
Abstract
Leucine-rich repeat kinase 2 (LRRK2) G2019S (glycine to serine) is the most common mutation associated with sporadic and familial Parkinson's disease (PD) with 80% penetrance by age 70. This mutation is found worldwide, with up to 40% of individuals in the North African Arab population carrying the mutation. Induced pluripotent stem cells derived from fibroblasts of patients carrying the LRRK2 G2019S mutation have been a critical source of cells for generating dopaminergic neurons and studying G2019S-related pathology. These studies have elucidated LRRK2-related mechanisms of mitochondrial dysregulation, increased reactive oxygen species, truncated and simplified neurites, and cell death. These phenotypes are thought to result from the G2019S mutation increasing substrate access and therefore increasing the catalytic rate of the serine/threonine kinase. In this article, we critically review the contributions of in vitro modeling to the current knowledge on LRRK2 G2019S. We also analyze the role of patient-derived cell lines for the identification and validation of therapeutic targets, emphasizing their importance as part of a 3R approach to translational research and personalized medicine.
Introduction
P
LRRK2 mutations, in particular G2019S, are of great interest due to their high incidence in patients with PD. Several pathogenic or susceptibility mutations have been identified in the LRRK2 protein, most of them residing in, or affecting, the GTPase or kinase domains such as N1437H, R1441C|G|H, Y1699C, G2019S, and G2385R (Fig. 1). N1437H is located in the GTPase domain and seems to increase GTP binding [9]. R1441C|G|H are also located in the GTPase domain and decrease GTP hydrolysis [10,11]. All of these mutations present neuronal loss in the SN. Y1699C is located in the COR domain and also decreases GTP hydrolysis [12]. G2385R is located in the C-terminal WD40 domain and decreases kinase activity [13]. G2019S is located in the kinase domain and increases kinase activity, which has also been demonstrated in the R1441C|G and Y1699C mutations [14]. Genome-wide association studies have implicated additional mutation sites on the LRRK2 locus that are associated with PD and may affect LRRK2 expression (e.g.: [15]).
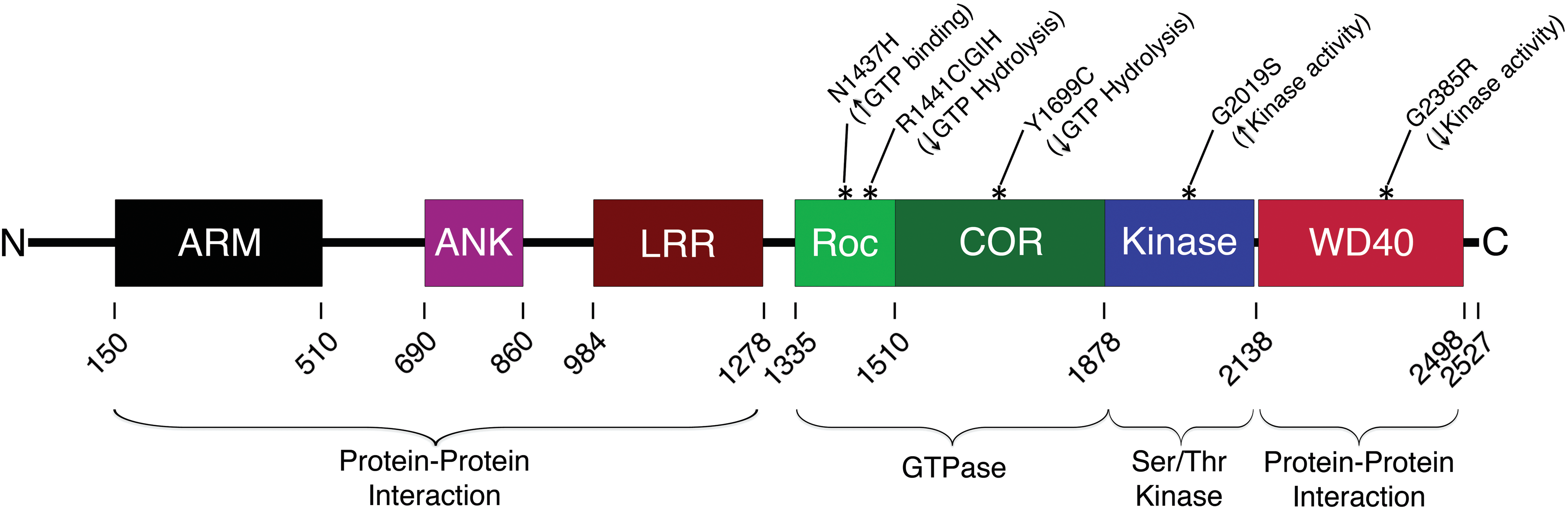
LRRK2 is a 2527 amino acid protein with several protein–protein interaction domains, as well as two catalytic domains. The mutations that are currently implicated the most in Parkinson's disease lie within the Roc/COR GTPase domain and the serine/threonine kinase domain. LRRK2, leucine-rich repeat kinase 2. Color images available online at
The discovery of SNPs that are associated with disease manifestation has provided a more direct route for utilizing in vitro modeling strategies because it pinpoints specific proteins involved in the neurodegeneration. Almost in parallel, the discovery of reprogramming adult cells to pluripotent stem cells in 2007 [16,17] has further expanded research methods for evaluating SNP-mediated cellular dysfunction in patient-derived cells. Using patient-derived cells as a platform for understanding diseases allows for more critical evaluation of intracellular mechanisms as well as cell-type-specific susceptibilities after differentiation to midbrain dopaminergic neurons in the case of PD.
In this article, we critically review the contributions of in vitro modeling to the current knowledge on LRRK2 G2019S. We also analyze the role of patient-derived cell lines for the identification and validation of therapeutic targets, emphasizing their importance as part of a 3R approach to translational research and personalized medicine.
LRRK2 in Normal Conditions
LRRK2 is a 2,527 amino acid, 285 kDa protein that spans 51 coding exons (Fig. 1). LRRK2 contains a Ras-of-Complex (ROC) GTPase domain adjacent to a C-terminal-of-ROC (COR) linker region, a serine/threonine protein kinase domain, N-terminal leucine-rich repeat (LRR) motifs, and WD40 repeats near the C-terminus that forms a β-propeller structure. It is expressed in most organs, including brain, heart, liver, kidney, and lungs.
LRRK2 mRNA and protein expression is found in rodent brain areas that relate to PD pathology, including the cortex, striatum, and SN, as well as other brain regions (e.g. hippocampus and cerebellum) [18,19]. Likewise, humans with and without PD present LRRK2 mRNA and protein expression in similar brain regions as described in rodents [20].
LRRK2 is proposed to have several intracellular functions. LRRK2 seems to modulate synaptic endocytosis through Rab5b, which is a member of the rab family of GTPases. Overexpression or siRNA-mediated knockdown of LRRK2 in rat primary hippocampal cells disrupts endocytic function [21], whereas overexpression of functional Rab5b rescues this phenotype. Interestingly, wild-type (WT) LRRK2 and Rab5b overexpression have negative effects in neurite outgrowth [22]. It has also been found that LRRK2 G2019S affects lysosomal transport through an inability to inhibit Rab7-induced clustering of lysosomes [23]. Evaluation of other proteins in the Rab family has identified some of them that act as substrates and are phosphorylated by LRRK2 [24,25]. In addition, several groups have found interactions between Rab7L1 and Rab29 with LRRK2 [26 –30].
LRRK2 has also been reported to interact with SNARE complex proteins [31] and when silenced, leads to an increase in excitatory postsynaptic current despite alterations in presynaptic vesicle pool distribution and decreased visualization of docked vesicles. LRRK2 seems important for maintaining autophagy and lysosomal homeostasis [32,33]. Several cellular and animal models of LRRK2 dysfunction have pointed to autophagy pathways being involved in the neurodegenerative pathway but specific molecular mechanisms are not yet known [34].
A number of studies found that LRRK2 undergoes autophosphorylation [35 –38]. Eighteen serine and threonine residues can autophosphorylate under normal conditions [35]. Many of these phosphorylated sites are clustered around two regions, the ROC domain and the region immediately upstream of the LRR regions. The LRR region autophosphorylation sites modify GTP-binding activity of the GTPase domain [38]. Serine residues 910, 935, 955, and 973 are constitutively phosphorylated, but not necessarily by autophosphorylation [33].
LRRK2 G2019S Phenotypes
LRRK2 G2019S is the most common mutation associated with familial PD with 80% penetrance by age 70 [39 –41]. This mutation is found worldwide and is most prevalent in North African Arabs and Ashkenazi Jews [42] with up to 40% of individuals in the North African Arab population carrying the mutation [39]. G2019S leads to middle to late onset PD and the symptoms are generally levodopa responsive. Located in the kinase region of LRRK2, the G2019S mutation facilitates substrate access, but not affinity to the kinase, thus increasing the catalytic rate of the enzyme. The G2019S mutation has been found to increase phosphorylation of α-syn, the main component of LBs, at serine 129 (S129) [43]. Age-dependent loss of SN DAergic neurons as well as identification of LBs and LNs is found in patients who carry the LRRK2 G2019S mutation.
In many ways, the manifestation of clinical signs in patients with LRRK2 G2019S is indistinguishable from IPD. Recent reports from the LRRK2 Cohort Consortium shed light on several of the similarities and differences between these two PD cohorts. A study evaluating motor and nonmotor signs reported patients with G2019S exhibiting increased susceptibility to postural instability gait disorder compared with IPD [44]. However, the frequency of motor fluctuations and dyskinesias was not different between the groups. LRRK2 G2019S PD patients had lower geriatric depression scale scores and better UPSIT (University of Pennsylvania Smell Identification Test) scores. There was no difference in SCOPA-AUT (Scale for Outcomes of Parkinson's Disease-Autonomic Symptoms), RBDSQ (REM Behavior Disorder Screening Questionnaire), MoCA (Montreal Cognitive Assessment), or Epworth Sleepiness Scale scores. A caveat to the study, noted by the authors, was the lack of power for statistical analysis. These results largely replicated those of an earlier study that found no differences in anxiety or SCOPA-AUT scores, but increased UPSIT scores in LRRK2-PD females compared with males [45]. In addition, the mean Bristol stool scale score was significantly higher in LRRK2-PD patients compared with IPD. Evaluation of the prodromal phase of LRRK2-PD patients, found no significant differences between G2019S nonmanifesting carriers (NMCs), and nonmanifesting noncarrier relatives in nonmotor symptoms, except for a small group of NMCs with mild motor signs, olfactory dysfunction, and slight decreases in DAT SPECT (Dopamine transporter single-photon emission computed tomography) uptake [46].
Evaluation of cerebral spinal fluid (CSF) and blood biomarkers in LRRK2 patients are producing intriguing results. In one study the levels of mitochondrial DNA in the CSF of LRRK2-PD patients was higher than NMCs and IPD [47]. Using a cutoff value of 19 copies/μL, the mitochondrial content within the CSF of the recruited subjects, could distinguish IPD from LRRK2-PD with a success rate of 71%. In addition, a second study using transcriptomics reported 850 differentially expressed genes in blood between LRRK2-PD and IPD patients [48].
Removal of functional LRRK2 and LRRK2 homologs in Drosophila, C. elegans, mice, and rats has consistently supported the notion that LRRK2 is not critical for proper development and survival of the dopaminergic system (See book chapter [49]). While the LRRK2 kinase domain of mice and rats is relatively well conserved compared with humans (97%); Drosophila LRRK and C. elegans LRK-1 are not (36% and 37% respectively) [50]. However, overexpression of human mutated LRRK2 has provided results that in part recapitulate disease phenotypes.
Studies in transgenic LRRK2 G2019S rodents suggest that amount of mutated protein being expressed and age are critical factors for developing pathological changes. Physiological levels of LRRK2 G2019S in mice 12–13 months of age did not lead to DAergic nigral cell loss or decreases in neurite length or branching [51], whereas another group reported impaired dopamine release by 12 [52] and 22–24 months of age [53]. DAergic nigral cell loss has been observed in G2019S transgenic mice (3–5-fold increased expression) after 19–20 months, but not when examined after 1–2 months of age [54]. G2019S transgenic mice 18–24 months of age, but not after 6–12 months, were also found to have significant levels of phosphorylated tau [53].
Patient-Derived Stem Cells for In Vitro Evaluation of DAergic Neurodegeneration
The use of induced pluripotent stem cells (iPSCs) from healthy subjects and patients with PD are invaluable for in vitro assessment of genetic mutations in the human population. A number of pathological mechanisms triggered by the G2019S mutation have been identified using patient-derived iPSCs differentiated into DAergic neurons (Fig. 2). Findings include increased expression of oxidative stress genes, namely HSPB1, NOX1, and MAOB, by day 35, but not at day 50 or 55 [55] and higher monomeric α-syn at day 60. G2019S neurons also present increased cell death, CASP9 expression, and susceptibility to 6-OHDA-induced cell death and MG-132 (a proteasome inhibitor). In an independent study, diffuse α-syn, vacuolated soma, fragmented nuclei, increased CASP3, and simplified neurites were observed uniquely in LRRK2 G2019S patient-derived dopaminergic neurons after 75 days of culture [56]. Neurite simplification has since been reported at earlier stages of DAergic maturation in patient-derived LRRK2 G2019S neurons [57]. Interestingly, kinase inhibition with LRRK2-IN-1, inhibition of pERK with PD0325901, addition of nerve growth factor (NGF), or gene correction was observed to mitigate many of the G2019S-mediated phenotypes previously observed [58,59]. Gene correction was also observed to reverse mitochondrial DNA damage resulting from LRRK2 G2019S and R1441C in patient iPSC-derived neurons [60].
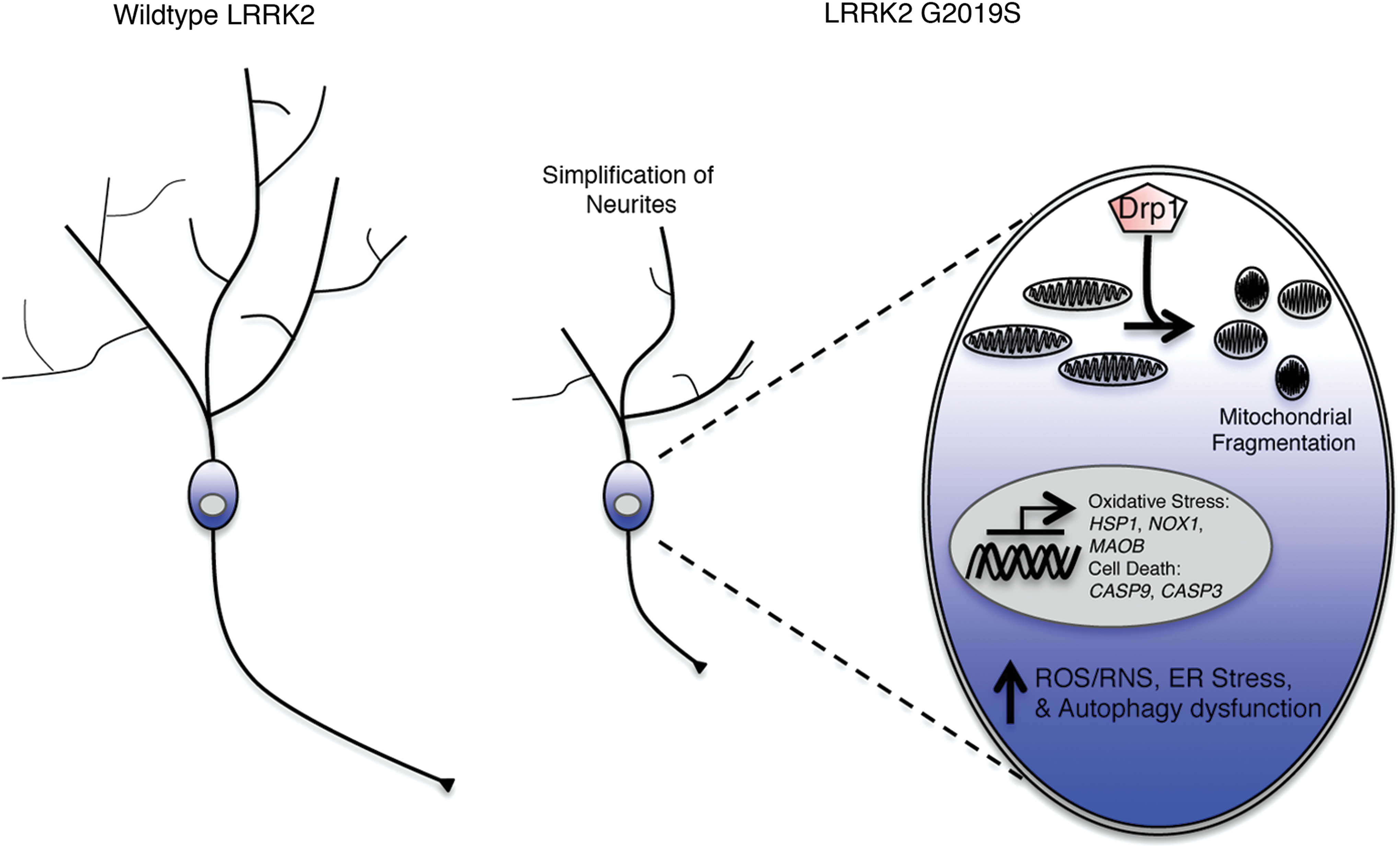
Schematic illustrating the major phenotypic alterations resulting from the LRRK2 G2019S mutation in patient-derived DAergic neurons compared with wild-type samples. Color images available online at
Degeneration of DAergic neurons in the SN is proposed to be induced by way of hyperphosphorylation of dual specificity mitogen-activated protein kinase kinase 4 (MAP2K4), an activator of MAPK, which deregulates mechanisms of cell proliferation, differentiation, and survival. In human fibroblasts, mutations such as G2019S increase overall levels of autophagy by overactivation of the MAPK1/3 pathway [61,62]. LRRK2 G2019S also leads to abnormal mitochondrial morphology, fragmentation, and damaged mitochondrial DNA resulting in elevated reactive oxygen species (ROS).
Although iPSCs derived from patients with PD have proven effective at modeling pathology in vitro, one caveat is the potential need for “aged” cells. The process of reprogramming adult fibroblasts removes much of the epigenetic information as well as age-related processes. The use of progerin, a truncated protein form of Lamin A that is associated with premature aging, can induce aged effects to iPSC lines [63]. Cellular factors of aging were analyzed based on decrease in LAPα, formation of double-stranded DNA breaks (γH2AX), loss of heterochromatin markers (H3K9me3 and HP1γ), and shortening of telomeres. Expression of progerin induced dendritic degeneration, progressive tyrosine hydroxylase loss, inclusions, and premature neuromelanin deposition. However, these data are yet to be reproduced by other investigators to further validate the use of progerin as a true model of aged cells in culture.
Therapeutic Targets of LRRK2 Dysfunction
The increase in kinase function resulting from the G2019S mutation has led to efforts to inhibit phosphorylation of LRRK2 substrates, as well as targeting downstream pathways of cellular degeneration.
Kinase inhibition
The compounds H-1152 and sunitinib were among the first discovered to inhibit LRRK2 [64]. Administration to both Swiss 3T3 and LRRK2 G2019S overexpressing HEK-293 cells induced dose-dependent dephosphorylation of S910 and S935 within LRRK2, which reduces the binding capacity of 14-3-3, an important regulator of localization. This inhibitory capacity was also observed in LRRK2 G2019S patient-derived Epstein-Barr virus (EBV)-transformed lymphoblastoid cells. In addition, reduced 14-3-3 interactions led to increased accumulation of LRRK2 in the cytoplasm, which was not found in untreated WT and G2019S patient-derived cells. LRRK2 accumulation was not observed in T-REx cells expressing a dual mutated LRRK2 species with both G2019S and a novel, inhibitor-resistant, A2016T mutation. Thus, H-1152 and sunitinib's interaction with LRRK2 is in some way a dependent factor of inhibition of kinase activity.
Through in vitro kinase assays of isolated recombinant LRRK2 protein, the indolinone compounds, GW5074 and indirubin-3′-monoxime, were observed to inhibit LRRK2 autophosphorylation as well as phosphorylation of the LRRK2 substrate, myelin basic protein [65]. Loss of DAergic neurons in mice, resulting from herpes simplex virus amplicon-mediated delivery of LRRK2 G2019S, was attenuated by injections (twice daily; 2.5 mg/kg i.p.) of GW5074 or indirubin-3′-monoxime. Overexpression of WT LRRK2 and a kinase-dead D1994A mutant LRRK2 in separate animals did not produce neuronal cell loss suggesting the cell death observed was dependent on the increased kinase activity of LRRK2 G2019S.
Due to concerns regarding GW5074's specificity, CZC-25146 and CZC-54252 were identified as potential candidates with high LRRK2 inhibitory potency and minimal toxicity [66]. Both compounds attenuated LRRK2 G2019S-induced neuronal cell death and related mechanisms of dysfunction in LRRK2 G2019S overexpressing rat cortical neurons, and human fetal primary cortical neurons; yet, pharmacokinetic studies suggested poor blood–brain barrier (BBB) penetration. Another screened compound with minimal off-target inhibition and strong binding affinity to WT and G2019S LRRK2 was LRRK2-IN-1 [67]. Similar to H-1152 and sunitinib, LRRK2-IN-1 also showed dose-dependent inhibition of S910 and S935 phosphorylation with subsequent loss of 14-3-3 binding in WT and G2019S LRRK2-transfected HEK-293 cells. It also induced aggregate localization of the LRRK2 protein that recovered two hours after removal of the compound from the culture medium. LRRK2-IN-1 inhibition was further validated in the mouse kidney (100 mg/kg; i.p.), but also presented poor BBB penetration.
In an effort toward clinical translation, a study evaluated BBB penetration and safety of the LRRK2 kinase inhibitors GNE-7915 or GNE-0877 in rodents and cynomolgus monkeys [68]. After oral administration of either GNE-7915 or GNE-0877 to male C57BL/6 mice, the authors reported that both compounds were found in similar concentrations in total brain homogenates as in the lung and kidney. Although there was no phenotype observed in rodents, when GNE-7915 was administered to cynomolgus monkeys over a seven-day period, increased vacuolation in type-II pneumocytes was observed. A follow-up 29-day toxicity study in monkeys replicated the initial finding and demonstrated that GNE-0877 also presented the vacuolation of type-II pneumocytes. These data strongly support testing first-in-class LRRK2 inhibitors in whole systems, especially primates, for the safe transition into the clinic.
A recent study utilized the compound 3-(4-pyrimidinyl) imidazole (MLi-2), an orally available and selective LRRK2 inhibitor that can penetrate the BBB [69]. Preliminary data in rats demonstrated dose-dependent reduction of phosphorylated S935 after oral dosing. Altogether, these studies demonstrate the feasibility of identifying and developing compounds targeting LRRK2 kinase activity. Careful screening could lead to a promising therapeutic strategy for patients with LRRK2 overactivity.
Inhibition of dynamin-related protein 1
Targeting the GTPase dynamin-related protein 1 (Drp1) is proposed as a different approach to preventing LRRK2 G2019S neurodegeneration [70]. Drp1 is a regulator of mitochondrial fission, in contrast to the fusion regulator GTPases: optic atrophy 1 and mitofusin 1/2. Overexpression of LRRK2 G2019S has been reported to increase activation of Drp1; subsequently leading to fragmentation of mitochondria in both rat primary cortical neurons and the human SH-SY5Y neuroblastoma line [71]. To evaluate if Drp1 inhibition would be sufficient for phenotypic recovery, the inhibitor P110 was applied to LRRK2 G2019S expressing HEK-293 cells [70]. P110 abolished Drp1 translocation to mitochondria, and subsequently reduced fragmentation, which led to recovery of mitochondrial membrane potential, mitoROS production, and ATP levels. Similar results were obtained in patient G2019S-derived DAergic neurons [70]. Interestingly, both P110 and a lysosome inhibitor NH4Cl corrected neurite shortening. Validation of Drp1 targeting with P110 in vivo has not yet been reported.
Conclusions and Discussion
The discovery of monogenic mutations linked to PD etiology has led to a focus on better understanding potential mechanisms involved in neurodegeneration. Further evaluation that the G2019S mutation within the kinase domain of LRRK2 produces higher catalytic output indicates that overphosphorylation of LRRK2 substrates is the logical first tier of the degenerative cascade.
The shared etiology of both familial and IPD inherently suggests a commonality of mechanistic dysfunction. A better understanding of these downstream pathways disrupted as a result of a known mutation may provide therapeutic targets that more broadly attack neurodegeneration across disparate PD risk factors. Unfortunately, validated LRRK2 substrates are not well described. Likewise, several of the now definitive in vitro neuronal phenotypes of LRRK2 G2019S have not been identified in vivo. By utilizing patient-derived stem cells for screening, many of these gaps are more likely to be worked out. In addition, cell lines derived from patients also present researchers with the ability to screen potential molecular therapies before in vivo validation, adhering to the 3Rs of animal research (i.e., replacement, reduction, and refinement), and provide an opportunity for personalized medicine approaches aiming to assess individual responses to treatment.
Although the in vitro evaluation of patient-derived neurons has provided researchers with insights of intracellular dysfunction, there are several challenges with this modeling approach. In vitro differentiation strategies for obtaining A9 DAergic differentiation have improved, yet it is unclear to what extent the differentiated cells are truly similar to SN DAergic neurons in vivo, or how they compare across disparate differentiation regimens. In that regard, little investigation into non-DAergic neuronal subtypes has been reported and is needed to understand the underlying mechanisms of susceptibility to LRRK2 G2019S-mediated dysfunction. Ultimately, PD affects several neuronal populations in the central and peripheral nervous system with corresponding motor and nonmotor symptoms. In vivo validation of cellular dysfunction observed in culture can be difficult, but it is important to identify which cell types and phenotypes are affected in the animal system as a whole.
With optimization in mind, it is critical not to rush into clinical translation without first understanding the ectopic side effects systemic administration of an inhibitor of LRRK2 activity drug may produce. LRRK2 is highly expressed in tissues, such as the lung and kidney, but is also expressed throughout the body [72]. As a result, strategies aiming to remove LRRK2 expression from the brain could have severe peripheral effects. In that regard, the identification of kinase-dependent pathology resulting from the increased catalytic rate of LRRK2 with G2019S has led to a search for LRRK2-specific kinase inhibitors.
Crossdisciplinary research is an advantageous approach to mechanism discovery. Peripheral examination of SNP-mediated mutations in proteins such as LRRK2 that are not specific to the CNS could shed light into what particular mechanisms are involved in the cell death process or cell-type susceptibilities. To the best of our knowledge, there has been only one study of patient-derived cell lines to evaluate the impact of the G2019S mutation in non-DAergic neurons (i.e., sensory neurons) [59]. New reports on iPSC-derived enteric or sympathetic ganglia for in vitro modeling are urgently needed as they could facilitate discovery of therapeutics that target nonmotor symptoms resulting from peripheral neurodegeneration.
With any disease, existing models need to be transformed and improved to better replicate the intended phenotypes. The use of genomic editing strategies for SNP production for instance, allows for modeling physiological levels of mutant protein without the insertion of an exogenous transgene. The use of nonhuman primates that have nearly identical genetic homologs could potentially produce the first reproduction of PD. If the disease could be sufficiently replicated, then information as to the effects species differences may have on protein structure and compensation of SNPs can be gained.
In addition to improving current models, evaluating the translatability of phenotypes across different PD-associated SNPs could further validate the mechanisms that require interference. With advancements such as RNAseq, samples from control, IPD, familial, and mutation-specific patients can be crossevaluated for dysregulated genes.
In conclusion, patient-derived cell lines have played a critical role to advance our understanding of LRRK2 G2019S-associated nigral DAergic neurodegeneration. This emerging work facilitates the next generation of in vitro studies aiming to further understand G2019S pathology in the central and peripheral nervous system, identify therapeutic targets, and screen novel compounds.
Footnotes
Acknowledgments
This research was supported by NIH grants R24OD019803, P51OD011106, NINDS T32-Neuroscience Training Program (S.C.V.), and the UW-Madison Office of the Vice Chancellor for Research and Graduate Education.
Author Disclosure Statement
No competing financial interests exist.