
Abstract
Intravenously administered mesenchymal stromal cells (MSCs) are rapidly entrapped in the lungs, where they display an anti-inflammatory phenotype. Intramuscular (IM) delivery provides an increased MSC dwell-time, which could result in a sustained modulation of an inflammatory milieu. We studied the therapeutic effects of IM delivered MSCs to treat a distant (contralateral) inflammation, and compared the efficacy of neonatal (umbilical cord) and adult bone marrow MSCs (BMMSCs). Inflammation decreased over 48 h, but neonatal cells showed an earlier response than BMMSCs. Tumor necrosis factor-induced gene-6 (TSG-6) was released at the site of MSC delivery, while neutrophil infiltration was abrogated and inflammation reduced at the contralateral site. MSCs did not distribute to the organs or to the site of inflammation. Thus, IM delivery presents a promising alternative for the treatment of inflammation, and neonatal MSCs may represent a stronger candidate than those derived from adult BM to treat inflammatory diseases.
Introduction
T
One important advantage of IM delivery of MSCs is their demonstrated increase in dwell-time in situ. This was first shown by Bartholomew et al. [7], who delivered baboon MSCs, engineered to express human erythropoietin (EPO), IM to NOD/SCID mice and reported serum EPO levels, above those of controls, up to 1 month. An 8-month dwell-time was reported by Vilalta et al. [8] for human adipose-derived MSCs transplanted in the thigh muscle of BALB/c homozygous nude nu/nu mice, although the majority of cells disappeared from the muscle within the first week. Braid et al. [9] also showed that a depot of IM delivered human umbilical cord perivascular cells (HUCPVCs), genetically modified to secrete an antiviral monoclonal antibody, provided protection against exposure to Venezuelan equine encephalitis virus, with secretorily active MSCs detectable for ∼4 months. More recently, Braid et al. [6], compared the dwell-time of human MSCs (hMSCs) when delivered IM (5 months), IP or SC (3–4 weeks), and IV (3 days) in healthy athymic mice.
While several authors have reported that the number of IM injected cells decreases with time, there is little evidence of distribution to other organs. Ramot et al. [10] reported that placental-derived hMSCs remained at the thigh musculature injection site in NOD/SCID mice for 3 months. Similar results were reported by Creane et al. [11], who detected human bone marrow MSCs (hBMMSCs) 3 months after injection in the thigh and calf musculature of BALB/c nude mice, but no migration of MSCs to the brain, heart, lungs, kidneys, spleen, or liver. On the contrary, it has been frequently shown that small numbers of IV delivered MSCs, which are rapidly entrapped in the capillary beds of lungs [2], lodge in other organs, mainly the liver and spleen [12 –15]. Nevertheless, during entrapment in the lungs, MSCs have been shown to alter the tissue microenvironment of a remote injured/inflamed site through their secreted soluble factors [2]. Thus, it is clear that the physical presence of the delivered MSCs at the target site is not essential for therapeutic immune modulation. Indeed, it is now generally accepted that trophic factors released by MSCs are responsible for the majority of their therapeutic effects. Thus, IM delivery of MSC to the hamstrings has been shown to increase cardiac function [16,17], predominantly through the release of vascular endothelial growth factor (VEGF) [18], although other trophic factors may also play an important role [19].
Nevertheless, IM delivery of MSCs has mainly been used for treatment of focused pathologies, including myocardial ischemia [20,21], muscular dystrophy [16], and peripheral nerve lesion-associated muscular atrophy [22]. The output measures in these studies focused on local effects in the affected muscle, cardiac muscle, or limb, and the systemic sequelae of the IM MSC administrations were not explored [21,22]. However, IM delivery of MSCs may also represent a valuable alternative to treat systemic conditions where the long dwell-time of secretorily active cells would provide an advantage over the rapid disappearance of cells from the lungs following IV delivery. Indeed, Consentius et al. [23] showed that mismatched placental-derived MSCs, while administered locally into critical limb ischemia (CLI) patients, also inhibited immune responses by modulating dendritic/natural killer cell interactions. Furthermore, Liu et al. [24] demonstrated a prophylactic reduction in joint inflammation in an antibody-induced/lipopolysaccharides (LPS)-challenged murine rheumatoid arthritis model, when gene-modified hBMMSCs were delivered IM (bilateral hind limbs). However, they also showed that untransduced MSCs were not effective in abrogating inflammation, which would apparently contradict the findings of Shabbir et al. [16] who reported reduced circulating plasma myeloperoxidase (MPO) levels following IM hBMMSC administration.
The lack of uniformity between reported MSC source, delivery route, and animal model complicates comparison of available preclinical data and has hindered the development and adoption of IM MSC delivery in clinical studies. To begin addressing this issue, we compared the efficacy of three MSC sources—hBMMSCs, HUCPVCs, and mouse bone marrow MSCs (mBMMSCs)—in an established murine hind paw carrageenan-induced inflammation model [25] in immune-competent mice. To avoid the local effects of delivering cells into the ipsilateral limb, in each case the cells were delivered, postinflammation induction, into the contralateral quadriceps. Then, we monitored paw circumference, MPO levels, and ingress of both neutrophils and macrophages into the paw tissue. As tumor necrosis factor-alpha (TNF-α) is a potent inflammatory mediator, we measured circulating TNF-α levels and the level of tumor necrosis factor-induced gene-6 (TSG-6) at the IM injection site, and tracked biodistribution and dwell-time of the delivered cells. We hypothesized (1) that IM transplanted MSCs could effectively downregulate such a remote source of acute inflammation and (2) that the efficacy of the MSC population, in abrogating the inflammation, would depend on the tissue source of the cells.
Materials and Methods
HUCPVC culture and source
Human umbilical cords were collected under a protocol approved by the Health Sciences Research Ethics Board of the University of Toronto and the Research Ethics Board of Mount Sinai Hospital, Toronto (#s 28546 and 13-0066-E respectively). The human umbilical cord perivascular cells (HUCPVCs) were isolated by, and using a proprietary serum and xeno-free process of, Tissue Regeneration Therapeutics, Inc., Toronto, Canada. Passage 1 cells were seeded at 1,333 cells/cm2, incubated at 37°C in a 5% CO2 atmosphere in a humidified incubator, and expanded in Lonza TheraPEAK™ MSCGM-CD™ (Lonza; Cat. No. 00192125) medium, which was changed every 3 days. The cells were enzymatically dissociated from the culture dish at ∼70% confluency using TrypLE Select CTS (Invitrogen; Cat. No. A12859-01). HUCPVCs derived from five donors were pooled and expanded to passage 3 (P3).
hBMMSC culture and source
hBMMSCs, isolated from hBM aspirates, were obtained as frozen vials at passage 1 from the Center for the Preparation and Distribution of Adult Stem Cells (
mBMMSC culture and source
mBMMSCs, isolated from mBM aspirates, were obtained as frozen vials at passage 1 (donated by the Phinney Group at Scripps Research Institute). Frozen vials were thawed at 37°C in water bath followed by resuspension in complete culture medium consisting of α-MEM (Gibco; Cat. No. 32561-037), 10% fetal bovine serum (Atlanta Biologicals, Optima; Cat. Nos. S12450, Lot. M13174), and 1% penicillin/streptomycin (Gibco; Cat. No. 15140122). mBMMSCs were seeded at 5,000 cells/cm2, incubated at 37°C, under hypoxic conditions (5% O2, 5% CO2 atmosphere) in a humidified incubator. The medium was changed every 4 days followed by enzymatic dissociation of cells from the culture dish at 70% confluency using 0.25% trypsin-EDTA (1 × ) (Sigma; Cat. No. T3924). Cells were then reseeded for growth up to P3.
TNF-α treatment in cultures
Only mBMMSCs were primed (pretreated) before IM delivery. mBMMSCs were harvested at P3 and plated at 12,000 cells/cm2 in T175 flasks in complete media. After 18–24 h, when cells had adhered to the culture plates, the medium was changed to that supplemented with recombinant mouse tumor necrosis factor-alpha—rmTNF-α (80-235; R&D Systems; Cat. No. 410-MT-050/CF). The priming condition was 1 ng/mL of rmTNF-α and a 1-h incubation time.
MSC fluorescence labeling
Post-trypsinization, cells were washed 1× with phosphate-buffered saline (PBS) (Gibco; Cat. No. 10010023) and then incubated in fresh media containing 3.5 μg/mL 1,1′ dioctadecyltetramethyl indotricarbocyanine iodide (DiR) (Invitrogen; Cat. No. D12731) for 19 min at 37°C. The labeled MSCs were then centrifuged for 5 min at 1,000 rpm at 4°C and washed twice with PBS. A sample of labeled MSCs was checked for viability, fluorescence, and MSC surface markers postlabeling with a flow cytometer.
Gaussia luciferase transfection of HUCPVCs
A premade Gaussia luciferase (GLuc) recombinant adenovirus was purchased from Vigene Biosciences (Rockville, MD). GLuc is a secreted luciferase protein that is used to monitor biological processes in vivo [26 –29]. HUCPVCs were seeded at a cell density of 23,000 cells/cm2 and 24 h later were exposed to the virus at a multiplicity of infection (MOI) of 100 in 100 μL/cm2 of culture medium. After 24 h of incubation at 37°C, 5% CO2, cells were washed with PBS three times and fresh growth medium was added. Engineered HUCPVCs were harvested 3 days later for experimental use.
Flow cytometry on MSC surface markers postlabeling
Briefly, 1 × 105 of freshly harvested HUCPVCs, unlabeled or labeled with DiR, were washed in PBS containing 1% bovine serum albumin (BSA) and 2 mM EDTA (flow buffer) and incubated for 30 min at 4°C in the same buffer containing the following conjugated anti-human antibodies (at 1:5–1:20 dilutions): HLA-DR-FITC (eBioscience; Cat. No. 11-9952-42), CD31-APC (eBioscience; Cat. No. 17-0319-42), CD45-FITC (eBioscience; Cat. No. 11-9459-42), CD105-PE (BD Biosciences; Cat. No. 560839), and MHC I-APC (BD Biosciences; Cat. No. 555555). The cell suspensions were washed with the flow buffer and then resuspended in the flow buffer. Immediately before analysis on the Cytomix FC 500 flow cytometer (Beckman Coulter), cells were stained with propidium iodide (PI) to exclude dead cells. Next, 5,000 live or PI-negative events were collected. Labeling efficiency was assessed by measuring DiR fluorescence in FL5, while surface marker detection via antibodies was measured in FL1 for FITC and FL2 for PE. Flow cytometry data were analyzed using Kaluza Software (Beckman Coulter) (Supplementary Fig. S1; Supplementary Data are available online at
In the case of GLuc-transfected HUCPVCs, flow cytometry was used first to determine the efficiency of transfection by intracellular detection of the GLuc protein. Surface marker profile was then assessed following the same procedure previously described. Control and GLuc-transfected cells were washed in the flow buffer and fixed in 2% paraformaldehyde solution. Cells were then washed twice with the flow buffer and permeabilized in prechilled (−20°C) 100% methanol at −20°C. This was followed by washing twice with the detergent solution (PBS +0.5% BSA +0.5% Tween 20) and resuspension in the detergent solution. A rabbit anti-human primary antibody against GLuc protein (New England Biolabs; Cat. No. E8023S) was added at 1:50 dilution and incubated for 30 min at 4°C. Cells were again washed twice and resuspended in the detergent solution for incubation with a secondary goat anti-rabbit antibody conjugated to Alexa Fluor 546 (Life Technologies; Cat. No. A-11010). Finally, cells were washed twice with the detergent solution and resuspended in the flow buffer for flow cytometric analysis.
Gating of HUCPVCs was based on forward scatter (FS) and side scatter (SS) characteristics and 5,000 of the gated events were counted. Transfection efficiency was determined by measuring the fluorescence signal of Alexa Fluor 546 in the FL2 channel. The efficiency of GLuc-HUCPVCs was measured as 85.4%. For phenotyping, control and GLuc-transfected HUCPVCs were incubated with the following conjugated anti-human antibodies (at 1:5–1:50 dilutions): HLA-DR-FITC (eBioscience; Cat. No. 11-9952-42), CD45-FITC (eBioscience; Cat. No. 11-9459-42), CD10-FITC (eBioscience; Cat. No. 11-0106-42), CD142-APC (eBioscience; Cat. No. 17-1429-42), CD31-APC (eBioscience; Cat. No. 17-0319-42), CD34-APC (eBioscience; Cat. No. 17-0349-42), CD90-FITC (BD Biosciences; Cat. No. 561969), CD73-PE (BD Biosciences; Cat. No. 550257), CD105-PE (BD Biosciences; Cat. No. 560839), CD166-PE (BD Biosciences; Cat. No. 559263), CD146-PE (BD Biosciences; Cat. No. 550315), CD140b-PE (BD Biosciences; Cat. No. 558821), and MHC I-APC (BD Biosciences; Cat. No. 555555). PI was added before analysis and 5,000 live events were collected (Supplementary Fig. S2).
Animal studies
The animal experiments were performed in accordance with the Canadian Council on Animal Care and the University of Toronto Animal Care Committee under protocol number 20011728. Eight-week-old adult CD1 female mice (Charles River Laboratories, Gatineau, Canada) were acclimatized for 1 week before animal studies. Mice were then randomized into groups. Mice were fed with alfalfa-free pellets (Harlan Teklad; Cat. No. TD97184) during acclimatization and throughout the study period. Lack of chlorophyll in Alfalfa-free pellets reduces autofluorescence in the stomach and intestines and hence improves fluorescent optical imaging.
Carrageenan induction in mice hind paw
We used a well-developed model of inflammation (carrageenan-induced hind paw) and induced 9-week-old CD1 mice with 1% w/v carrageenan (unilaterally). Mice were anesthetized with isoflurane in nitrous oxide and oxygen (900 mL total flow rate; 3%–4% induction and 2% maintenance). Twenty-five microliters of 1% w/v λ-carrageenan (Sigma; Cat. No. 22049) solution in 0.9% saline was injected (needle gauge 30) subcutaneously into the plantar region of the right hind paw.
MSC delivery and measurement of inflammation
At the first peak of inflammation, 4 h postinduction, mice were anesthetized to have control over the site of injection. Next, different sources of MSCs (ie, mBMMSCs, hBMMSCs, and HUCPVCs) at a density of 1.3 × 106 cells resuspended in 0.9% saline were delivered contralaterally in the posterior thigh musculature, n = 6–7/group. Note: Due to insufficient total cell number at P3, we were only able to deliver mBMMSCs for the designated 48-h time point. The circumference of the inflamed paw was measured using a cotton thread prior and post-MSC delivery at 4, 24, and 48 h postinduction. Paw measurements were taken at 360° from digit 1 plantar to digit 1 dorsal, as illustrated with black ink in Fig. 1C.

Experimental design.
Assessment of biodistribution of IM delivered MSCs
Real-time longitudinal assessment (in vivo imaging)
To assess the biodistribution of MSCs, mice were imaged prior and post-MSC delivery, at 24 h, 48 h, and up to 33 days. Mice were imaged under anesthesia. The fluorescent flux emitted by the DiR-labeled HUCPVCs was quantified using an IVIS® imaging system. Images were acquired using 2D epi-illumination to identify the region of interest (ROI) where fluorescence signal was detected. Next, each mouse was imaged with 3D FLIT (with an exposure time of 200 ms), which includes a computed tomography data set of the mice's surface tomography and multiple fluorescent transillumination images to allow 3D reconstruction (excitation/emission at 745/840 nm). For bioluminescence, we aimed to correlate the in vivo cell number to the luminescence flux, for which 1 × 106, 8.75, 7.5, 6.25, 5, 3.75, 2.5, and 1.25 ( × 105) cell densities of HUCPVCs were suspended in PBS in black clear-bottomed tissue culture-treated 24-well plates (Visiplate) (PerkinElmer; Cat. No. 1450-605). Coelenterazine (Nanolight; Cat. No. 3031) (300 μg/300 μL sterile ddH2O/well) was added using a multichannel pipette immediately before open filter imaging using the Xenogen IVIS system (Supplementary Fig. S3A, B). Then, we assessed the kinetics of Gaussia-coelenterazine for which we IM delivered 1 × 106 GLuc-HUCPVCs, followed by IV perfusion of 250 μg/100 μL coelenterazine in sterile ddH2O and acquired open filter images every 1 min up to 15 min (Supplementary Fig. S3C) on auto-exposure.
The quantification of the ROI encircled in red is computed in Supplementary Fig. S3D. For the primary study, mice were intravenously injected with coelenterazine (250 μg/100 μL sterile ddH2O per 23 g mouse) and in vivo GLuc activity was measured at different time points (24, 48, 72, 96, and 120 h post-HUCPVC delivery). Data acquisition was obtained 1 min postsubstrate administration (exposure time = 9 s. Fluorescence and luminescence 3D reconstruction was performed using the Living Image software. Anatomical coregistration was performed using the Xenogen digital mouse atlas included in the IVIS software.
PCR and droplet digital PCR
Custom primers were designed against mouse SRY, murine actin, human FARS2, and human/mouse glyceraldehyde 3-phosphate dehydrogenase (GAPDH) using the National Center for Biotechnology Information (NCBI) website (Supplementary Table S1). To assure primer specificity, we blasted against human/mouse genome (NCBI-BLAST). Reverse transcription quantitative polymerase chain reaction (RT-qPCR) was the carried out on the synthesized complementary DNA using SsoAdvanced™ Universal SYBR Green Supermix (Bio-Rad; Cat. No. 1725272) according to the manufacturer's instructions (10 μL reaction volume) at an annealing temperature of 65°C. Each sample was run in triplicate and Bio-Rad CFX384 Touch™ System was used for fluorescence measurement. Murine actin and h/m GAPDH were used for normalization of expression level of mouse SRY and human FAR2. For more accuracy, we assessed the copy number of human/male mouse genomic DNA using droplet digital PCR (ddPCR) technique on organs and the induced paw. PCR containing QX200 ddPCR EvaGreen Supermix (Bio-Rad; Cat. No. 186-4034) with various concentrations of DNA—as low as 50 ng and as high as 1 μg (using HindIII restriction enzyme; New England Biolabs; Cat. No. R0104T). Bio-Rad Automated Droplet Generator was used following the manufacturer's recommendations followed by amplification in a thermal cycler for ddPCR. Using QX200 droplet reader and QuantaSoft software, the positive and negative droplets for target DNA were quantified.
Blood and tissue collection
Terminal blood collection was performed through intracardiac puncture while mice were under deep anesthesia. Terminal blood samples were collected at 4, 24, and 48 h postinduction of inflammation. The induced paw and the injected muscle, as well as organs (kidneys, lungs, liver, heart, and spleen), were immediately excised postsacrifice, frozen in liquid nitrogen, and further cryogenically ground into fine powder with mortar and pestle. Fine and well-homogenized powders were immediately stored at −80°C for further analysis.
Protein isolation and purification
Approximately 40 mg frozen tissue powder from both muscle and paw were weighed followed by an immediate addition of 1 mL mammalian cell lysis buffer supplemented with Benzonase Nuclease and protease inhibitor solution provided in the protein extraction kit, Qproteome Mammalian Protein Prep Kit (Qiagen; Cat. No. 37901). Tissue dissociation was performed by means of ceramic beads (Omni International; Cat. No. 19-646) and Mini-BeadBeater (MIDSCI; Cat. No. 607), 2 × 45 s. Samples were then centrifuged at 1,000g for 5 min at 4°C to remove tissue debris and the supernatant was aliquoted and stored at −80°C for further use.
DNA isolation and purification
Approximately 100 mg frozen tissue powder from paw, kidneys, lungs, liver, heart, and spleen were weighed followed by an immediate addition of 720 μL PureLink Genomic Digestion Buffer supplemented with 80 μL Proteinase K provided in the DNA purification kit (Invitrogen; Cat. No. K182001). Tissue dissociation was performed by means of ceramic beads and Mini-BeadBeater, 2 × 30 s. Samples were then incubated on a heat block overnight at 55°C to obtain maximum lysis (recommended by the kit). Samples where then centrifuged at 14,000g for 3 min at room temperature to remove debris. DNA lysis was further purified using the aforementioned kit and the recommended protocol. DNA samples were then aliquoted and stored at −80°C for further use.
Enzyme-linked immunosorbent assay and quantification
Blood serum supplemented with protease inhibitor cocktail (BioShop; Cat. No. PIC001.1) that contains 2 mM AEBSF HCL, 130 μM bestatin, 14 μM E-64, 1 μM leupeptin, and 0.3 μM aprotinin. Samples stored at −80°C were then tested for TNF-α levels using the TNF-α Quantikine ELISA (enzyme-linked immunosorbent assay) Kit (R&D Systems; Cat. No. MTA00B). Before measurements, protein samples were centrifuged at 1,000g for 5 min at 4°C to remove cellular debris. A standard curve was generated using the recombinant TNF-α protein in the kit and all samples were tested in triplicate and measurements followed the recommended procedure. Sample absorbance was measured at 450 nm using a plate reader (Molecular Devices, SpectraMax i3x Multi-Mode Detection Platform).
Paw lysates that were extracted from tissue powders (Qproteome Mammalian Protein Preparation Kit) contained protease inhibitor and were stored at −80°C. MPO activity in the paw lysates was measured using the MPO ELISA Kit (Hycult Biotech; Cat. No. HK210). Before measurement, protein samples were centrifuged at 1,000g for 5 min at 4°C to remove cellular debris. A standard curve was generated using the protein standard provided in the kit. Sample absorbance was measured at 450 nm. Total protein of the same samples (ie, same aliquots) was measured using the Pierce BCA Protein Assay Kit (Thermo Fisher Scientific; Cat. No. 23225) and normalized to the total measured protein. All samples were tested in triplicate and measurements followed the recommended dilution and procedures.
Western immunoblotting
Protein lysates from muscle tissue were quantified using Pierce Bovine Serum Albumin Standard (Thermo Fisher Scientific; Cat. No. 23208). Total protein concentration of 5 μg was chosen to run on 4%–15% Mini-PROTEAN® TGX stain-Free™ Protein Gels (Bio-Rad; Cat. No. 4568083). Protein samples, including the positive control recombinant TSG-6 protein, before running on gel, were added to 4× Laemmli sample buffer (Bio-Rad; Cat. No. 1610747) that contained 2-mercaptoethanol (Bio-Rad; Cat. No. 1610710) with a ratio of 3 parts of sample with 1 part of Laemmli. Samples were then incubated on a heat block at 95°C for 5 min, followed by 5 min on an ice bath. Next, the denatured protein samples were centrifuged at 14 × g for 2 min at 4°C followed by gel electrophoresis at 165V for 35–40 min. Then, 10 μL all Blue prestained protein ladder (Bio-Rad; Cat. No. 1610373) was loaded on the first column followed by the positive control standard recombinant TSG-6 protein (Bio-Rad; Cat. No. 2326-TS) and protein samples in the consecutive columns. The running buffer used was 1× Tris/glycin/SDS (Bio-Rad; Cat. No. 1610732) that contained 25 mM Tris, 192 mM glycine, 0.1% SDS, pH 8.3.
The gels were then activated for 1 min using the ChemiDoc™ MP Imaging System (Bio-Rad). Gels were then transferred to LF PVDF blots following the manual for Trans-Blot Turbo™ RTA Mini LF PVDF Transfer Kit (Bio-Rad; Cat. No. 1704274) using the Trans-Blot® Turbo™ Transfer System (Bio-Rad) at 25 V, 1.3 A, for 7 min. The blots were then rehydrated in 100% methanol followed by transfer to 1× Tris-buffered saline, 0.1% Tween 20 (TBST). Protein blots were then imaged in the ChemiDoc™ MP Imaging System (Bio-Rad) for chemiluminescent detection of total protein used for data normalization and loading control. The blots were then blocked with 3% BSA at room temperature for 90 min. Primary antibody to human/mouse TSG-6 (R&D Systems; Cat. No. 259820) was diluted 1:1,000 in 1% BSA and incubated for 60 min at room temperature with mild agitation, followed by 4 × 15-min washes with TBST. The blots were then incubated in horseradish peroxidase-linked secondary antibody, goat anti-mouse (Jackson ImmunoResearch Laboratory; Cat. No. 115-035-146), 1:2,000 dilution in 1% BSA, for 30 min followed by 4 × 15-min washes with TBST. Bands were detected using Clarity Max Western ECL Substrate (Bio-Rad; Cat. No. 1705062) according to the manufacturer's protocol. Quantification was performed digitally using Image Lab™ Software (Bio-Rad). Band signals were normalized to the digitally quantified total protein (Bio-Rad stain-free system) (Supplementary Fig. S4). Note: Protein levels of TSG-6 were only measured in mice treated with human MSCs since the mice treated with mBMMSCs showed little improvement in downregulation of inflammation.
GLuc measurement in blood circulation
To assess the secretory activity of IM xenotransplanted HUCPVCs, we collected about 20 μL blood samples from the tail vein of mice in EDTA-treated cuvettes at 24, 48, 72, 96, and 120 h post-HUCPVC delivery. Bioluminescence was tested using BioLux Gaussia Luciferase Assay Kit (New England Biolabs; Cat. No. E3300L). Five microliters of whole blood × 3 replicates of each sample was tested in white luminescence Greiner 96-well flat-bottomed plates, using BMG PHERA star injector-equipped luminometer with the following setting: 2–10 s of signal integration, and 50 μL of injection volume, gain 4,000, 400 rpm double orbital shaking for 30 s to mix the whole blood with the substrate before reading. Data were normalized to controls.
Giemsa and immunofluorescence labeling
To assess the activity of neutrophils and macrophages in the inflamed hind paw of mice prior and post-MSC delivery, we used Giemsa staining to identify neutrophils and anti-F4/80 to immunofluorescently (IF) label macrophages in cryosections. Postsacrifice, mice paw was fixed in 4% paraformaldehyde (PFA) in PBS for 24 h at 4°C. Samples were then washed, decalcified using Immunocal (StatLab; Cat. No. 1414-32), and decalcification was confirmed by MicroCT scout-view. Samples were then embedded in an optimal cutting temperature compound. Transverse plantar cross sections were consistently obtained from the same area of the hind paw. Sections are taken from the soft tissue layers before reaching the bones. When choosing the sections, to be consistent, the difference in the thickness of the inflamed paw in the control group in comparison with the paws from the MSC-treated group with minimal edema was taken into consideration. Eight to 10 μm cross sections of the plantar hind paw were stained either with Giemsa or IF using anti-F4/80 (ABD-Bio-Rad; Cat. No. MCA497R) 1:300 for 1 h at room temperature and goat anti-rat cross-adsorbed Alexa Fluor 647 (Invitrogen; Cat. No. A21247) 1:400 for 30 min at room temperature. Antigen retrieval was performed on the cryosections used for IF staining, using 20 μg/mL Proteinase K (Thermo Fisher; Cat. No. EO0491) in TE buffer (50 mM Tris and 1 mM EDTA at pH 8) for 15 min at room temperature, followed by permeabilization with 0.3% Triton in PBS and blocking using 10% goat serum for 2.5 h at room temperature in humidified chamber. Nucleic acid staining was performed using Hoechst 1:1,000 for 5 min at room temperature.
To investigate the fate of GLuc-HUCPVCs in the muscle tissue, the injected muscle tissues were cryosectioned (5–7 μm) and fluorescently labeled using anti-Gluc (E8023S) 1:400 for 1 h at room temperature and goat anti-rabbit cross-adsorbed Alexa Fluor 568 (Invitrogen; Cat. No. A11011) 1:600 for 30 min at room temperature. Antigen retrieval, permeabilization, blocking, and nucleic acid staining protocol was the same as previously mentioned.
Statistical analysis
Statistical analysis of groups/time points was performed by means of one-way or two-way analysis of variance (ANOVA) and multiple comparison by Tukey's statistical test between the means of replicates at significance level of 95% confidence interval. Each figure legend indicates details of the statistical analysis performed. Statistical analysis was performed using GraphPad Prism 6.01 (GraphPad Software, Inc., La Jolla, CA).
Results
Reduced edema in the mouse paw
As shown in the time line in Fig. 1A, MSCs were delivered IM in the contralateral quadriceps muscle (Fig. 1B) at 4 h postinduction of inflammation. This time was chosen because acute inflammation developed by 1% carrageenan has been shown to elicit a biphasic response with an initial peak at 4 h with a second peak of inflammation starting from 6 h and rising to 72 h before resolving around 96 h [30]. Changes in the paw circumference post-treatment were measured as shown in Fig. 1C.
The change in the paw circumference (Fig. 2), as a metric of the amount of edema, substantially increased throughout the 48-h study in the untreated group, compared to normal values. On the contrary, edema in both the hBMMSC and HUCPVC groups reduced over a period of 48 h. The greatest effect was seen with the HUCPVC group at 24 h with a statistically significant difference compared with controls at the same time point. The hBMMSC group only achieved this level of edema reduction by 48 h. Edema reduction further decreased in the HUVPVC group from 24 to 48 h where the significance of the difference between controls increased to P < 0.001 due, in part, to the continuing increase in circumference of the control group paws. The mBMMSC group showed little difference but not significant (P = 0.87) from the controls at 24 h although this was somewhat increased by 48 h (P < 0.05).

Circumference measurements at 24 and 48 h. Data for each animal are normalized to their baseline measurements of inflammation (4 h postinduction). Two-way ANOVA and multiple comparison with Tukey's post hoc (5 mice/group and 4 measurements/animal/time point). Significance level of 95% CI. Data are mean ± SEM. *Represents P ≤ 0.05; **P ≤ 0.01; ***P ≤ 0.001, and ****P ≤ 0.0001. Green, control (); gray, mBMMSCs ( ); purple, hBMMSCs (
); purple, hBMMSCs ( ); navy, HUCPVCs (
); navy, HUCPVCs ( ). ANOVA, analysis of variance; CI, confidence interval; hBMMSCs, human bone marrow mesenchymal stromal cells; HUCPVCs, human umbilical cord perivascular cells; mBMMSCs, mouse bone marrow mesenchymal stromal cells; SEM, standard error of the mean. Color images available online at
). ANOVA, analysis of variance; CI, confidence interval; hBMMSCs, human bone marrow mesenchymal stromal cells; HUCPVCs, human umbilical cord perivascular cells; mBMMSCs, mouse bone marrow mesenchymal stromal cells; SEM, standard error of the mean. Color images available online at
Neutrophil and macrophage activity abrogation in the mouse paw
To assess the effect of IM transplanted MSCs on immune cell extravasation to the carrageenan-induced paw, we first assessed the degree of neutrophil infiltration into the tissue surrounding the vasculature in cryosections prepared from a representative animal from each group (Fig. 3A). At 48 h, the number of neutrophils in the paws of untreated mice was qualitatively higher (∼60%) than the MSC-treated ones (data not shown). Moreover, the HUCPVC group showed a lower number of neutrophils compared with the hBMMSC group at both 24 and 48 h.

Neutrophil and macrophage activity in acute paw inflammation. ); gray, mBMMSCs ( ); purple, hBMMSCs (
); purple, hBMMSCs ( ); navy, HUCPVCs (
); navy, HUCPVCs ( ).
).
To quantify these differences, we measured MPO in homogenized paw tissue lysates (5 mice/group). MPO is an enzyme released from the neutrophil azurophilic granules that is known as a hallmark for the neutrophilic activity. As shown in Fig. 3B, neutrophilic activity in untreated mice paws was primarily increased to
Reduced level of TNF-α in the blood serum
To assess the quantity of circulating inflammatory mediators, we measured the amount of TNF-α, as a principal inflammatory moderator, at the 48-h time point. As shown in Fig. 4, the level of TNF-α in the control group (

Serum level of TNF-α in acute inflammation. ELISA range of 10.9–700 pg/mL. Values below the dash-line are considered at the same level. One-way ANOVA and multiple comparison Tukey's post hoc (3–4 mice/group). Outlier in Ctrl group not included in statistical tests. Significance level of 95% CI. Data are mean ± SEM. **Represents P ≤ 0.01 and ***P ≤ 0.001. Light blue, healthy (); green, control ( ); gray, mBMMSCs (
); gray, mBMMSCs (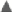 ); purple, hBMMSCs (
); purple, hBMMSCs ( ); navy, HUCPVCs (
); navy, HUCPVCs ( ). Ctrl, control; TNF-α, tumor necrosis factor-alpha. Color images available online at
). Ctrl, control; TNF-α, tumor necrosis factor-alpha. Color images available online at
Increased level of TSG-6 in the MSC-transplanted muscle
Next, we measured the level of TSG-6—a key anti-inflammatory protein released by activated MSCs in response to TNF-α stimulation—in muscle tissue lysate from the MSC transplant site. As shown in the chemiluminescent blots (Fig. 5A), the intensity of the bands increased in the MSC-transplanted muscle tissues compared to controls. In Fig. 5B, TSG-6 in the HUCPVC-transplanted muscle tissues, normalized to stain-free blots of total protein/column, showed a relative intensity unit of

TSG-6 protein level in lysates of MSC-delivered muscles. ); purple, hBMMSCs ( ); navy, HUCPVCs (
); navy, HUCPVCs ( ). IM, intramuscular; TSG-6, tumor necrosis factor-induced gene 6. Color images available online at
). IM, intramuscular; TSG-6, tumor necrosis factor-induced gene 6. Color images available online at
Biodistribution of IM transplanted MSCs
To assess MSC biodistribution, HUCPVCs were fluorescently labeled with the membrane dye DiR—prior IM delivery. To evaluate the stability of the MSC phenotype postlabeling, a panel of surface markers were tested using flow cytometry (Supplementary Fig. S1). DiR labeling of HUCPVCs did not result in any change in the expression levels of CD73+, CD105+, CD90+, MHCI+, and HLA-DR− surface receptors (Supplementary Fig. S1A). DiR-labeled HUCPVCs (1.3 × 106) were then delivered into the hind limb thigh musculature and followed by IVIS imaging (Fig. 6). Although the carrageenan-induced inflammation was resolved after 96 h (Δ paw circumference = 0), the DiR fluorescence signal was still detectable for at least 33 days posttransplantation. Interestingly, no fluorescence signal was detected in other organs within the course of the study. We further investigated the presence of xenotransplanted male MSCs (genomic DNA-sex determining region Y) in the female mice organs (lungs, heart, kidneys, spleen, liver, and inflamed paw). Using both RT-qPCR and ddPCR, no cells were detected in the lungs, heart, liver, kidneys, spleen, or paws, except in one inflamed paw from the hBMMSC group.
Longitudinal study of DiR-labeled HUCPVCs, xenotransplanted intramuscularly in CD1 mice. Representative images of n = 3 mice/time. DiR, 1,1′ dioctadecyltetramethyl indotricarbocyanine iodide. Color images available online at
Flash kinetics of Gaussia coelenterazine
The photon flux of GLuc is linearly correlated with cell densities (Supplementary Fig. S3A, B) and it is reported to linearly correlate with the concentration of secreted protein in blood [31]. However, a limitation of GLuc is the flash kinetics and fast signal decay due to inactivation (degradation and auto-oxidation) of the substrate coelenterazine [32] (Supplementary Fig. S3C, D). This disadvantage also limited transformation of in vitro photon flux data of cell densities to in vivo 3D images acquired for biodistribution and xenoreactivity studies. The algorithms in the Living Image software® of IVIS system by PerkinElmer assume consistent light output, which is not applicable to flash kinetics. Coelenterazine binds to serum proteins and therefore also has fast systemic clearance rate [33]. To evaluate the kinetics, images were acquired every minute from the time of coelenterazine perfusion and up to 15 min. Note that it is easy to make an erroneous conclusion on the biodistribution of cells with the noise generated as a result of higher exposure time (Supplementary Fig. S3C). This preliminary study suggested that all animals must be imaged within the first minute post-IV perfusion of coelenterazine to produce comparable longitudinal data (Supplementary Fig. S3D). Hence, preassessment of the kinetics of decay, consistency in the imaging time postperfusion of substrate, and constant exposure time among all animals were all requirements for this study. Supplementary Figure S3D illustrates the computed decay of luminescence flux over time.
Temporal host immune response to xeno-IM transplanted MSCs
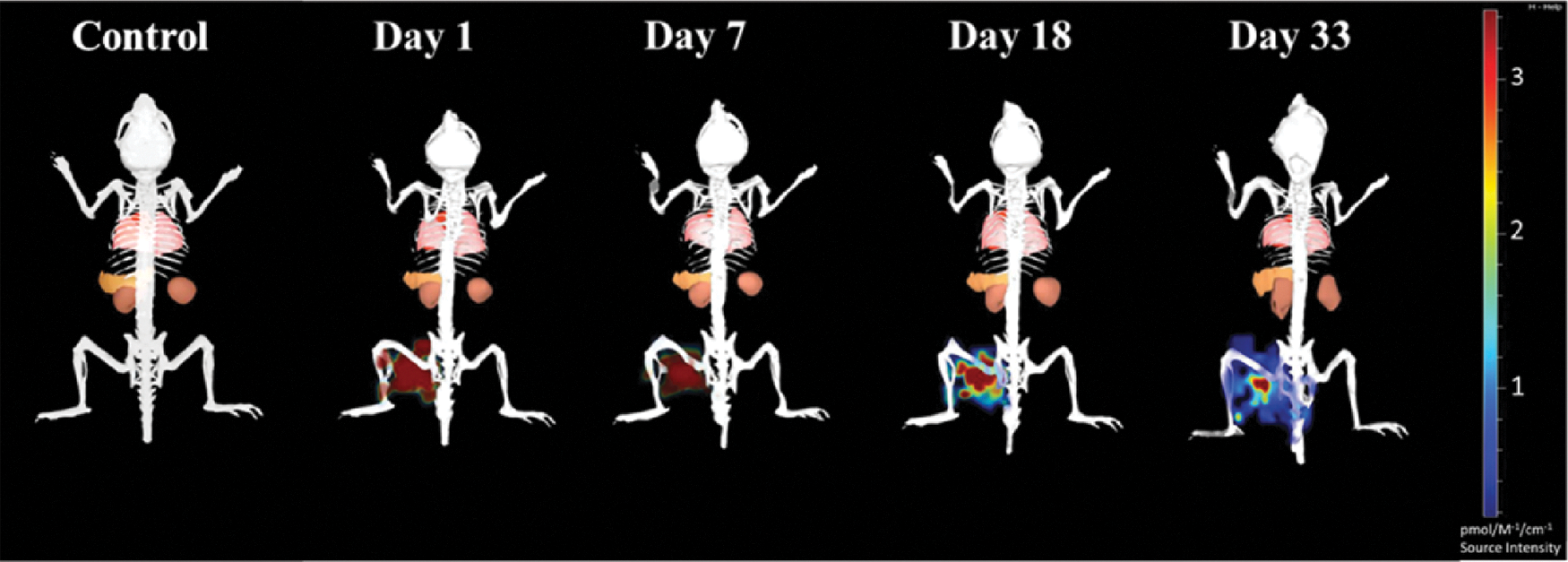
To further verify the bioactivity of the IM transplanted HUCPVCs in the CD1 immune-competent mice, we adeno-transfected the HUCPVCs with GLuc (Fig. 7). To evaluate the stability of the MSC phenotype posttransfection, a panel of surface markers were tested using flow cytometry (Supplementary Fig. S2). GLuc transfection of HUCPVCs resulted in a reduction in the expression level of CD10 by 44.11%, CD146 by 11.84%, MHCI+ by 16.25%, and an increase in the expression level of CD142 surface receptors by 27.43%. Other markers CD90+, CD73+, CD105+, CD166+, CD140b+, and HLA-DR− did not significantly change posttransfection (Supplementary Fig. S2A). As shown in Fig. 7A–C, 48 h post–IM transplantation of GLuc-HUCPVCs, the signal was reduced by 62.5%; and 89.5% by 72 h; 97.36% by 96 h; and 99.3% by 120 h.
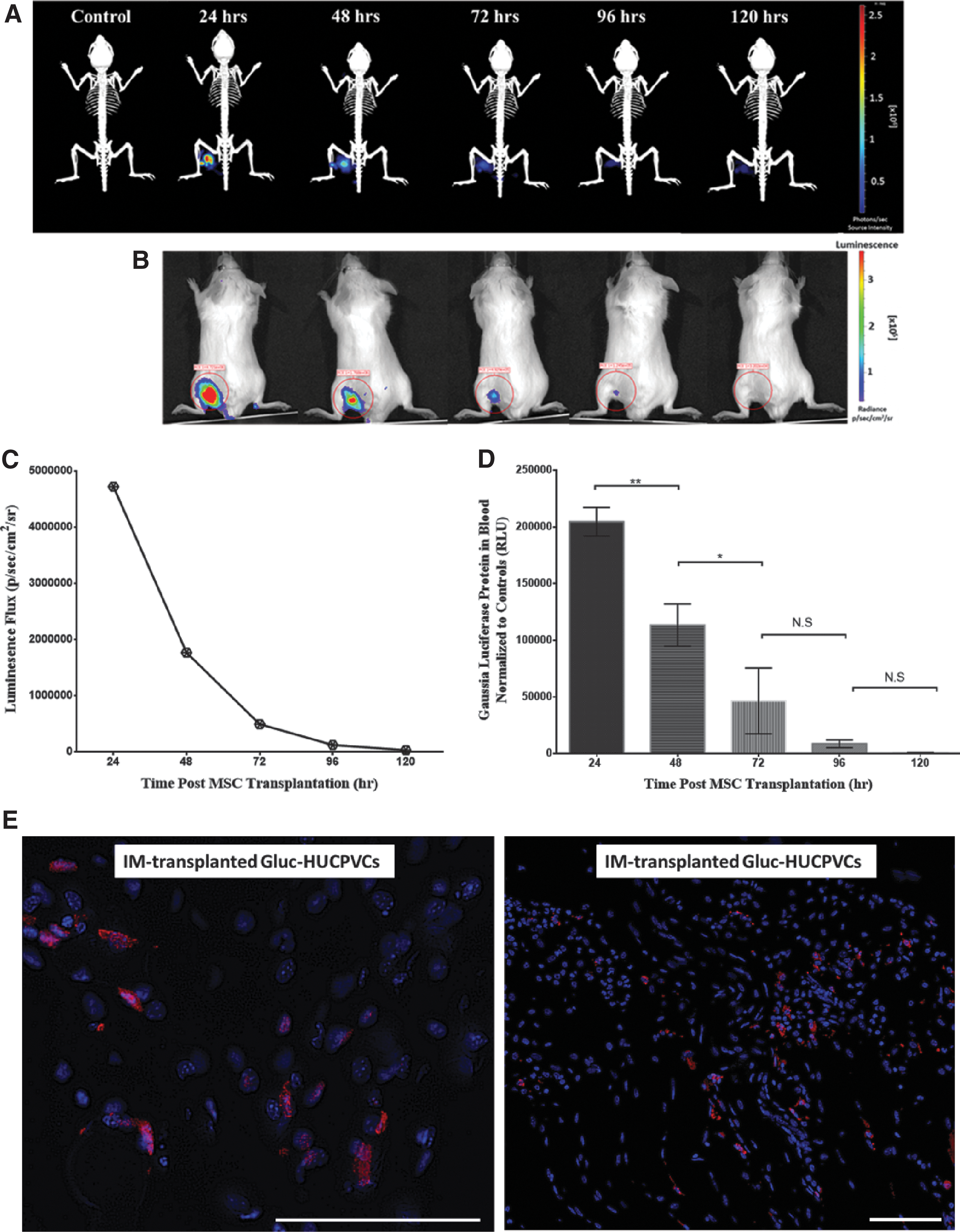
Longitudinal study of Gaussia luciferase expressing HUCPVCs. CD1 mice were intravenously injected with coelenterazine, and in vivo GLuc activity was measured at different time points. Data acquisition was obtained 1-min postsubstrate administration, longitudinal representative images (n = 5 mice/time).
To assess the rate at which the secreted proteins in the muscle enter the blood circulation, we measured the secreted GLuc protein in the whole blood (Fig. 7D). As shown in Fig. 7D, there is a strong correlation between the density of IM residing HUCPVCs and the circulating GLuc level. Circulating GLuc protein decreased by 44.5% 48 h after transplantation; 77.2% after 72 h; 95.7% after 96 h; and 99.7% after 120 h. We further investigated the fate of GLuc HUCPVCs in the muscle tissue 5 days posttransplantation. The MSC-transplanted muscle cross sections in Fig. 7E show that GLuc-HUCPVCs were detectable in the muscle after 5 days of xenotransplantation, although at low density, which was below the detection limit of the IVIS system. The low level of GLuc protein detected in the blood at day 5 correlates with the low density of MSCs detected in the muscle. On the contrary, the membrane dye (DiR) (Fig. 6) survived beyond 33 days.
In addition, we should emphasize that, during IM delivery, care must be exercised to avoid puncture of major vessels. In one of our animals, we observed an immediate extravasation of blood on “IM” delivery in the hind limb musculature, indicating that a major blood vessel was inadvertently punctured. Although we excluded this mouse from our studies, we observed a systemic distribution of MSCs, including signal from the site of ear tag injury used to identify this mouse following MSC delivery (Supplementary Fig. S5).
Discussion
In this study, we demonstrated that unstimulated and nonengineered IM delivered MSCs have the potency to diminish an existing acute source of inflammation in an anatomically distant location through systemic distribution of anti-inflammatory factors. We have further demonstrated that a neonatal source of MSCs (HUCPVCs) exhibits higher sensitivity to inflammatory stimuli, in vivo, than an adult source (BMMSCs), resulting in more effective downregulation of inflammation. These findings corroborate our in vitro results, which showed that HUCPVCs exhibited higher sensitivity, and a prompter response, to TNF-α stimulation compared with hBMMSCs [34].
Clinical studies have already confirmed the safety of IM delivery of MSCs in patients with CLI [35 –37], with no signs of tumorigenesis or necrosis at the IM site of MSC delivery. Despite this proven safety, MSC IM delivery has been mainly used to treat focal pathologies. However, even in such therapies, Laurila et al. [38] reported a local increase in VEGF that was of host (rat) rather than human origin. This observation further emphasizes the paracrine effect of the exogenously transplanted cells rather than their engraftment and differentiation [38]. One of the main concerns in local IM therapy is identifying the best time for delivery with respect to the stage of inflammation, to avoid MSC survival and potency being impaired by harsh environmental tissue cues [39,40]. These observations and concerns further condone transplanting MSCs away from the site of injury.
Some studies have used the IM delivery route to target cardiac myopathy. Shabbir et al. [17] reported significant cardiac improvement in a hamster model of heart failure by hind limb IM transplanted MSCs. Increased systemic levels of hepatocyte growth factor (HGF), leukemia inhibitory factor (LIF), and granulocyte/macrophage colony stimulating factor (GM-CSF) were measured in the MSC-treated groups that resulted in myocardial repair. Similarly, Mao et al. [19] showed improved cardiac function following fore- and hind limb IM delivery of MSCs in a rat model of dilated cardiomyopathy, with increased levels of HGF, insulin-like growth factor-1 (IGF-1), LIF, GM-CSF, and VEGF compared with controls. Both studies reported plasma levels of cytokines and growth factors related to angiogenesis (IGF-1 and VEGF), apoptosis (HGF and GM-CSF), or support of growth and differentiation of stem and progenitor cells (LIF) [41]. However, to our knowledge, the study by Liu et al. [24] is the only one that has evaluated the potency of IM delivered MSCs to downregulate inflammation in a mouse model of antibody-mediated/LPS challenged arthritis. The MSCs were delivered before induction of inflammation and no immunomodulatory effect was observed for IM transplanted MSCs that were not engineered to express human soluble tumor necrosis factor receptor II (hsTNFR).
On the contrary, in our studies, we have demonstrated that MSCs, IM delivered postinflammation induction, significantly downregulated macrophage activity, systemic levels of TNF-α, and neutrophil infiltration at the inflammatory site. As a counterpoint to Liu et al. [24], our findings show the importance of the time frame within which MSCs should be administered to attain their immunomodulatory action.
In this study, we delivered MSCs 4 h after induction of inflammation, to not only coincide with the peak inflammatory response [30] but also to assess the effect of MSCs on alleviation of such acute inflammation. PGE2, as a potentiator of acute inflammation, has been shown to cause vasodilation followed by the release of histamine and bradykinin from tissue-resident sentinel leukocytes leading to formation of edema that is involved in the first phase of acute inflammation [42]. Following vasolidation, neutrophils are recruited to the inflamed tissue and, as the inflammation resolves, the cells die apoptotically and the remnants are then removed by macrophages and dendritic cells [43]. Our results demonstrate that HUCPVCs have abrogated neutrophil infiltration more effectively than adult sources of MSCs at earlier time points (confirmed by MPO levels and histology of the induced paw tissue). This could partly be attributed to the higher inherent expression level of TSG-6 in unstimulated HUCPVCs compared with hBMMSCs and their prompt response to low-grade inflammation that was observed in our in vitro studies [34].
TSG-6 protein has been shown to abrogate neutrophil migration and infiltration [44] through interference with chemokine/GAGs and chemokine CXCL8 interactions [45]. In addition, TSG-6 protein has been shown to interact with macrophages through CD44 receptor to decrease translocation of nuclear factor-κB [46] and inhibit inflammation. Our results provide additional insights to the previously demonstrated therapeutic advantages of neonatal MSCs [47 –50], including higher MSC frequency, growth rate, and life span, and also superior immunomodulatory properties, compared with adult tissue-derived MSCs [51 –55].
It should be noted that we did not observe MSC migration to the site of inflammation that has been reported for IV delivered MSCs. Vilalta et al. [8] reported detection of 10% of MSCs at the IM delivery site and colonization of 1.5% MSCs in the liver over an 8-month study. However, in our study, in agreement with other reported studies of IM delivered MSCs [9,11,20], we did not observe migration to, or colonization of, distant organs—delivered MSCs remained in the muscle mass (except in the one case when we were aware of vasculature puncture). Our assessments of the genomic DNA of inflamed paw tissue and organs (ie, the heart, lungs, liver, spleen, and kidneys), together with both fluorescence and luminescence imaging in two consecutive studies, confirmed that IM delivered MSCs remained in the skeletal muscle.
Assessment of the therapeutic efficacy of human-derived MSCs xenotransplanted in immunocompetent mice has mainly been conducted in short-term studies over a period of a few days due to the rapid host immune rejection. Nevertheless, Prockop et al. [56] have recently strongly endorsed the validity of such models in preclinical studies. However, the poor response of mBMMSCs compared with hMSCs in our experiments begs additional comment. First, as discussed by Prockop et al. [56], mouse MSCs are known to exhibit genomic instability and grow slowly in culture. On the contrary, hMSCs can undergo 35–55 population doublings without undergoing spontaneous transformation [57] and are thus more stable. Second, mMSCs are known to effect immune suppression by different mechanisms to hMSCs [56,58]. Both of these differences may have contributed to the greater efficacy of hMSCs in our assays.
Indeed, in this study, we have used TSG-6, which is a key anti-inflammatory protein secreted by MSCs and has been proposed as a biomarker to predict the efficacy of MSCs [59], as a surrogate for the library of anti-inflammatory mediators secreted by MSCs. Our objective was not to prove the mechanism of TSG-6 action—which has been done so convincingly by others, or illustrate the plethora of anti-inflammatory agents released by MSCs, but to compare the potency of the MSCs harvested from different tissue sources in this experimental model. Nevertheless, we believe that future mapping of comprehensive cytokine responses in such models could inform the current debate on the therapeutic functional phenotypes of MSCs harvested from diverse tissue sources.
We showed that the delivered cells were only secretorily functional for up to 4 days and undetectable after 5 days. On the contrary, DiR-labeled human cell membrane fragments were still detectable at 33 days, potentially resulting from remnant phagocytosis by host macrophages, as has previously been described [60]. Thus, longer in vivo survival of a xenogeneic stromal cell would require pharmacological immunosuppression.
Finally, current systemic MSC therapy is described as a “hit and run” phenomena [14], primarily owing to the short dwell-time of infused MSCs, which are transiently effective. To extend the period over which sustained modulation of an inflammatory milieu is attained, multiple timed doses of MSCs have been administered [61 –63]. An alternative would be to use MSCs at a site that allows extended dwell-time of secretorily active MSCs for optimal therapeutic efficacy per dose. Although the experiments reported herein were, of necessity, of short duration in an immune-competent murine inflammation model, in a clinical setting the extended MSC dwell-time afforded by IM delivery could potentially be an advantage over the transitory efficacy of IV delivery. Indeed, human skeletal muscles account for about 40% of total adult body weight and comprise large striated muscle fibers [64] that may provide suitable adherence sites for exogenously delivered MSCs. In addition, the high level of vascularization present in skeletal muscle, together with the contractile movement of muscles, provides an efficient putative route for the transport of therapeutic agents into the systemic circulation [65].
Conclusions
The MSC response to TNF-α stimulation, in vivo, is MSC population and source dependent. Neonatal MSCs, delivered IM at a distant site, exhibited higher levels of secreted TSG-6 protein, and an earlier response, to a contralateral acute inflammation than MSCs derived from adult BM. Thus, neonatal MSCs may represent a stronger candidate than those derived from adult BM to treat inflammatory diseases.
Footnotes
Acknowledgments
S.H.J. gratefully acknowledges receipt of Ontario (OGS), Queen Elizabeth II, and Harron Graduate Scholarships during the course of this work. The authors are grateful to the Centre for Microfluidic Systems in Chemistry and Biology, University of Toronto, for the use of the Bio-Rad CFX384 Touch System as well as the 3D (Diet, Digestive Tract, and Disease) Centre funded by the Canadian Foundation for Innovation and Ontario Research Fund, project number 19442 and 30961, for the use of IVIS imaging system and BMG PHERAstar system. The mBM stromal cells were kindly donated by Dr. Donald G Phinney at the Scripps Research Institute, Florida.
The work was, in part, presented as a poster at the recent ISSCR meeting, June 14–17, 2017, in Boston.
Author Disclosure Statement
S.H.J., C.E., Y.L., and E.C. declare no conflicts. J.E.D. is the founding president and officer of Tissue Regeneration Therapeutics Inc (TRT), Toronto, which provided the HUCPVCs.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.