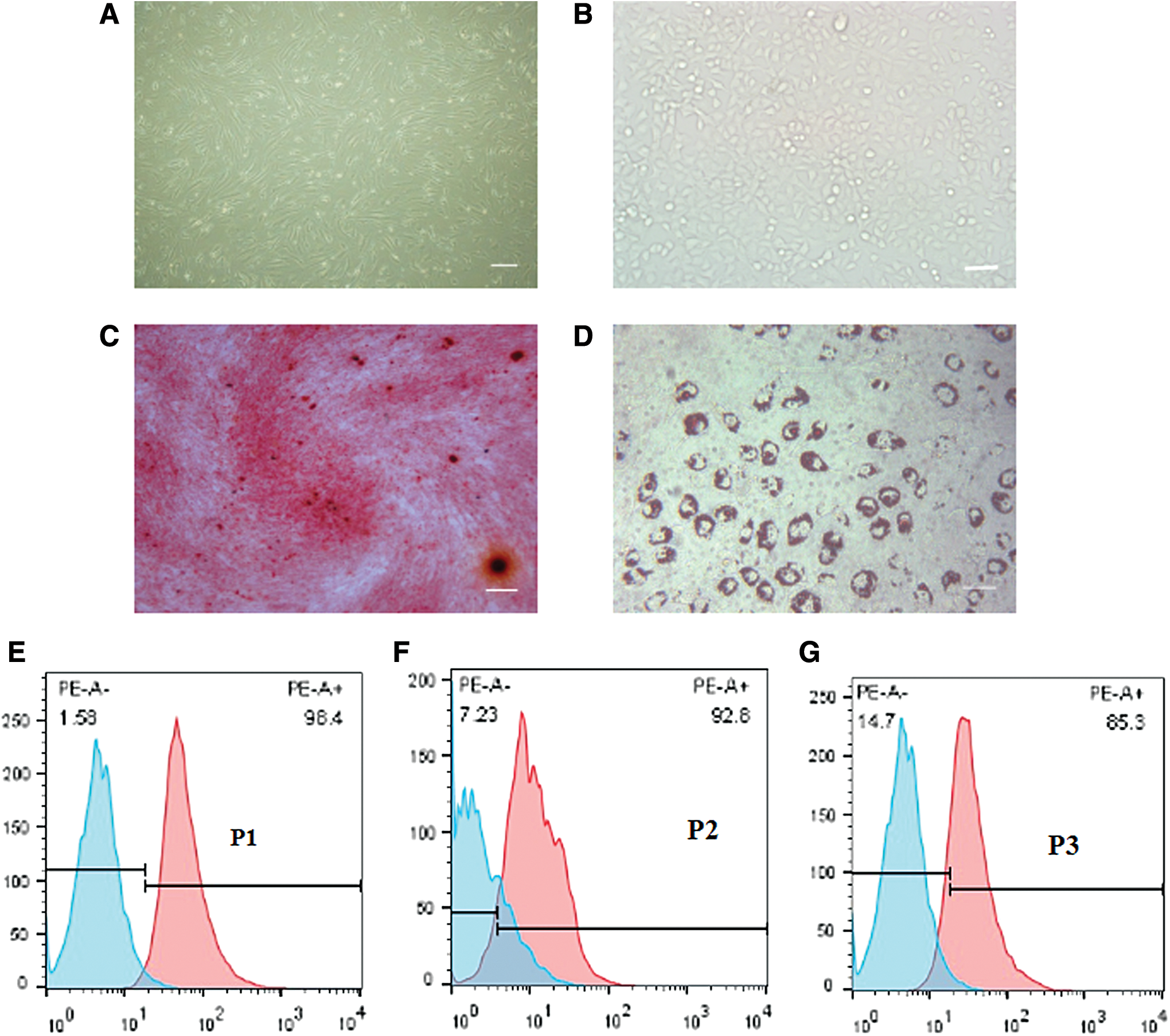
Morphology of AFSCs and osteogenic and adipogenic differentiation.
Characterization of mRTECs
Two or three days later, mRETCs were covered with the bottom of the plate, and then passaged at a dilution of 1:3 at 80%–90% confluence. The cells were polygonal in shape and had cobblestone appearance after cell conjugation (Fig. 1B).
Osteogenic and adipogenic differentiation of AFSCs
To evaluate the differentiation ability of clonal AFSCs, cells were induced into osteoblasts and adipocytes using lineage-specific induction media in vitro. After incubation in osteogenic or adipogenic induction medium for 3 weeks, cells demonstrated calcium mineralization and lipid droplets following staining with Alizarin Red S and Oil Red O, respectively (Fig. 1C, D).
Lentivirus transfection
Lentivirus vectors were constructed by Shanghai Jikai Gene Technology Con. (Fig. 2A), and a MOI of 20 was used. Fluorescence microscopy was performed to observe GFP green fluorescence, which was indicative of transfection efficiency. There was no visible green fluorescence in untransfected AFSCs, while faint green fluorescence was observed after transfection on the first day. Fluorescence was obviously enhanced by the fifth day (Fig. 2B).

Lentivirus vector production and transfection.
Assessment of GDNF by ELISA and RT-PCR analysis
GDNF gene incorporation was analyzed by ELISA and RT-PCR, and the results were shown in Fig. 3. For the ELISA analysis, secretion of the GDNF protein by the Lv-GDNF-AFSCs group began on the first day following transfection, and GDNF gene expression increased over time; the GFP-AFSCs group exhibited a trend similar to AFSCs (Fig. 3A, P < 0.05 for each). In the RT-PCR analysis, we obtained corresponding results indicating enhanced expression of the GDNF gene with increased transfection time (Fig. 3B, P < 0.05 for each). Based on these results, GDNF-AFSCs successfully and stably expressed GDNF and were appropriate for further investigation.

GDNF assessments.
H/R mRTECs
Flow cytometry was performed to detect the apoptosis of mRTECs subjected to H/R for 0, 24, 48, and 72 h, and normal mRTECs were used as controls. The apoptosis rates at 0, 24, 48, and 72 h were 10.47% ± 1.43%, 16.93% ± 4.81%, 42.77% ± 7.32%, and 19.77% ± 7.59%, respectively, whereas the apoptosis rate of the control was 5.30% ± 0.44% (Fig. 4). The highest apoptosis rate was achieved at 48 h, followed by a reduction (P < 0.05 vs. Control, P < 0.05 vs. H/R for 24 h and P < 0.05 vs. H/R for 72 h). H/R induced apoptosis of RTECs was significantly increased. The time of hypoxia was the same, but with the increase of reoxygenation time, the apoptosis rate of RTECs increased, and the peak of 48 h was reached, and then the apoptosis rate decreased, which might be related to the initiation of self repair by the injured cells.

mRTEC H/R.
Renal tubular epithelial-like differentiation of AFSCs in vitro
Some cells transformed into the morphology of epthelia-like cells after coculture for 8 days, and these cells exhibited triangular or polygonal shapes (Fig. 5A).
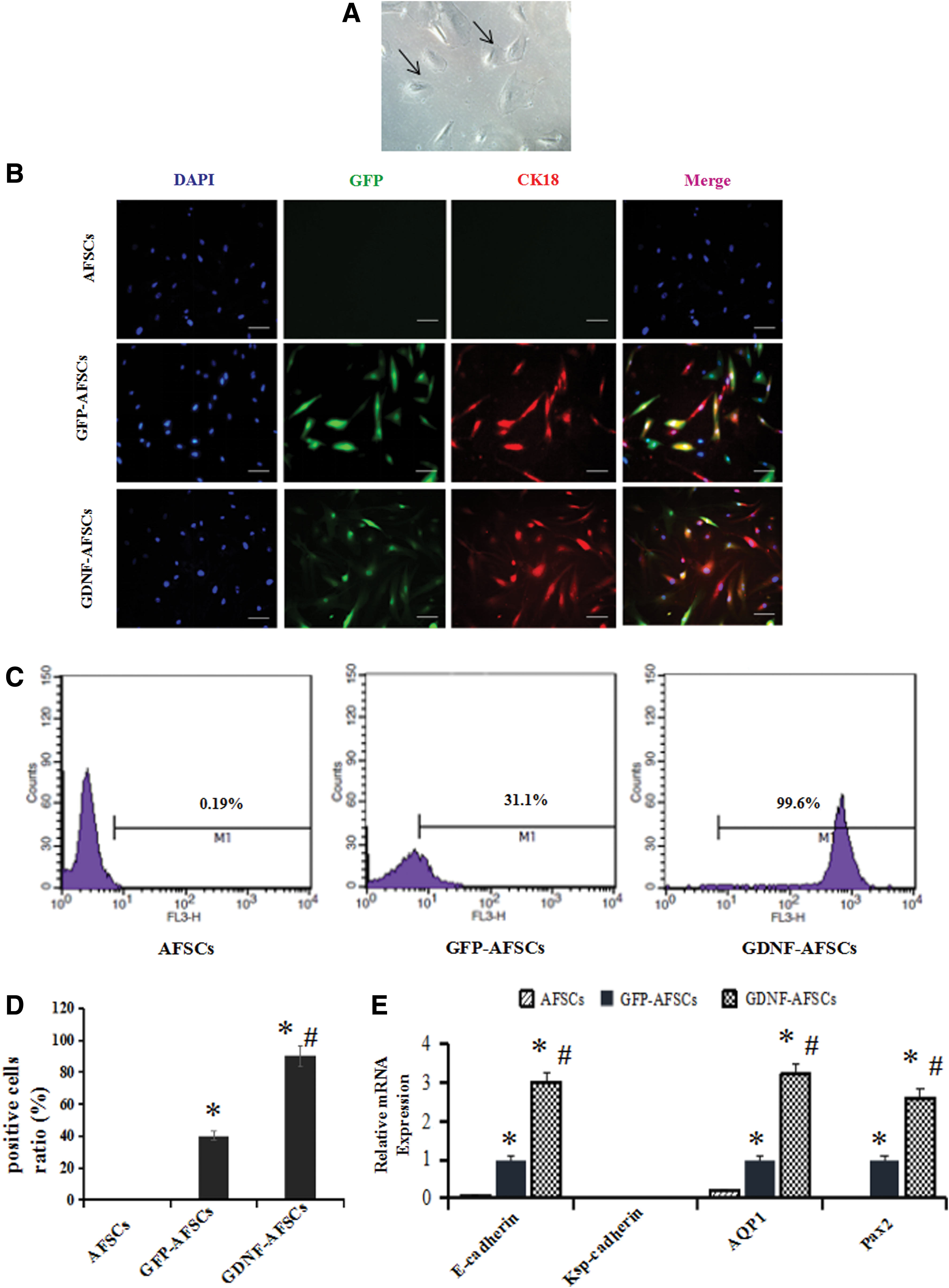
Epithelial-like differentiation of AFSCs. AFSCs were cocultured with H/R mRTECs in a transwell system for 8 days.
After 8 days of coculture, an immunofluorescent assay and flow cytometry results both showed CK18 was significantly increased in the GDNF-AFSCs group compared with the GFP-AFSCs group and the control group (P < 0.05 vs. AFSCs, P < 0.05 vs. GFP-AFSCs) (Fig. 5B–D). E-cadherin, aquaporin-1 (AQP1), and paired box 2 (Pax2) expression were increased in GFP-AFSCs and further enhanced in GDNF-AFSCs based on quantitative RT-PCR results (Fig. 5E, P < 0.05 vs. AFSCs, P < 0.05 vs. GFP-AFSCs). CK18, E-cadherin, AQP1, and Pax2 expression were higher in GDNF-AFSCs than in GFP-AFSCs and control cells. Thus, GDNF gene enhanced the ability of AFSCs to differentiate into epithelial-like cells.
Discussion
Based on our study, hypoxia leads to RTEC apoptosis, for which the highest rate was observed at 48 h. The GDNF gene was successfully transfected into AFSCs using a lentiviral vector. AFSCs and H/R mRTECs coculture resulted in the expression of CK18 and E-cadherin of AFSCs, which was further promoted by the GDNF gene. Thus, AFSCs represent a novel source of human stem cells for regenerative treatment of the kidney, and recombinant GDNF contributes to the progression of differentiation.
In recent years, with the development of tissue engineering, stem cell treatment has become a hot topic in life science research due to characteristics such as a high self-renewal ability and multi-differentiation capacity. At different developmental stages, stem cells are divided into embryonic stem cells and somatic stem cells. Embryonic stem cells with high regenerative potential are capable of differentiating into nearly all cell lines and organs, but these stem cells are derived from the fetus and their use is restricted by ethical considerations. The differentiation ability of somatic stem cells is relatively weaker; these cells only differentiate into certain cell lines and organs. Amniotic fluid cells have previously been used in prenatal genetic testing. Prusa first demonstrated the presence of stem cells among the mixture of cells in amniotic fluid, thus representing a source of stem cells for research into stem cell biology and the development of new tissue engineering approaches [18]. AFSCs differ from embryonic stem cells and somatic stem cells but possess the functions of both cell types [19]. AFSCs express two types of stem cell label proteins, suggesting AFSCs may represent a transitional stage between embryonic stem cells and adult stem cells. The high proliferation potential, remarkable plasticity, and low tumor risk of AFSCs position these cells as a powerful tool in the field of regenerative medicine and cell-based therapy [20]. AFSCs possess strong proliferation and differentiation potential. In this study, AFSCs collected from human amniotic fluid obtained in the second trimester differentiated into osteocytes and adipocytes in appropriate differentiation media. Osteogenic induction medium contained 100 nM/mL dexamethasone, 50 μM/mL vitamin C, and 10 mM/mL β sodium glycerophosphate, and adipogenic induction medium contained 200 μM/mL indomethacin, 0.5 mM/mL 3-isobutyl-1-methylxanthine, 10 μg/mL insulin, and 1 μM/mL dexamethasone.
RTECs constitute the major proportion of renal interstitial cells, which are involved in the absorption, secretion, and excretion functions of the kidney, and acid-base and water-ion equilibrium of bodily fluids. RTEC injury is the direct cause of damage to renal structure and function, which subsequently cause acute kidney injury and lead to renal tubular atrophy and eventual end-stage renal failure [21]. According to several studies, stem cells exert therapeutic effects in a kidney injury model by promoting cell proliferation and decreasing apoptosis. Hauser investigated whether cells isolated from human amniotic fluid contributed to tubular regeneration after experimental acute kidney injury [22]. In a study by Morigi et al., mesenchymal stem cells (MSCs) induce a regenerative process in a model of cisplatin-induced acute kidney injury by repairing damaged RTECs and improving kidney structure and function [23]. Sedrakyan et al., used a mouse model of Alport syndrome to demonstrate the ability of AFSCs to improve the progression of renal interstitial fibrosis and confirm their benefit for the treatment of chronic kidney disease [24]. Hypoxia induces RTECs to become lesioned target cells and promotes renal interstitial fibrosis [25]. The renal tubular cell is the most obvious site of renal ischemia injury, while MSCs are enriched at sites of injury to repair renal function [26,27].
GDNF, a distantly related member of the transforming growth factor-beta superfamily, plays roles in a number of biological processes, including cell survival, neurite outgrowth, cell differentiation, and cell migration. GDNF is widely used in the treatment of neuronal disorders. According to Pichel, GDNF is expressed not only in the nervous system but also very prominently in the metanephric kidney, and mutant mice exhibit kidney agenesis or dysgenesis after GDNF gene knockout [28]. Moreover, GDNF induces ureter bud formation and branching during metanephros development. Rota et al., preconditioned AFSCs with GDNF to improve their regenerative potential by enhancing AFSCs homing to the interstitium of injured kidneys, and in vitro experiments indicated GDNF promotes AFSCs mobility, survival, and growth-promoting factor secretion [16]. GDNF plays a role in the formation of an organization of morphogenesis. It can regulate the tendency and differentiation of stem cells and reverse the pathological changes. Ahdjoudj et al., showed that the diseased tissues and organs secreted a series of cell growth factors and chemokines, such as transforming growth factor (TGF-β) [29]. GDNF has a wide range of interactions with these factors [30].
CK18 is a type of cytoskeletal filament that exists in a variety of epithelial cells and in RTECs. CK18 was expressed in both proximal RTECs and distal tubular epithelial cells, especially indistal tubular epithelial cells [31]. E-Cadherin is a type of cell adhesion molecue that is important in the formation of adherens junctions to bind cells with each other. In epithelial cells, E-cadherin-containing cell-to-cell junctions are often adjacent to actin-containing filaments of the cytoskeleton. E-Cadherin mainly expressed in the distal renal tubules and collecting ducts, which played an important role in maintaining epithelial cell polarity and function [32,33]. AQP1 is a water channel protein that is expressed in renal proximal tubule [33]. Pax2 is the earliest marker of the intermediate mesoderm, from which the renal epithelial cells arise. Subsequently, Pax2 is expressed in all epithelial precursors of the kidney [34].
According to our previous results, AFSCs transfected with GDNF more effectively differentiate into endothelial-like cells in vitro [35]. Therefore, is it possible to induce AFSCs to differentiate into RTECs in vitro to provide a new approach for kidney disease therapy? In this study, a cell coculture system was employed to induce AFSCs to differentiate into RTEC-like cells, and RTECs were preconditioned with H/R to enhance differentiation. We also used a lentivirus as a GDNF gene vector to enhance AFSCs differentiation. Immunofluorescence, flow cytometry, and quantitative RT-PCR were performed to detect CK18, E-Cadherin, Ksp-Cadherin, AQP1, and Pax2 expression.
In this study, lentiviral vectors were efficiently transfected into AFSCs, and GDNF was stably expressed compared with GFP-AFSCs and GDNF-AFSCs. AFSCs were characterized by the expression of the renal tubular epithelial molecular markers CK18, E-cadherin, AQP1, and Pax2. In summary, the results of this study are consistent with the ability of the GDNF gene to promote AFSCs differentiation into RTEC-like cells. But further researches are needed to complete the structure and function integrality of the induced cells.
Footnotes
Acknowledgments
This study was partially supported by the project of the National Natural Science Foundation of China (81270769); project of Jiangsu Provincial Natural Science Foundation (BK20161172); project of Jiangsu Provincial Commission of Health and Family Planning (H201628); project of “Liu ge yi Gong Cheng” of Jiangsu high-level personnel (LGY2016043); project of 7th “Liu Da Ren Cai Gao Feng” of Jiangsu Province, China (2010-WS043); project of the Technology Development Foundation of Kuitun City (201134); Jiangsu Overseas & Training Program for University Prominent Young & Middle-aged Teachers and Presidents; and project of “shi er wu ke jiao xing wei” Key Medical Personnel of Jiangsu Province (RC2011116).
Author Disclosure Statement
No competing financial interests exist.