
Abstract
TP53 is a widely studied tumor suppressor gene that controls various cellular functions, including cell differentiation. However, little is known about its functional roles in smooth muscle cells (SMCs) differentiation from embryonic stem cells (ESCs). SMC differentiation is at the heart of our understanding of vascular development, normal blood pressure homeostasis, and the pathogenesis of vascular diseases such as atherosclerosis, hypertension, restenosis, as well as aneurysm. Using retinoid acid (RA)-induced SMC differentiation models, we observed that p53 expression is increased during in vitro differentiation of mouse ESCs into SMCs. Meanwhile, suppression of p53 by shRNA reduced RA-induced SMC differentiation. Mechanistically, we have identified for the first time that Myocardin, a transcription factor that induces muscle cell differentiation and muscle-specific gene expression, is the direct target of p53 by bioinformatic analysis, luciferase reporter assay, and chromatin immunoprecipitation approaches. Moreover, in vivo SMC-selective p53 transgenic overexpression inhibited injury-induced neointimal formation. Taken together, our data demonstrate that p53 and its target gene, Myocardin, play regulatory roles in SMC differentiation. This study may lead to the identification of novel target molecules that may, in turn, lead to novel drug discoveries for the treatment of vascular diseases.
Introduction
T
P53 is a transcription factor that is widely expressed in embryos and tumor tissues [4]. It functions through binding to DNA-specific sites to activate transcription of target genes such as p21, Bax, Tsp1, and Mdm2 [5,6]. Activation of p53 further induces cell growth arrest, apoptosis, DNA repair, and cell differentiation [7]. As a tumor suppressor, p53 plays an important role in the regulation of tumor cell proliferation and apoptosis [8]. Recently, accumulating studies have confirmed that p53 is also involved in the process of cell differentiation. Lin et al. reported that p53 promotes mouse embryonic stem cell (ESC) differentiation by inhibiting Nanog expression through direct binding to the Nanog promoter [9]. In addition, knocking out of p53 in stem cells or in transgenic mouse was reported to further affect the differentiation of mesoderm [10], neural progenitor cells [11,12], and mesenchymal stem cells [13].
Other transcription factors such as GKLF [14] and cAMP [15] were reported to inhibit the proliferation of SMCs through activating p53. In ApoE knockout mice, endogenous p53 inhibits the apoptosis of vascular SMCs and slows the symptoms of atherosclerosis [16]. Nevertheless, whether p53 could directly work on the differentiation of SMCs remains unknown. In this study, by using an in vitro ESC/SMC differentiation model [17], we found that the expression and activation of p53 were significantly increased during RA-induced ESC/SMC differentiation, accompanied by a large number of cell apoptosis. Knocking down the expression of p53 by shRNA led to a significant reduction of RA-induced ESC/SMC differentiation. In addition, SMC-selective p53 overexpression in transgenic mouse facilitated the switch of vascular SMC phenotypic characteristics from a migratory synthetic phenotype to a quiescent, contractile phenotype, which may restrain injury-induced neointimal formation. Further mechanistic studies by using chromatin immunoprecipitation (ChIP), PCR, and luciferase assay confirmed that p53 was involved in SMC differentiation by binding directly to the promoter of Myocardin and regulating Myocardin expression.
Experimental Procedures
SMC in vitro differentiation system
The mouse ESC (mESC) line D3 (ATCC) was cultured on mitomycin-C-treated mouse embryonic fibroblasts (MEFs) in Dulbecco's modified Eagle's medium (DMEM; Gibco) supplemented with 15% fetal bovine serum (FBS; Gibco), 2 mM
A404 cells were induced to differentiate into SMCs following the previous procedure, with minor modification [19]. Briefly, after an 8-h 0.5 μM doxorubicin treatment, A404 cells were treated with 1 μM RA for 4 days. On day 2, cells were lifted, plated in two 10-cm dishes, and incubated under the presence of 0.5 μg/mL puromycin (Clontech) for another 2 days.
Lentiviral constructs and p53 knockdown ESCs
Lentiviruses encoding p53 shRNA (#sc-29436-V) or control shRNA (#sc-108080) were purchased from Santa Cruz Biotechnology. D3 ESCs were infected with control shRNA lentivirus or p53 shRNA lentivirus for 24 h. The cell clones stably expressing p53 (mESCp53-shRNA) and control shRNA (mESCCtrl-shRNA) were selected for 5 days by puromycin. Western blot analysis was used to determine the expression levels of p53 in these cells.
RNA extraction and gene expression analysis by quantitative real-time PCR
Total RNA from cultured D3 ESCs was extracted and purified by using the RNeasy mini kit (Qiagen) following the manufacturer's instructions. cDNA was synthesized from 1 μg of total RNA with Superscript III first-strand synthesis system (Invitrogen). For quantitative real-time PCR (qRT-PCR) analysis, amplification reaction was performed using SYBR Green master mix (Bio-Rad) in Bio-Rad MyIQ PCR machine by following the manufacturer's protocol. The melting curve of each sample was measured to ensure the specificity of the products, and samples with an unexpected melting curve were excluded from further analysis. The following primer pairs designed by primer analysis software Oligo V7 were used for qRT-PCR: Myocardin, 5′-gtgggcccagcattttcaac-3′ (forward) and 5′-tttccggtatcgtgctttcctc-3′ (reverse); SM α-actin, 5′-ggcatccacgaaaccacctat-3′ (forward) and 5′-agccaccgatccagacagagta-3′ (reverse); SMMHC, 5′-atgctgggaaggtggactacaa-3′ (forward) and 5′-gtgcggaacatgcccttttt-3′ (reverse); p21, 5′-cgagaacggtggaactttgac-3′ (forward) and 5′-ccagggctcaggtagacctt-3′ (reverse); Mdm2, 5′-ggatcttgacgatggcgtaag-3′ (forward) and 5′-aggctgtaatcttccgagtcc-3′ (reverse); Bax, 5′-ccggcgaattggagatgaact-3′ (forward) and 5′-ccagcccatgatggttctgat-3′ (reverse); p53, 5′-cccctgtcatcttttgtccct-3′ (forward) and 5′-agctggcagaatagcttattgag-3′ (reverse); and 18S, 5′-ggaagggcaccaccaggagt-3′ (forward) and 5′-tgcagccccggacatctaag-3′ (reverse). Relative gene expression was calculated using the 2−ΔΔCT method. All qRT-PCR experiments were performed in triplicate, and 18S RNA was served as an internal standard.
Western blot analysis
Total proteins were extracted using the M-PER mammalian protein extraction reagent (Pierce) supplemented with a protease inhibitor cocktail (Roche Diagnostics) and separated by SDS-PAGE. The resolved proteins were transferred to PVDF membranes (Millipore). Antibodies against p53 (Santa Cruz; 1:1,000), SM α-actin (Millipore; 1:3,000), SM myosin heavy chain (SMMHC; Abcam; 1:2,000), Myocardin (Abcam; 1:500), and β-tubulin (Abcam; 1:10,000) were used for testing individual protein expression. Protein detection was achieved with the enhanced chemiluminescence system (Amersham Biosciences).
ChIP PCR assay
P53 ChIP samples were prepared from differentiating ESCs, and ChIP DNA quality was verified. RA-treated or control cells were fixed sequentially with 1% formaldehyde in PBS and then lysed, sonicated, and immunoprecipitated as described previously [18] using material from 5 × 107 cells per sample. All antibodies used had been previously tested for ChIP and validated for their specificity. Biological replicates are defined as p53 ChIP DNA prepared from distinct cell cultures grown, harvested, and processed on separate days. Sonicated chromatin was immunoprecipitated with mouse monoclonal anti-p53 antibodies. The primers used to amplify the area containing the predicted p53-binding site I were 5′-gcgcgttctggagatgtacc-3′ (forward) and 5′-aacagccggctcttaactctt-3′ (reverse), resulting in a 227-bp fragment (P1). The primers used to amplify the area containing the predicted p53-binding site II were 5- gaattcctggtcctcctgcc-3(forward) and 5-actaatggagcccacatgca -3(reverse), resulting in a 180-bp fragment (P2). Amplification was carried out using the following conditions: one cycle at 95°C for 2 min, followed by 37 cycles at 95°C for 30 s, 55°C for 30 s, and 72°C for 30 s and one final incubation at 72°C for 5 min.
Immunocytochemistry and apoptosis assay
For immunocytochemistry, cells were fixed in 4% paraformaldehyde (PFA) in PBS and blocked in 0.3% Triton X-100 and 2% BSA in PBS for 1 h at room temperature. The cells were washed three times in PBST (0.1% Triton X-100/PBS), blocked in 5% BSA for 30 min, and incubated with the anti-SM α-actin antibody (Millipore; 1:200) at 4°C overnight. Rhodamine-conjugated secondary antibodies (Millipore; 1:200) were used for antibody localization, and the nucleus was counterstained with Hoechst 33258 or DAPI (Abcam). The cells were washed three times with ice-cold PBS for 5 min each.
For DNA ladder assay, fragmented and intact DNA fractions were extracted from cultured cells by using the apoptotic DNA Ladder Detection kit (Abcam) according to the manufacturer's instructions. DNA ladder assay using 1.5% gel electrophoresis showed DNA fragmentation in the treated cells.
Fluorescence-activated cell sorting analysis
Fluorescence-activated cell sorting analysis (FACS) was performed following a previous report [18]. Briefly, cell pellets were fixed, permeabilized using a BD Biosciences Cytofix/Cytoperm™ kit, and incubated overnight with the anti-SM α-actin antibody (Millipore). Mouse IgG2a served as isotypic control (DakoCytomation). Rhodamine-conjugated IgG antibody was used as the secondary antibody (Millipore). Finally, fluorescence was analyzed using the FACSCalibur™ system (BD Biosciences) following the manufacturer's instructions.
Construction of reporter plasmids and luciferase reporter assays
The pcDNA3 p53 WT (p53) and pcDNA3 p53 S15A (p53-Mut) plasmids were gifts from David Meek (Addgene plasmids #69003 and #69004) [20]. To generate the Myocardin luciferase reporter plasmid, a 2,115 bp fragment (−1,865 to 250 bp) containing the p53 putative binding site was amplified from C57/B6 L mouse genomic DNA and then cloned into KpnI and HindIII sites of the pGL4.10 [Luc2] vector (Promega). The following primers were used for cloning the Myocardin promoter: 5′-cttttctctggagtcgcccc-3′ (forward) and 5′-gctttccaccagaaactggc-3′ (reverse). For the construction of the mut-Myocardin luciferase plasmid, the p53 binding site I was mutated from the Myocardin-luciferase construct using the QuikChange XL mutagenesis kit (Stratagene) following the manufacturer's instructions. Primers used were as follows, resulting in the mutation of the putative p53-binding element (capitalized is the partial mutated region from the predicted Myocardin promoter): 5′-gtaagctggggacaccagggaTCGCcacacctaggacaagcagata-3′ (forward) and 5′-tatctgcttgtcctaggtgtgGCGAtccctggtgtccccagcttac-3′ (reverse). The final sequences of all constructed plasmids were confirmed by DNA sequencing. Relative plasmids were transfected into HEK 293 cells using Lipofectamine 2000 (Invitrogen) for 4 hrs. Luciferase activity was measured 48 hrs after transfection using the Dual-Luciferase assay kit (Promega) with an EnSpire Multimode Plate Reader (PerkinElmer). Individual luciferase activity was normalized to the responding TK promoter Renilla luciferase activity.
Animals and wire injury of the femoral artery
For transgenic mouse construction, SM myosin heavy chain (SMMHC)-p53 plasmid was constructed and then microinjected into mouse-fertilized eggs. These eggs were transplanted into the oviduct of pseudopregnant mice. Transgenic mice were identified by PCR and Southern blot. In vivo studies were performed using male SMMHC-p53 transgenic mice and wild-type (WT) mice that were 18–25 weeks of age and weighed 25–30 g. All operational procedures were approved by the Hangzhou Normal University Animal Care and Use Committee. Mouse femoral injury model was done as described by Sata et al. [21]. Briefly, mice were anesthetized by isoflurane inhalation, and surgery was carried out using a Leica dissecting microscope on a heated surgical pad. The femoral artery was exposed and a 0.36 mm straight spring wire was readily inserted into the femoral artery via a muscular branch artery. The wire was left in the femoral artery for 1 min, and after it was removed, the muscular branch artery was ligated to restore the blood flow in the injured femoral artery. After 2 weeks, mice were euthanized by inhalation of anesthesia and quickly perfused with PBS solution. Femoral arteries were removed, fixed overnight in 4% PBS-buffered formalin, and embedded in paraffin.
Statistics
All experiments were performed in triplicate. Data were analyzed by SPSS12.0 and expressed as mean ± SD. Statistical comparisons between the two groups were made using an unpaired Student's t test, and probability values (P) < 0.05 were considered significant.
Results
P53 is upregulated during SMC differentiation from mouse ESCs
Previously, we have established an RA-induced in vitro differentiation model of SMCs from ESCs [17,18], illustrated in Fig. 1A. In the present study, we observed a large number of apoptotic cells in 10 μM RA-treated ESCs by Hoechst 33258 staining and apoptotic DNA ladder assay. During the process of SMC differentiation, RA-treated cells showed considerably condensed chromatin by Hoechst 33258 staining (Fig. 1B). These dense fragments are known as apoptotic bodies, which indicate an early apoptotic event. Meanwhile, DNA fragments that are roughly 180–200 bp intervals, which are a key feature of apoptosis, can only be observed in RA-treated cells (Fig. 1C).
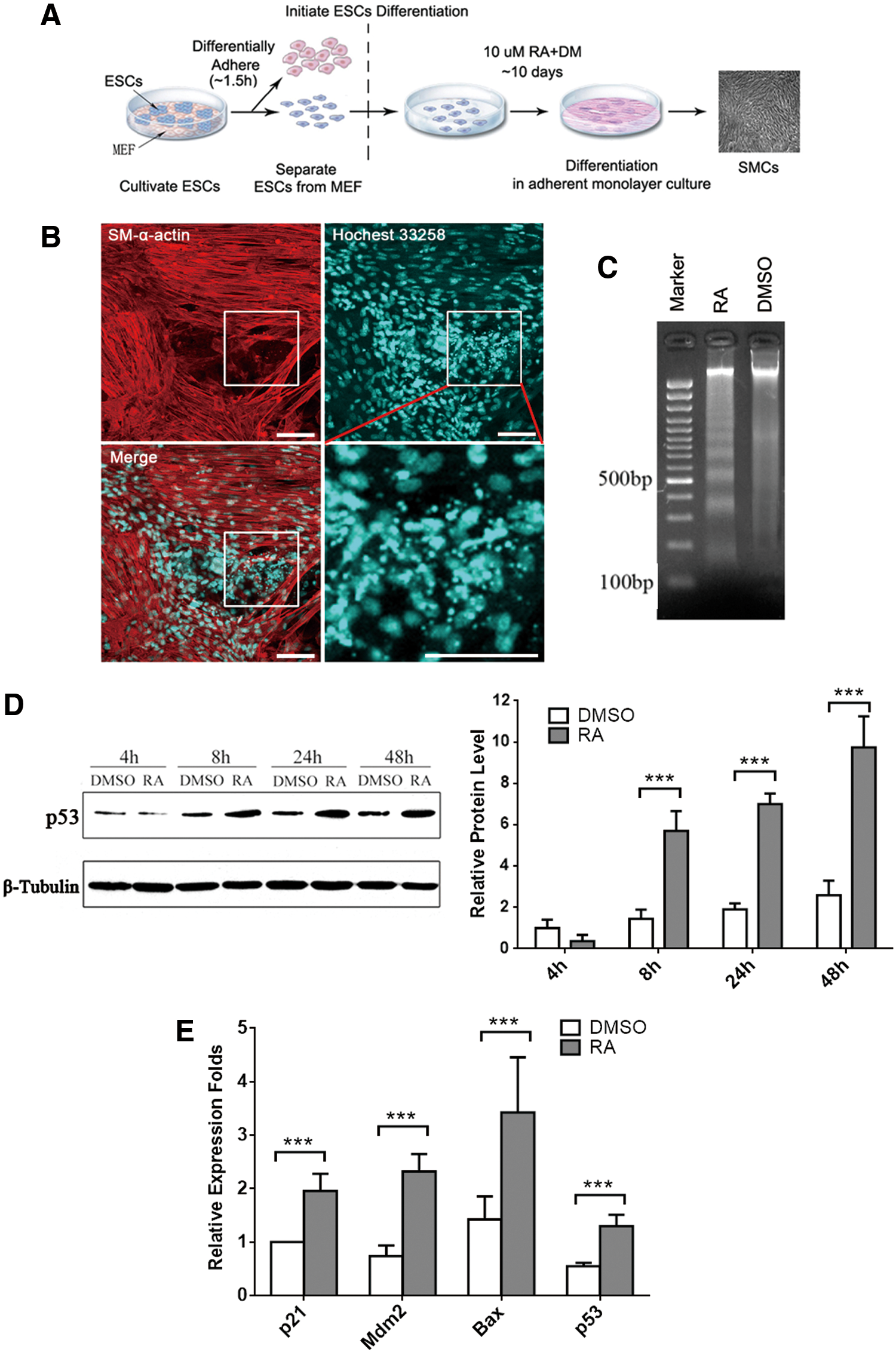
Elevated p53 expression following cell apoptosis is induced during mESC/SMC differentiation.
Furthermore, as the key regulator of cell apoptosis, p53 accumulates and transactivates downstream target genes such as Mdm2 (responsible for the feedback degradation circuitry of p53), p21 (responsible for cell cycle control), and Bax (responsible for mitochondrial membrane remodeling) in response to stress [22]. Here we found the protein levels of p53 were gradually elevated in the RA-treated group during ESC/SMC differentiation by western blot analysis (Fig. 1D). P53-targeted genes p21, Mdm2, and Bax were found to be upregulated at the mRNA level at day 3 after RA treatment compared to DMSO-treated groups (Fig. 1E). Our results demonstrated that both expression and transcriptional activity of endogenous p53 were consistently increased during RA-induced ESC/SMC differentiation.
Suppression of p53 inhibits RA-induced ESC/SMC differentiation
Previous studies demonstrated that p53 regulates various cellular processes, including cell differentiation in vitro and in vivo [23]. To uncover the roles of p53 in SMC differentiation, we establish an mESC line with stably p53 gene silencing by lentivirus-mediated shRNA interference. Western blot analysis showed that p53 protein expression was significantly inhibited in the mESCsh-p53 group compared with the mESCCtrl group at day 4 post-RA treatment (Fig. 2A). At day 9, immunofluorescence staining images indicated that the number of smooth muscle-like cells was remarkable declined after knocking down p53 protein (Fig. 2B). Correspondingly, the expression of SM α-actin and SM-MHC, the two key SMC markers, was decreased at both mRNA and protein levels in the mESCsh-p53 group (Fig. 2C, D). Furthermore, FACS analysis revealed that the proportion of SM α-actin-positive cells was considerably reduced from 90.7% to 71.1% at day 10 post-RA treatment (Fig. 2E). All indicated that knocking down of p53 impairs RA-induced ESC/SMC differentiation.
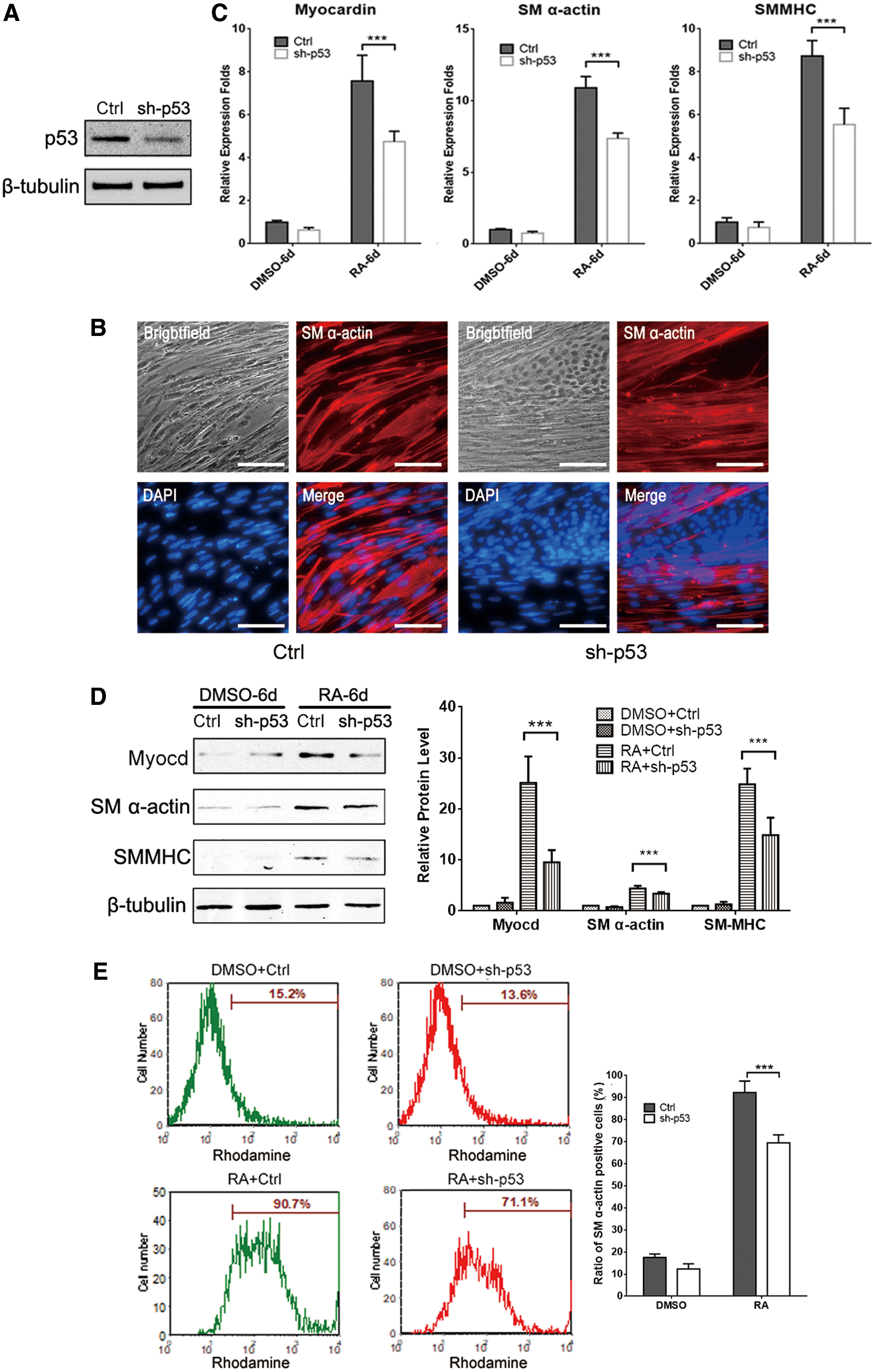
Suppression of p53 by shRNA attenuates SMC differentiation from ESCs.
P53 activates myocardin expression by binding directly to myocardin promoter
To better understand the molecular mechanism of p53 regulatory role during ESC/SMC differentiation, we performed a bioinformatic analysis for the p53-targeted genes in the Genomatix database (
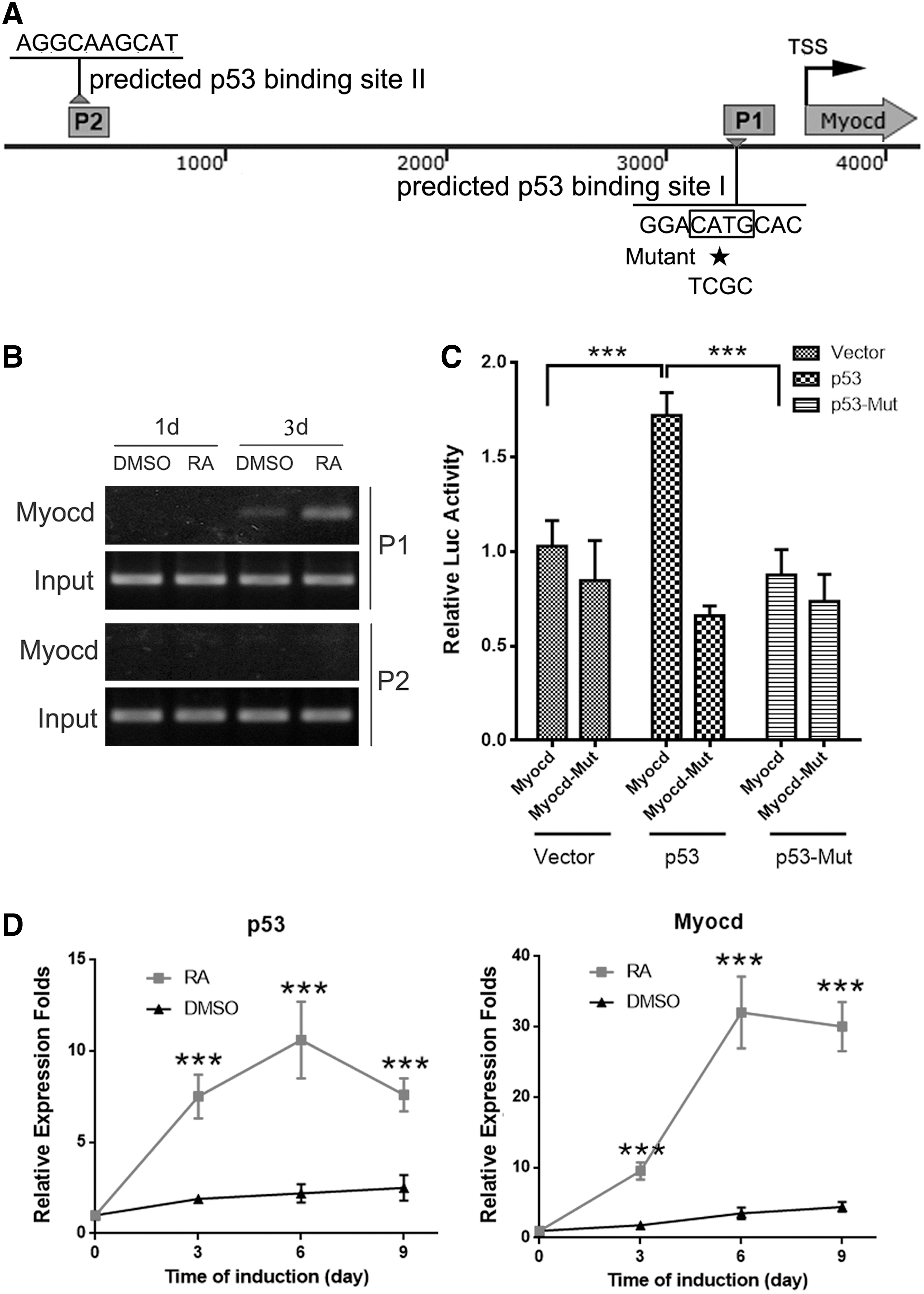
P53 activates Myocardin transcription by binding to the promoter directly.
Upregulating p53 promotes A404 cell differentiation into SMC
A404 is a mouse embryonic carcinoma cell (P19)-derived cell line, commonly used for SMC differentiation studies [19]. We further used this in vitro model to verify the role of p53 in SMC differentiation. During the RA-induced SMC differentiation, treatment with 0.5 μM doxorubicin upregulates p53 expression by doxorubicin-induced DNA damage repair [9]. As shown in Fig. 4A, the protein level of p53 was significantly increased in A404 cells after an 8-h doxorubicin treatment. At the same time, elevated expression of SMC differentiation markers SM α-actin and SMMHC was also observed at both mRNA (Fig. 4B) and protein levels (Fig. 4C). Interestingly, doxorubicin treatment also increased Myocardin mRNA level compared to vehicle-treated A404 cells (Fig. 4B), consistent with the results from the ESC/SMC differentiation model. As summarized in Fig. 4D, high concentration of RA activates p53, which then binds to the Myocardin promoter and enhances Myocardin expression, and consequently modulates SMC differentiation.
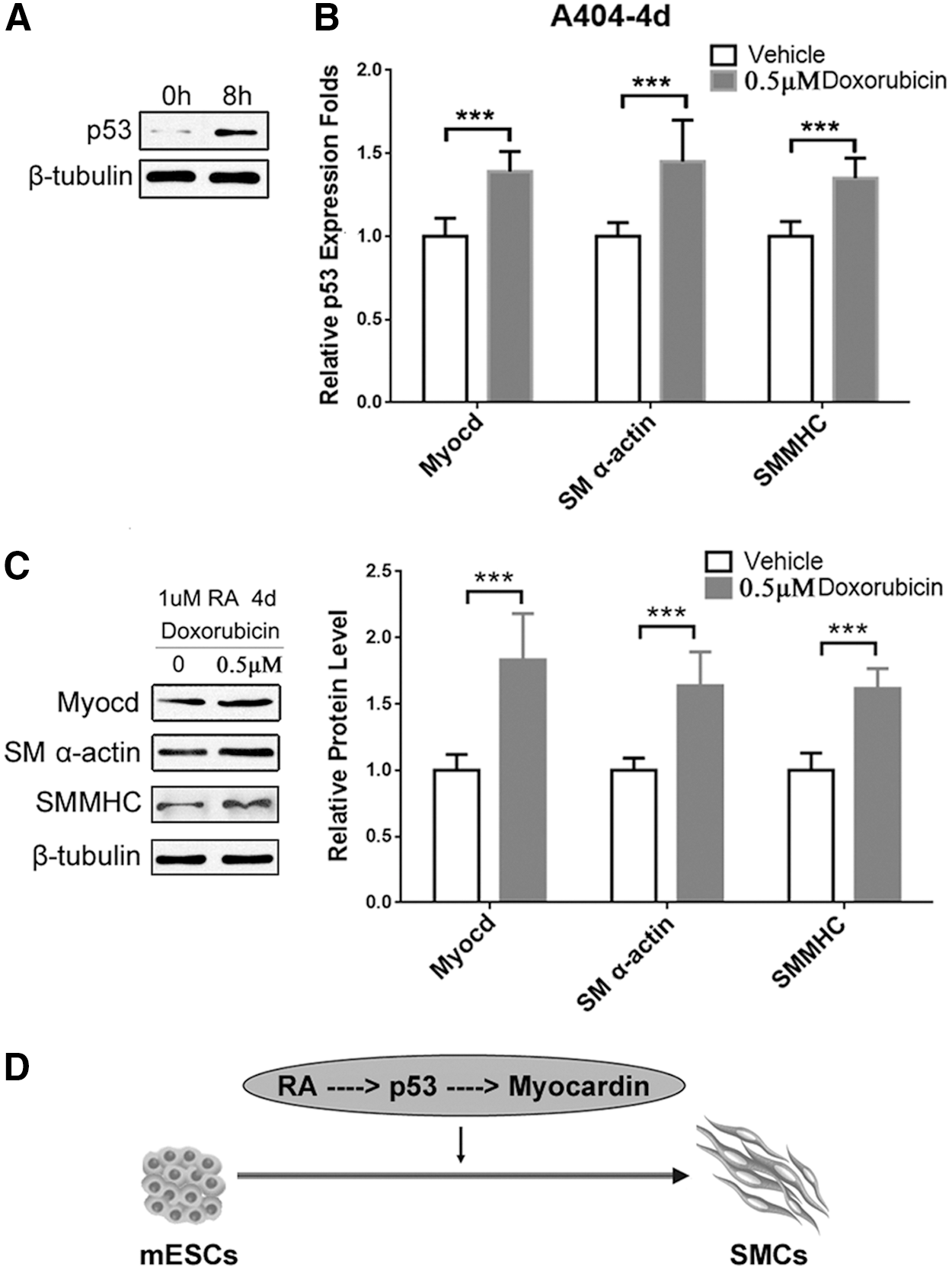
Increased p53 expression promotes A404 differentiation into SMC.
Transgenic overexpression of SMMHC-p53 in vivo inhibits injury-induced neointimal formation
To determine the function of p53 in SMC differentiation in vivo, we generated transgenic mice in a C57BL/6J background with SMC-selective overexpression of p53. The transgenic construct contains the SMMHC genomic region (containing the first intron) from −4.2 to +11.6 kb, the 1,173 bp full-length cDNA sequence, and the ployA element (Fig. 5A). There was no distinct effect on normal development and reproduction of the mice by over-expressing p53 in mature SMCs. RT-PCR analysis of different smooth muscle tissues from the third generation of transgenic mice showed that p53 was found overexpressed in five different smooth muscle tissues of SMMHC-p53 mice compared with WT mice (Fig. 5B). Ten-week-old WT mice and SMMHC-p53 transgenic mice were subjected to wire-mediated vascular injury. After 14 days, van Gieson staining showed that neointimal thickness and relative intima/medial ratio were remarkable lower in SMMHC-p53 transgenic mice than WT mice, indicating that SMC-selective overexpression of p53 significantly inhibits the formation of neointimal membrane during vascular injury in vivo (Fig. 5C).
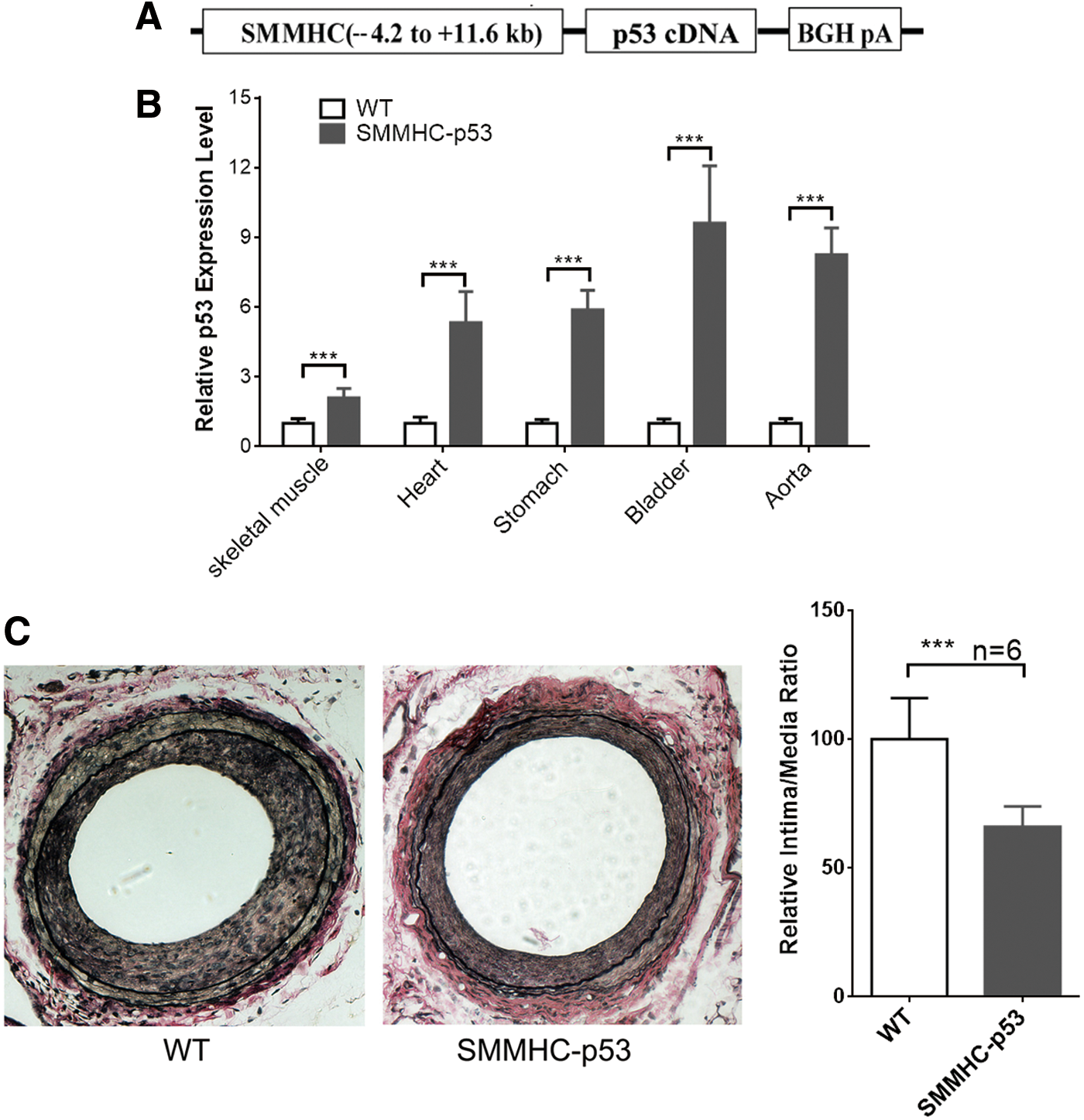
SMC-selective p53 transgenic overexpression inhibits injury-induced neointimal formation in vivo.
Discussion
SMC differentiation plays essential roles during vascular development and in regulating normal blood pressure homeostasis. Abnormal proliferation of SMC is key to the pathogenesis of vascular diseases such as atherosclerosis, hypertension, restenosis, as well as aneurysm. However, the progress in understanding the mechanisms of SMC differentiation was hindered, given SMCs have different embryological origins and can undergo phenotypic modulation in cell culture. A number of in vitro models have been developed for studying SMC differentiation, and thus, careful consideration should be taken so that the model chosen fits the questions being raised [24]. For example, SMC lines derived from adult tissue such as PAC1 cells (a pulmonary artery-derived SMC line) are not suitable for the study of SMC development [25]. In our present study, we used the RA-induced ESC/SMC differentiation system (Fig. 1A), with which each stage of early SMC development is recapitulated in the process of ESC/SMC differentiation. In brief, ESCs are separated from the MEF feeder layers and cultured in monolayer in the presence of RA (10−5 M) leading to SMC differentiation with 90.7% efficiency for SM α-actin expression. Morphological change into SMC-like phenotype is accompanied by elevated expression of SMC marker genes such as Myocardin, SM α-actin, and SMMHC. It is also accompanied by the appearance of functional SMC properties, including contraction in response to the muscarinic agonist carbachol, autonomous SM-like contraction frequency after prolonged culture, and functional calcium responses to the vasoconstrictors caffeine, endothelin, and the depolarizing agent KCl [17,26].
Cellular differentiation is often executed concomitantly with cell cycle arrest and the acquisition of a specialized lineage type by a highly coordinated set of events. On the contrary, the halted differentiation and enhanced proliferation are considered to be contributing to cell transformation and tumor formation [23]. Fittingly, the key tumor suppressor p53 is also a general regulator that controls cell differentiation [27]. Several studies have supported this concept, for example, in situ hybridization studies of mouse embryos revealed high p53 mRNA levels in all tissues until midgestation [28]. While in p53-deficient Xenopus laevis embryos, halted mesodermal differentiation and severe gastrulation defects were observed [29]. In addition, inhibiting p53 expression in Salamander resulted in the inhibition of limb regeneration [30]. In our study of SMC differentiation, we also found elevated expression of p53 and its targeting genes, initiating cell apoptosis rather than cellular senescence following differentiation induction (typical features of apoptosis can be observed in RA-treated ESCs, shown in Fig. 1B, C). Knockdown of p53 in the ESC/SMC differentiation system resulted in attenuated expression of SMC markers, including SM-MHC, SM α-actin, and Myocardin. These results are in line with previously published data, which indicated that knocking out of p53 in ESC impaired SMC and adipocyte formation [31]. It was reported in another study that resveratrol induced p53 expression, whereas suppressed Myocardin-mediated differentiation in adult SMC line [32]. These studies suggest that p53 may exert either a positive or a negative effect depending on the specific cell type and the specific differentiation stage.
Myocardin, a cotranscriptional activator of serum response factor (SRF), stimulates the expression of SMC genes, including SM α-actin, SMMHC, calponin, SM22, h-caldesmon, and SM myosin light chain kinase (MLCK) [33,34]. Transforming growth factor-beta1-induced transcript 1 (TGFβ1I1), a novel biomarker of the SM contractile phenotype, is also regulated by Myocardin/SRF [35]. Due to its central role in SMC generation, whether p53 functions through Myocardin in SMC differentiation is worth further study. Through bioinformatic analysis we identified two binding sites of p53 in the upstream area of Myocardin promoter, implying that p53 may affect SMC differentiation by directly acting on Myocardin. In support of this hypothesis, we performed ChIP-PCR assay and luciferase reporter assay, and confirmed that about 2 kb fragment (−1,865 to 250 bp) of the Myocardin promoter is the p53 working site. Taken together, we confirmed that a high concentration of RA stimulates the differentiation of ESCs in the expression of p53, after which p53 binds directly to the Myocardin promoter to promote its transcription, thereby inducing the expression of SMC genes.
In addition to p53 as a regulator of ESC/SMC differentiation, using another cell model (A404/SMC differentiation model), we provided compelling evidence that p53 promotes SMC differentiation in vitro. Our findings that upregulation of p53 by doxorubicin enhanced expression of the key SMC transcription factor Myocardin might represent a general mechanistic basis for the p53-modulated SMC differentiation. Although Myocardin plays pivotal roles, as both a necessary and a sufficient factor during SMC differentiation, there would be other target genes of p53 or p53 downstream genes such as p21, Mdm2, and Bax that may exert regulatory functions on SMC differentiation [36]. Furthermore, as mentioned above, knocking out of p53 can affect the differentiation of mesoderm. In Fig. 3D, the upregulation of Myocardin level was consistent with the increased p53 expression, which further confirmed our conclusion that p53 works directly on Myocardin for SMC differentiation. However, it does not preclude the possibility that p53 may first initiate the mesoderm differentiation (which is also required for efficient SMC differentiation) and then induce the SMC differentiation.
Interestingly, further in vivo study revealed that SMC-selective overexpression of p53 is associated with inhibition of injury-induced neointimal formation (Fig. 5). The reduction in neointimal area was attributable to a diminished accumulation of neointimal SMCs, as neointimal SMC accumulation after wire-induced vascular injury is predominantly attributable to the recruitment of SMC progenitor cells [37,38]. While increased p53 expression in injured vessels may be involved in SMC-mediated vascular repair by inducing cell cycle arrest, apoptosis, and differentiation. This is the first in vivo evidence supporting the conclusion that p53 promotes SMC differentiation. It is known that vascular SMC can switch phenotypic characteristics from a migratory synthetic phenotype in embryonic tissue to a quiescent, contractile phenotype in mature vessels to maintain the vascular tone. Importantly, during vascular remodeling in response to injury, VSMCs can switch back to an active synthetic phenotype characterized by increased VSMC proliferation, extracellular matrix synthesis, and migration [39]. Overexpressing p53 in vessels reduced vascular injury-induced VSMC phenotypic switch and intimal hyperplasia, which indicated that consistent with our experimental results in vitro, p53 may play essential roles in the regulation of SMC differentiation in vivo.
In summary, our study demonstrated that p53 acts as a general regulator in SMC differentiation. In agreement with previously published data in skeletal muscle cells [40], we found that p53 accelerates differentiation of SMCs. Interestingly, we observed that p53 facilitates SMC differentiation from mouse ESCs by binding to the promoter of Myocardin and increasing its expression.
Footnotes
Acknowledgments
This work was partially supported by the Natural Science Foundation of China grant (31101046), Natural Science Foundation of Zhejiang Province grant (LY15C070005), and Initiative Design Project of Agricultural Research of Hangzhou (20162012A07) to HH; the National Institute of Health grants (NS094930, NS091175, NS086820) to KY; Social Development Scientific Research Project (20140533B17) and Hangzhou Health Science and Technological Plan (2014A66) to ZT.
Author Disclosure Statement
No competing financial interests exist.