
Abstract
Mesenchymal stem cells (MSCs) are well known for their regenerative potential. Even though the ability of MSCs to proliferate and differentiate has been studied extensively, there remains much to learn about the signaling mechanisms and pathways that control proliferation and influence the differentiation phenotype. In recent years, there has been growing evidence for the utility of non-neuronal cholinergic signaling systems and that acetylcholine (ACh) plays an important ubiquitous role in cell-to-cell communication. Indeed, cholinergic signaling is hypothesized to occur in stem cells and ACh synthesis, as well as in ACh receptor (AChR) expression, has been identified in several stem cell populations, including MSCs. Furthermore, AChRs have been found to influence MSC regenerative potential. In humans, there are two major classes of AChRs, muscarinic AChRs and nicotinic AChRs, with each class possessing several subtypes or subunits. In this review, the expression and function of AChRs in different types of MSC are summarized with the aim of highlighting how AChRs play a pivotal role in regulating MSC regenerative function.
Introduction
Acetylcholine (ACh) and its receptors (AChRs) regulate cholinergic signaling in neurons. However, there is now sufficient evidence to confirm that cholinergic communication occurs in almost all the mammalian non-neuronal cells [1]. However, non-neuronal cells use ACh and AChRs in a different manner to neuronal cells. Unlike the neural cells, the uptake and synthesis of ACh and its mediated effects in non-neuronal cells occur in an ambiguous manner and have been reviewed elsewhere [1]. Nevertheless, the non-neuronal cholinergic system has been proven to be a powerful intercellular communication tool that plays a pivotal role in numerous cellular processes. ACh, through its receptors, can modulate gene expression, cell viability, cell proliferation, cell migration, and cell differentiation [1 –4]. Despite being known to regulate cell differentiation, the role of the non-neuronal cholinergic system in stem cells is still relatively unexplored.
Several types of stem cell express components of the non-neuronal cholinergic signaling system including functional AChRs. This includes non-neural stem cells, such as embryonic stem cells [5], hematopoietic stem cells [6], skeletal muscle stem cells [7], and mesenchymal stem cells (MSCs) [8,9].
There is sufficient evidence to conclude that MSCs express a functional cholinergic system, and studies suggest a role for ACh in regulating stem cell properties [8,9]. This review takes a holistic look at cholinergic signaling mechanisms in MSCs and their influence on function with the aim of demonstrating that these cells have cholinoceptive properties, which play important roles in determining MSC fate.
ACh Receptors
There are two major classes of receptors that bind ACh and transmit its signal, namely, muscarinic AChRs (mAChRs) and nicotinic AChRs (nAChRs) (Fig. 1). Apart from ACh, both classes of receptor bind to distinct secondary ligands that aided their identification; mAChRs bind muscarine and nAChRs bind to nicotine [10]. Both classes and their constituent subtypes permit communication between non-neuronal cells and activate signal-transduction pathways allowing maintenance of cellular function and ultimately organ homeostasis [1]. mAChRs and nAChRs have been shown to be expressed and functional in non-neuronal cells [11]. Both receptor families are membrane bound.
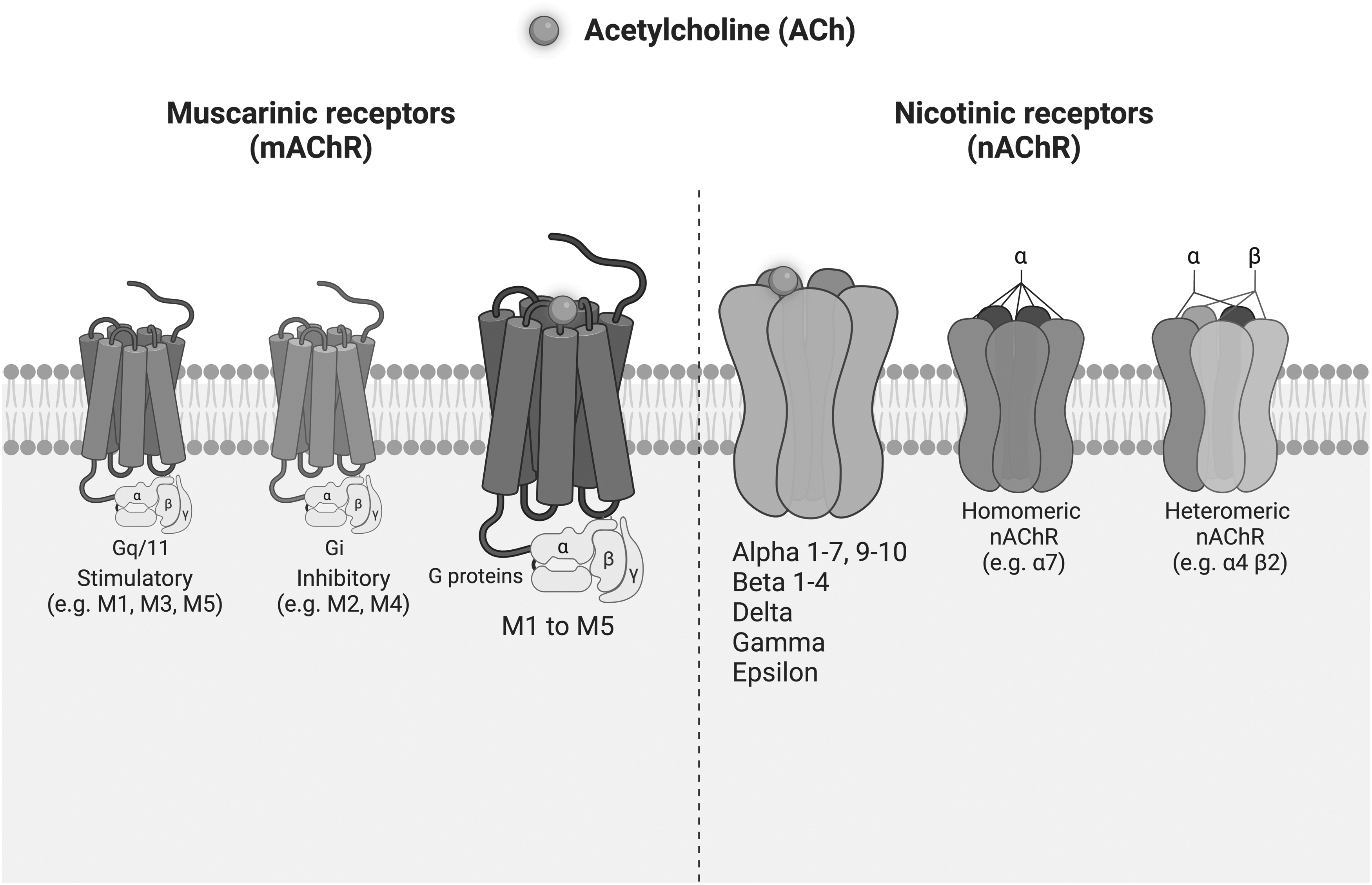
Schematic presentation of AChRs. Left: mAChRs are G-protein-coupled receptors. Based on downstream functionality of the coupled g proteins, they are commonly divided into two groups, stimulatory in nature (M1, M3, and M5) or inhibitory (M2 and M4). Right: nAChRs are pentamers from 16 possible subunits. They may present as either homopentamers (consisting of 5 identical subunits) or heteropentamers (consisting of combinations of different subunits). AChRs, acetylcholine receptors; mAChR, muscarinic acetylcholine receptor; nAChR, nicotinic acetylcholine receptors.
However, they are two inherently different classes of receptors, with structural differences, resulting in regulation of differential downstream effects [1] (Fig. 1). The mAChRs belong to the G-protein-coupled receptor (GPCR) family and mediate the metabotropic effects of ACh [12]. The nAChRs are ligand-gated ion channel receptors that mediate the ionotropic effects of ACh [13,14]. Both families include several subtypes or subunits, which again are expressed in a ubiquitous manner across a variety of non-neuronal cells [10,11,15,16].
The near-ubiquitous AChR expression across non-neuronal cell populations has made it challenging to evaluate their role. The expression of receptor classes or subtypes varies across different non-neuronal cell types and is influenced by cell state and environmental factors [1]. Both receptor classes form an auto- and paracrine loop of ACh activity in non-neuronal cells, which plays an important role in cell-to-cell communication. They may coexist in individual cells, with stimulation of one class potentially having a positive or negative effect on the other [17,18]. Furthermore, the wide-ranging influence of ACh on different types of non-neuronal cells adds to the complexity of this system [11].
Muscarinic Receptors
The mAChRs consist of five distinct subtypes referred to as type 1–5 (M1–M5). These receptors are members of the GPCR family [12] (Fig. 1). Once stimulated, mAChRs couple to distinct species of G proteins that in turn activate second-messenger signaling pathways, as well as activating gated ion channels [19]. Based on downstream functionality of the coupled G proteins, they are commonly divided into two groups; stimulatory (M1, M3, and M5) or inhibitory (M2 and M4) [20]. Thus, the cellular cascade of events depends on the species of G protein with which an mAChR interacts. This, arguably, is what makes these receptors relatively slower acting compared with their nAChR counterpart [12]. The resultant downstream effects of activated mAChRs are immensely complex and have widespread consequences. At almost all stages of development, mAChRs mediate the effects of ACh in almost all cells, both neuronal and non-neuronal [21].
In fact, abnormalities in mAChR signaling are a sign of a diseased state, such as in chronic obstructive pulmonary disease, overactive bladder, or neuronal diseases such as Alzheimer's disease [22]. This and the fact that they are GPCRs have led to their study from a pharmacological point of view. Indeed, there are commercial incentives to develop research into GPCRs as a whole [23]. To date, GPCRs, including mAChRs, are the most successful therapeutically targeted family of receptors [24 –26].
Nicotinic Receptors
The nAChRs are composed of multisubunit proteins that form ligand-gated ion channels within the cell membrane [22] (Fig. 1). An nAChR can be a pentamer based upon 13 possible subunits, which may present as either homopentameric (consisting of 5 identical subunits) or heteropentameric (consisting of combinations of different subunits) [10,27]. There are nine α-subunits (α1–7, α9, and α10) and four β-subunits (β1–4). In addition, other subunits such as delta (δ), epsilon (ɛ), and gamma (γ) have also been identified in humans [8,28]. The different subunit compositions of this receptor class allow for specialized properties and diverse functions, and thus mediate numerous downstream effects [10]. Multiple nAChR subunits have been identified in non-neuronal cell populations [29,30].
Generally, these receptors are rapid acting cationic receptors that mediate a temporal opening of ion channels to allow sodium, potassium, or calcium passage [10]. Consequently, an intracellular increase of such ions leads to activation of a series of signal transduction pathways. This, in turn, may lead to alterations in cell proliferation, cytoskeletal rearrangement, and differentiation [31].
Mesenchymal Stem Cells
MSCs are multipotent adult stem cells initially isolated from bone marrow [32]. MSCs have now been isolated from various other tissues, including adipose tissue [33], dental pulp [34], peripheral blood [35], salivary glands [36], skeletal muscle [37], skin [38], and placental tissue [39 –41]. The International Society of Cellular Therapy set three criteria to define stem cells as MSCs: (i) ability to adhere to plastic; (ii) expression of cell surface markers (e.g., CD73, CD90, and CD105) and lack of hematopoietic markers (e.g., CD14, CD34, and CD45) and class II major histocompatibility complex molecules; and (iii) ability to differentiate down mesodermal lineages [42 –44].
Generally, cultured MSCs present these features, however, some differences have been observed between MSCs of different origins [45]. MSCs tend to differentiate down mesodermal lineages, however, under appropriate stimuli, it has been suggested that MSCs are capable of differentiation into tissues of endodermal and neuroectodermal lineages [42]. As MSCs are self-renewing cells with immunomodulatory properties and the ability to be differentiated into several lineages [46 –49], they are a vital resource for tissue engineering, regenerative medicine, and cell-based therapy research [39,46,50].
Expression of mAChRs in MSCs
Bone marrow MSCs
All five mAChRs have been identified in human MSCs (hMSCs) (Table 1). Hoogduijn et al. were the first to investigate cholinergic signaling in MSCs [8]. Using polymerase chain reaction (PCR), the authors demonstrated that human bone marrow MSCs (BM-MSCs) express the M2 receptor gene (CHRM2). Moreover, the authors suggested that expression was dynamic given that only half of the BM-MSCs were positive for the M2 protein. Confirmation that BM-MSCs express a functional M2 receptor was demonstrated as stimulation with muscarine increased intracellular calcium (Ca2+) concentration and downregulated production of cyclic adenosine 3′,5′-monophosphate (cAMP). Intracellular Ca2+ and cAMP were previously proven to regulate MSC proliferation and differentiation [51].
Summary of Muscarinic Acetylcholine Receptors in Mesenchymal Stem Cells
AD-MSCs, adipose-derived MSCs; BM-MSCs, bone marrow MSCs; FM-MSCs, fetal membrane MSCs; MSCs, mesenchymal stem cells; RD-MSCs, reaming debris-derived MSCs; SGDCs, salivary gland-derived stem cells; UC-MSCs, umbilical cord-derived MSCs;
Furthermore, muscarine induced an increase in the levels of phosphorylation of the extracellular signal-regulated protein kinases 1 and 2 (ERK1 and ERK2) [8]; the ERK1/2 pathway has been linked to control of differentiation, phosphorylating the transcription factors PPARG and RUNX2, to switch of adipose differentiation, and turn on osteogenesis [52 –54]. These data imply that the M2 receptor activates downstream signaling pathways that govern MSC proliferation and differentiation.
The expression of M1, M2, and M3 receptor genes (CHRM1, CHRM2, and CHRM3) in BM-MSCs has also been reported [55]. Upregulated expression of these receptors after treatment with erythropoietin under both normoxic and hypoxic conditions was reported in BM-MSCs and likely marked the induced neuronal-like cell differentiation. Treatment of BM-MSCs with ACh led to an increase in concentration of intracellular Ca2+, which was hypothesized to be mediated by M1 and M3 receptors and further influenced the phospholipase C and inositol-1,4,5-triphosphate (IP3) signaling axis. Although it was hypothesized that the effects of ACh on BM-MSCs are mediated by the M1 and M3 receptors, the influence of other mAChR subtypes (e.g., M2) was not investigated in detail.
The M2 and M4 receptors, despite being thought to modulate inhibitory signaling pathways, have been shown to stimulate phospholipase C activity [56]. In addition, ACh is a universal cholinergic agonist, and therefore, the influence of the nAChRs could also not be excluded.
The expression of M2 and M3 receptor genes (CHRM2 and CHRM3) was reported in a study exploring mAChR expression in human BM-MSCs, induced pluripotent stem cells (iPSCs) and MSCs derived from human iPSCs (iPS-MSCs) [28]. Interestingly, the M2 gene (CHRM2) is expressed in native iPSCs and during the differentiation phase into iPS-MSCs, but it was not detected at the end of the differentiation period. This implied that MSCs generated from iPSCs lose M2 expression. Consistent expression of the M2 receptor gene (CHRM2) in BM-MSCs was, however, observed despite donor-dependent variability. Expression of the M3 receptor gene (CHRM3) also varied during the differentiation process into iPS-MSCs. M3 receptor gene (CHRM3) expression was detected in native iPSCs, decreased during the differentiation process, and increased again at the end stage of differentiation into iPS-MSCs, unlike the BM-MSCs, where the M3 receptor gene (CHRM3) was clearly expressed.
The authors suggest that the variation in the expression profile among the different cell types might contribute to different signaling capabilities, which in turn may lead to their differing biological characteristics. Variation in mAChR expression pattern between passages and upon differentiation of MSCs has indeed been reported in a study investigating human BM-MSCs [57]. Real-time PCR shows downregulation in the expression of the M1 (CHRM1) and M5 (CHRM5) receptor genes in consecutive passages as well as during both osteogenic and adipogenic differentiation. Furthermore, the study reported that treatment with atropine, a general muscarinic antagonist, significantly upregulated the expression of the M4 receptor gene (CHRM4) during adipogenic differentiation of BM-MSCs.
Expression of the M3 receptor was detected at both the mRNA and protein levels in mouse BM-MSCs [58]. The M3 receptor was localized primarily to the endoplasmic reticulum in the investigated BM-MSCs and as such was not competent to signal. This was confirmed in agonist studies. It may be the case that during differentiation, membrane translocation occurs and enables functional M3 receptor signaling. However, this hypothesis was not investigated further. Rat BM-MSCs have been shown to express M1 and M4 receptors at the protein level and the M1 receptor was found to be localized in both the cytoplasm and cell membrane [59]. Interestingly, fluorescence-activated cell sorting analysis only showed that a third of the rat BM-MSCs expressed the M1 receptor. Treatment with ACh, a universal cholinergic agonist, caused enhanced migration of rat BM-MSCs in a dose- and time-dependent manner with no effect on proliferation.
The effect of ACh on rat BM-MSC migration was hypothesized to be mediated by the M1 receptor using atropine, a general mAChR antagonist. Indeed, activation of the M1 receptor was shown to trigger the ERK1/2 and protein kinase C signaling pathways with release of Ca2+, which in turn regulated migration [59].
Adipose-derived MSCs
Human adipose-derived MSCs (AD-MSCs) express the M1 and M2 receptor genes (CHRM1 and CHRM2), and the expression of both was upregulated following cardiogenic differentiation and denoted as markers for cardiomyocytes [60]. Interestingly, native AD-MSCs only express the M2 receptor gene (CHRM2), while expression of the M1 receptor gene (CHRM1) was only detected once AD-MSCs were differentiated into cardiomyocytes. Another study demonstrated changes in the pattern of mAChR gene expression upon AD-MSC differentiation [61]. Expression of all five mAChR genes (CHRM1–CHRM5) fluctuated throughout the differentiation of AD-MSCs into cells that expressed neural proteins. Expression levels of the M1 (CHRM1), M3 (CHRM3), and M4 (CHRM4) receptor genes rose during the differentiation process.
In contrast, expression levels of the M2 (CHRM2) and M5 (CHRM5) receptor genes declined during the differentiation process; however, expression of the M2 (CHRM2) receptor gene recovered toward the end of differentiation. Furthermore, AD-MSCs isolated from rats express functional M2 receptor [9]. Rat AD-MSCs have been demonstrated to express the M1 (CHRM1), M2 (CHRM2), and M3 (CHRM3) receptor genes and expression of the M2 receptor was confirmed at the protein level. Stimulation of AD-MSCs with arecaidine propargyl ester hydrobromide (APE), a selective M2 agonist, caused autocrine upregulation of expression of the M2 gene (CHRM2). In addition, activation of the M2 receptor inhibited AD-MSC proliferation, migration, and the cell cycle.
However, these effects were reversed when the agonist was withdrawn. Selectivity of APE for the M2 receptor in AD-MSCs was also confirmed using methoctramine, an antagonist with preference for the M2 receptor. In addition, activation of the M2 receptor resulted in downregulated expression of key genes involved in cell proliferation and migration (cyclinD1, PCNA, c-jun, PDGFR-β, CXCR4, and CXCR7). These findings agree with the hypothesized role of the M2 receptor as an inhibitory mAChR and suggest that M2 receptor activation places AD-MSCs in a quiescent state.
Salivary gland-derived stem cells
Two studies isolated the M3 receptor protein from porcine salivary gland-derived stem cells (SGDCs) [62,63]. Both studies reported an increase in intracellular Ca2+ activity upon stimulating the porcine SGDCs with carbachol and suggested that this effect is mediated through the M3 receptor. However, carbachol is an ACh analogue that can mimic the effect of ACh on both mAChRs and nAChRs. Both studies viewed the M3 receptor as a salivary gland marker of generated salivary gland organoids and do not report expression of other AChRs, or present data for the selectivity of carbachol to the SGDC M3 receptor.
Reaming debris-derived MSCs
MSCs extracted from reaming debris (RD-MSCs) of male and female patients with osteoporosis, and MSCs from healthy donors, have been differentiated down osteogenic, chondrogenic, and adipogenic lineages, and the expression of mAChR genes was shown to be differential and dynamic [64]. Indeed, only the M4 (CHRM4) and M5 (CHRM5) receptor genes were expressed in RD-MSCs isolated from male donors, while female donors expressed the M2 (CHRM2), M4 (CHRM4), and M5 (CHRM5) receptor genes. RD-MSCs from female donors with osteoporosis showed no differences in mAChR expression profile to RD-MSCs from healthy female donors. However, expression of the specific subtype of mAChRs showed a degree of subject specificity in both undifferentiated RD-MSCs and RD-MSCs differentiated down specific lineages. This was hypothesized to be related to a donor-specific condition. However, the observed differences in mAChR expression by RD-MSCs pre- and postdifferentiation are compelling evidence for a role of mAChRs in regulating MSC differentiation.
Fetal membrane MSCs
In a study that investigated mAChR gene expression in MSCs isolated from human fetal membrane (FM-MSCs), the authors report variation in the expression pattern of mAChR genes between passages and upon differentiation of FM-MSCs [57]. Indeed, by passage 3, FM-MSCs demonstrated upregulated expression of the M1 (CHRM1) receptor gene in addition to differentiation down both osteogenic and adipogenic lineages. Expression of the M2 (CHRM2) receptor gene was downregulated during differentiation of FM-MSCs down an osteogenic lineage and expression of the M3 (CHRM3) receptor gene was maintained throughout the differentiation process. Treatment of FM-MSCs with atropine, a general muscarinic antagonist, enhanced their viability and upregulated expression of the M1 (CHRM1) receptor gene during osteogenic differentiation. However, atropine treatment had no effect on the ability of FM-MSCs to differentiate down adipogenic or osteogenic lineages.
The authors suggest that the M1 receptor may play an important role in differentiation of FM-MSCs. However, without selective stimulation or knockout experiments, it remains unclear which mAChRs are functional in FM-MSCs as atropine is a general muscarinic antagonist that can act on all mAChRs.
Umbilical cord-derived MSCs
Expression of the M2 (CHRM2), M3 (CHRM3), and M4 (CHRM4) receptor genes was detected in hMSCs derived from the umbilical cord (UC-MSCs) [65]. Stimulation of UC-MSCs with ACh induces an intracellular Ca2+ response. The authors indicated that the ACh-induced response is mediated by the M3 receptor through the phosphoinositide 3-kinase (PIK3) axis. The authors viewed the M3 receptor as the best candidate to investigate Ca2+ intracellular signaling mediated by the PIK3 axis. This is based on how mAChRs, naturally, couple to G protein and mediate downstream signaling. The M1, M3, and M5 receptors that couple to G proteins are known to influence Ca2+ mobilization, while M2 and M4 couple to G proteins that inhibit adenylate cyclase.
Indeed, the authors show data demonstrating that a selective M3-antagonist abolished the induced effects of ACh on UC-MSCs [65]. Interestingly, the authors also reported the ability of a PIK3 inhibitor to abolish the induced effects of ACh on UC-MSCs, suggesting that the PIK3 inhibitor might function by obstructing the ACh binding site of the M3 receptor.
In summary, the above studies suggest the involvement of mAChRs in activating signaling pathways that regulate MSC function. For example, the M2 receptor has been suggested to activate the ERK1/2 pathway in BM-MSCs [8], while the M1 and M3 receptors influence the IP3 signaling axis of the same MSC type [55]. These data imply that mAChRs can activate downstream signaling pathways that govern MSC function. However, only one study in AD-MSCs has to date provided direct evidence for a role of mAChRs (the M2 receptor) in inhibiting proliferation, migration, and the cell cycle [9].
Expression of nAChRs in MSCs
Bone marrow MSCs
Expression of the nAChR subunits has been reported in MSCs (Table 2). In BM-MSCs, Hoogduijn et al. detected gene and protein expression of the α3, α5, and α7 nAChR subunits [8]. Confirmation of functional nAChR expression was determined by stimulation with nicotine, which led to an increase in intracellular Ca2+ and an increase in the levels of phosphorylation of ERK1 and ERK2. However, this was observed in only half of the BM-MSC population upon stimulation with nicotine. It was, however, suggested that the nicotine-induced effects are mediated through the α7 nAChR in BM-MSCs as the study showed an increase in levels of phosphorylated ERK in C3H10T1/2 cells (functionally similar cells to MSCs), transfected with the α7 nAChR construct, after stimulation with nicotine. Although nicotine is a general nicotinic agonist, it remains unclear if the other nAChRs, the authors identified to be expressed by BM-MSCs, could have contributed to these observations.
Summary of Nicotinic Acetylcholine Receptors in Mesenchymal Stem Cells
iPS-MSCs, induced pluripotent stem cell-derived MSCs;
Variation in the expression profile of nAChR genes has been reported between human BM-MSCs, iPSCs, and iPS-MSCs [28]. Native iPSCs do not express the α1 subunit (CHRNA1) gene; however, both iPS-MSCs and BM-MSCs express transcripts of this gene. The genes for the α3, α4, α5, α7, α9, and β1 subunits (CHRNA3, CHRNA4, CHRNA5, CHRNA7, CHRNA9, and CHRNB1) were strongly expressed in iPSCs and during the generation of iPS-MSCs. However, they were only weakly expressed in generated iPS-MSCs. While BM-MSCs showed donor-dependent expression of α4, α5, α7, and β1 subunit genes (CHRNA4, CHRNA5, CHRNA7, and CHRNB1), the β2 and β4 subunit genes (CHRNB2 and CHRNB4) were only expressed at low levels in iPSCs and during the generation of iPS-MSCs. Differential expression profiles of nAChR genes have been reported in hMSCs [66].
Gene expression of the α1, α2, α3, α4, α5, α7, α9, β2, β3, and β4 subunits (CHRNA2, CHRNA3, CHRNA4, CHRNA5, CHRNA7, CHRNA9, CHRNB2, CHRNB3, and CHRNB4) was confirmed in hMSCs [66]. Further analysis confirmed protein expression of the α7, β2, and β4 nAChR subunits. The study also provided evidence of functional nAChRs in hMSCs through stimulation with nicotine. Indeed, treatment with 1 μM nicotine or less induced spontaneous migration of hMSCs; however, higher doses (>1 μM) caused cell death. Furthermore, the study provided evidence that nicotine inhibits the growth factor C3a- and basic fibroblast growth factor-induced migration in hMSCs. Moreover, the study provided data showing that the nicotine-induced effects are mediated through the α7 nAChR in hMSCs. Indeed, the α7 nAChR selective antagonist α-bungarotoxin (α-BTX) was shown to abolish the inhibitory effects of nicotine.
The study also provides in vivo data demonstrating impaired migration of transplanted hMSCs to the bone marrow and spleen in mice as a result of nicotine exposure. Indeed, in a separate study, it has been suggested that higher doses of nicotine cause apoptosis and impair proliferation, while at nontoxic concentrations it decreases the migratory potential of MSCs [67].
A functional heteropentameric α4β2 nAChR has been reported in rat BM-MSCs [68]. Indeed, stimulation with nicotine suppressed the osteogenic potential of rat BM-MSCs in a concentration-dependent manner. Nicotine (>0.1 μM) had a negative effect on the expression of osteogenesis markers such as Runx2, BSP, Col1, and OCN. Higher concentrations of nicotine (10 μM) significantly inhibited mineralization of differentiated rat BM-MSCs. The authors indicated that suppressed rat BM-MSC osteogenesis occurs due to nicotine promoting the activity of the angiotensin-converting enzyme (ACE) and activating the bone renin–angiotensin system (RAS). This was confirmed using dihydro-β-erythroidine, a selective inhibitor of the α4β2 nAChR, which partially counteracted the nicotine-induced expression of ACE and activation of the RAS.
In a separate in vivo study, expression of a functional α7 nAChR in rat BM-MSCs has been reported [69]. The study showed that nicotine impaired the ability of BM-MSCs to repair cartilage defects in rats. Indeed, nicotine suppressed chondrogenic differentiation of rat BM-MSCs as evidenced by reduced Safranin-O staining of newly formed cartilage tissue. In addition, nicotine inhibited expression of chondrogenic markers such as Col2A1 and Sox9 in rat BM-MSC regenerated tissue. The authors indicate that the α7 nAChR mediates nicotine's ability to downregulate Col2A1 expression by suppressing its upstream effector Sox9. Indeed, the study provides evidence of the involvement of the α7 nAChR in mediating the effect of nicotine as methyllycaconitine, a specific α7 nAChR antagonist, inhibited nicotine-induced Ca2+ influx in rat BM-MSCs. Furthermore, repressed BM-MSC chondrogenesis is thought to occur through the Ca2+/calcineurin/NFATc2 signaling pathway upon nicotine stimulation.
The study demonstrated that nicotine decreased cytoplasmic dephosphorylated NFATc2 with concomitant nuclear translocation of NFATc2 in response to an increase in intracellular Ca2+. NFATc2 is capable of binding to the Sox9 promoter, thus decreasing Sox9 expression. These nicotine-induced effects were abolished when BM-MSCs were pretreated with methyllycaconitine, indicating the involvement of the α7 nAChR in attenuating the Ca2+/calcineurin/NFATc2 signaling pathway.
Adipose-derived MSCs
Human AD-MSCs express both the α and β nAChR subunits [61]. Expression of the α7 and β4 nAChR subunits (CHRNA7 and CHRNB4) was upregulated, while the α3, α6, and β2 nAChR subunits (CHRNA3, CHRNA6, and CHRNB2) were significantly downregulated in neuronal differentiated AD-MSCs. Functional nAChRs were confirmed as nicotine induced an increase in intracellular Ca2+, most significantly when AD-MSCs underwent neuronal differentiation. However, no data eluding to which nAChRs mediate the effect of nicotine are reported. In another study, AD-MSCs derived from rats were found to express a functional α7 nAChR [70]. Indeed, stimulation with ICH3, a selective α7 nAChR agonist, inhibits rat AD-MSC proliferation. Further analysis confirms that the α7 nAChR inhibits AD-MSC proliferation by promoting cell cycle arrest through downregulation of Cyclin D1 expression.
However, activation of the α7 nAChR significantly enhanced rat AD-MSC migration through upregulation of CXCR4, a chemokine receptor that also mediates cellular migration. Both these effects could be counteracted using α-BTX, an α7 nAChR selective antagonist. Interestingly, ICH3 treatment of AD-MSCs also increased the protein expression of the M2 mAChR, suggesting a potential cross-interaction mechanism between mAChR and nAChR.
Periodontal ligament-derived MSCs
Human periodontal ligament-derived MSCs (PDL-MSCs) have been reported to express the α7 and β4 nAChR subunit genes (CHRNA7 and CHRNB4) and nicotine stimulation amplifies their expression [71]. Nicotine stimulation had a negative impact on PDL-MSC viability in a dose-dependent manner. Higher concentrations of nicotine (>100 μM) were associated with increased DNA fragmentation in PDL-MSCs and accumulation of cells in the subG1 phase of the cell cycle, the phase associated with apoptosis. Indeed, nicotine in millimolar levels activated apoptotic pathways in PDL-MSCs and increased expression of p53, a proapoptotic marker, was evident after only a 30-min treatment with 10 mM nicotine. This was associated with a decrease in levels of the Bcl-2 antiapoptotic protein and an increase in the well-known apoptotic marker caspase-3.
However, the nicotine-induced apoptosis was blocked when PDL-MSCs were pretreated with α-BTX, the aforementioned α7nAChR-specific antagonist. Thus, confirming the role of the α7 nAChR in mediating the nicotine-induced effects on apoptosis in PDL-MSCs. Data from a study by Zhou et al. confirmed that PDL-MSCs express a functional α7 nAChR and nicotine inhibited PDL-MSC proliferation in a dose-dependent manner [72]. Moreover, stimulation with nicotine dose dependently impaired osteogenic differentiation of PDL-MSCs. Indeed, differentiated PDL-MSCs showed significant decreases in bone mineralization associated with decreased expression of osteogenic genes and protein markers (ALP, OCN, BSP, and RUNX2). However, the nicotine-induced impairment of differentiation was partially reversed by α-BTX, suggesting that the α7 nAChR regulates, to an extent, PDL-MSC osteogenic differentiation. In fact, both gene and protein expressions of the α7 nAChR in osteo-differentiated PDL-MSCs are increased when nicotine is present.
The involvement of the α7 nAChR and cholinergic signaling in the process of osteogenesis has been suggested. In fact, several in vivo and in vitro studies in chick and mouse have reported the involvement of Ach-dependent pathways regulating skeletogenesis and bone development [73 –75]. In which it was suggested that α7 nAChR mediated the nicotine inhibitory effects on cartilage and bone formation [74]. All which are supportive of a role for cholinergic regulation in bone development. In addition, nicotine-stimulated nAChRs can initiate relevant downstream signaling pathways. Indeed, it was shown that the α7 nAChR mediates the downstream effects of nicotine through the Wnt/β-catenin pathway in PDL-MSCs [72]. Nicotine stimulation of PDL-MSCs leads to a decrease in protein expression of Wnt-related factors, DKK-1 and GSK-3β, and an increase in the expression of active-β-catenin protein. The latter has been previously shown in a separate study to suppress PDL-MSC osteogenic differentiation [76]. However, in the presence of α-BTX, all these effects were reversed, again providing further evidence of a functional α7 nAChR modulating the Wnt/β-catenin pathway in PDL-MSCs [72].
Wharton's jelly MSCs
Human Wharton's jelly MSCs (WJ-MSCs), derived from the mucous connective tissue between the amniotic epithelium and the umbilical vessels found in the umbilical cord, express genes encoding the α3, α5, α7, β2, and β4 nAChR subunits (CHRNA3, CHRNA5, CHRNA7, CHRNB2, and CHRNB4) [77]. WJ-MSCs, in response to nicotine treatment, demonstrate significantly decreased proliferation but no change in viability, apoptosis, or necrosis. The chondrogenic differentiation capacity of WJ-MSCs was impaired, to an extent, by nicotine. Indeed, while nicotine treatment did not affect the collagen output of differentiated WJ-MSCs, it did impair the quality of the collagenous matrix formed, as determined by the proteoglycan content. This was asserted to be due to the downregulated expression of chondrogenic markers, including Sox9, Col2a1, and aggrecan. The authors suggested that the α7 nAChR mediated the nicotine-induced effects in WJ-MSCs as it induced Ca2+ influx into the cells.
In a separate study, human WJ-MSCs were confirmed to express a functional α7 nAChR as well as the α3 and α9 nAChR subunit genes (CHRNA3 and CHRNA9) [78]. Furthermore, injection of human WJ-MSCs into α7 nAChR-deficient mice demonstrated improved episodic memory and suggests the increased regenerative potential of WJ-MSCs to improve cognitive functions through the α7 nAChR.
Reaming debris-derived MSCs
Expression profile data available for nAChRs provide interesting insight into variation dependent upon sex and health of RD-MSC donors [64]. For example, the α5, α7, and α9 nAChR subunit genes (CHRNA5, CHRNA7, and CHRNA9) were expressed in RD-MSCs isolated from all donor groups (male and female, healthy and diseased). In contrast, the α2, α6, and α10 nAChR subunits (CHRNA2, CHRNA6, and CHRNA10) were only expressed in RD-MSCs isolated from diseased female donors, and the α3 nAChR subunit gene (CHRNA3) was only expressed in RD-MSCs isolated from diseased male donors. The study also reported variations in the expression of certain nAChR subunits between different differentiated lineages. However, the α7 subunit gene (CHRNA7) was expressed in RD-MSCs differentiated down all lineages (osteogenic, chondrogenic, and adipogenic lineages).
Furthermore, the α3 subunit gene (CHRNA3) was expressed in adipocytes generated from female RD-MSC donors; but not by adipocytes generated from male RD-MSC donors. Therefore, it can be concluded that the expression profile of the nAChR subunits is dynamic in RD-MSCs.
In summary, the aforementioned studies provide evidence that MSCs express functional nAChRs that have been shown to mediate the impact of nicotine. These in turn activate signaling pathways such as the ERK1/2 [8], Ca2+/calcineurin/NFATc2 [69], and Wnt/β-catenin pathways that are involved in MSC function [72]. However, there is limited direct evidence for a role of a specific nAChR in translating the effect of nicotine. One example of a demonstrated direct effect is in the case of the α7 nAChR, which appears to mediate nicotine inhibitory effects on hMSC migration [66]. In addition, impaired chondrogenic differentiation of BM-MSCs in response to nicotine has been demonstrated to be mediated through α7 nAChR [69].
Interestingly, the α7 nAChR was shown to inhibit AD-MSC proliferation, however, enhances migration [70]. In PDL-MSCs, the α7 nAChR mediates nicotine-induced apoptosis [71], and to an extent impaired these cells' ability to undergo osteogenic differentiation [72]. However, in BM-MSCs, the α4β2 nAChR was shown to suppress cell osteogenic potential in response to nicotine [68].
Conclusion
The studies presented in this review show the widespread expression of AChRs in MSCs and demonstrate the involvement of these receptors in MSC function. It appears that AChR expression in MSCs is dynamic, dependent on the type of MSCs, and can be individually based on donor or differentiation lineage. However, expression of all mAChR subtypes has been identified in most of the MSCs studied, as well as in both the α and β nAChR subunits. Furthermore, consistent expression of a particular AChR subtype across different types of MSCs has been observed. For example, the M3 mAChR and α7 nAChR are expressed in multiple types of MSCs. This may suggest central roles of these subtypes in regulating MSC function.
Downstream signaling of AChRs in MSCs
Some of the presented studies examined the downstream effects of AChR activation on MSC regenerative potential. In stem cell therapy, the regenerative output is determined by the ability of the cells to migrate, proliferate, and differentiate. The presented findings show involvement of major pathways involved in regulating these functions. In the MAPK/ERK pathway, phosphorylation of ERKs is known to regulate proliferation and differentiation of stem cells [79]. As described herein, both mAChR and nAChR were shown to be involved in triggering this signaling pathway cascade in MSCs. Indeed, as AChR activation facilitates downstream signaling pathways that influence cell homeostasis, they have the capability to influence the regenerative potential of MSCs. However, it is the presence of a subtype functional AChR that determines specific downstream signaling cascade activation and determines cell fate. In the case of mAChRs, downstream signaling is mostly dependent on the coupled G protein (Fig. 2).
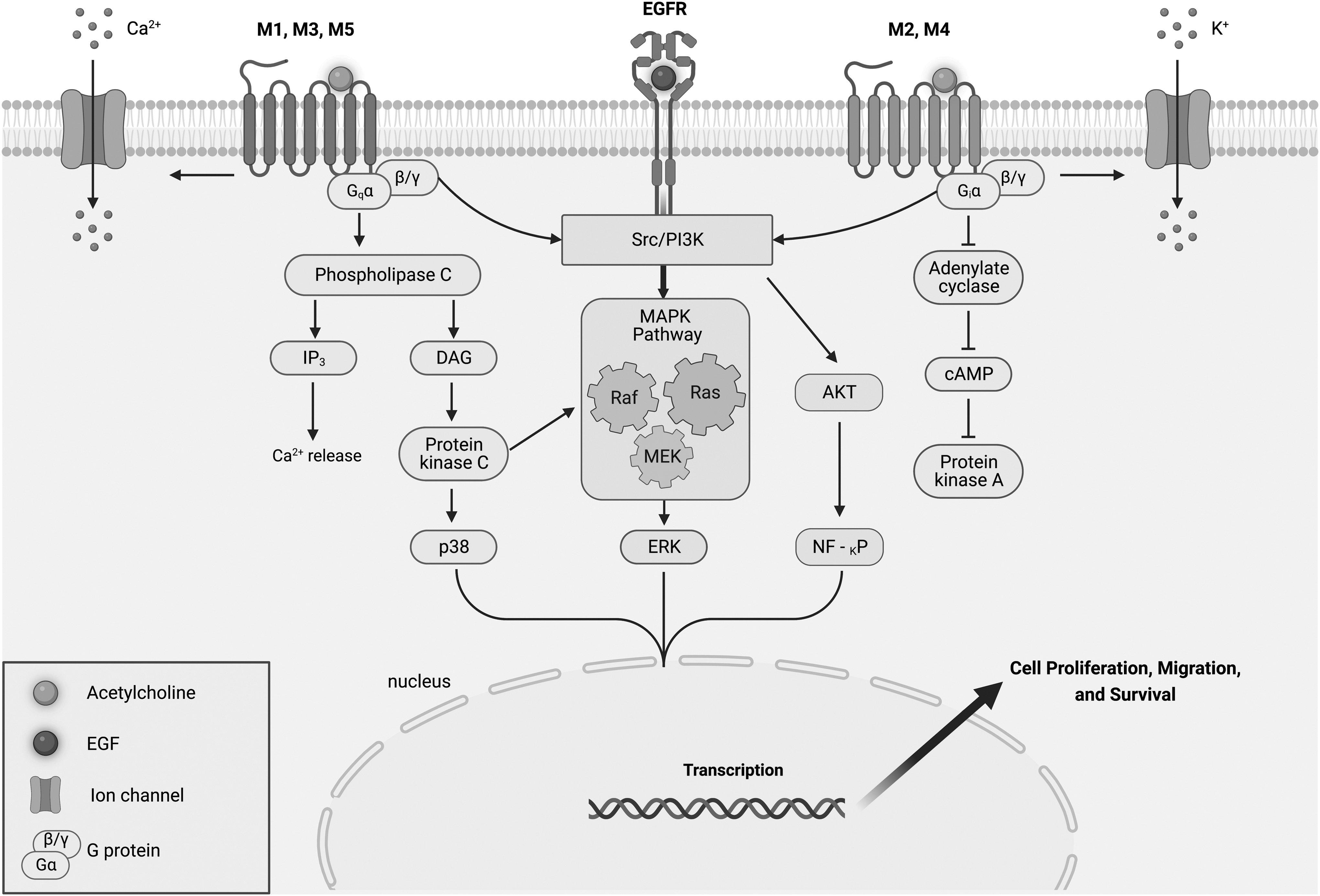
Metabotropic signaling of muscarinic receptors. Upon stimulation with ACh, or a subunit-specific agonist, M1, M3, and M5 receptors activate PLC resulting in downstream PKC activation and an increase in IP3 and Ca2+ levels. PKC can also activate the MAPK cascade and ERK1/2. M2 and M4 subtypes inhibit the activity of the adenylyl cyclase, leading to a decrease in intracellular cAMP. In addition, mAChRs can activate ion channels. Common pathways for all mAChRs are the activation of ERK1/2 through an Src/PI3K pathway. The M2 subtype may also modulate Akt signaling by means of upstream PI3K activation, influencing transcription factors regulating proliferation. cAMP, cyclic adenosine 3′,5′-monophosphate; ERK, extracellular signal-regulated protein kinase; IP3, inositol-1,4,5-triphosphate; PKC, protein kinase C; PLC, phospholipase C.
Indeed, the stimulatory group of mAChRs (i.e., M1, M3, and M5) coupled to Gq proteins mainly influences Ca2+ influx in MSCs. While the inhibitory group of mAChRs (M2 and M4) coupled to Gi proteins mainly influences cAMP production. Furthermore, both groups of mAChRs share a common downstream pathway for ERK1/2 activation. Dependent on the mAChR subtype, this can result in promoting MSC function, for example, M1 regulating BM-MSC migration [59], or inhibiting MSC growth, for example, M2 inhibiting AD-MSC proliferation [9].
In the case of nAChRs, these receptors are mainly gated ion channels, for example, the most characterized in MSCs is the α7 nAChR, which modulates intracellular Ca2+ concentration. Most of the reviewed studies examined downstream effects of nAChR activation through nicotine. It may have been the focus of these studies to determine the impact of nicotine on the overall potential of treated MSCs. Indeed, nicotine had an overall negative impact on MSC regenerative potential. The majority of the studies identified the α7 nAChR in mediating nicotine's effects on MSCs. The consequent changes in the intracellular Ca2+ concentrations can initiate several signaling cascades (Fig. 3). This can involve MAPK effectors, through the PI3K pathway or in conjugation with other pathways, such as the calcineurin/NFATc2 or Wnt/β-catenin pathways.
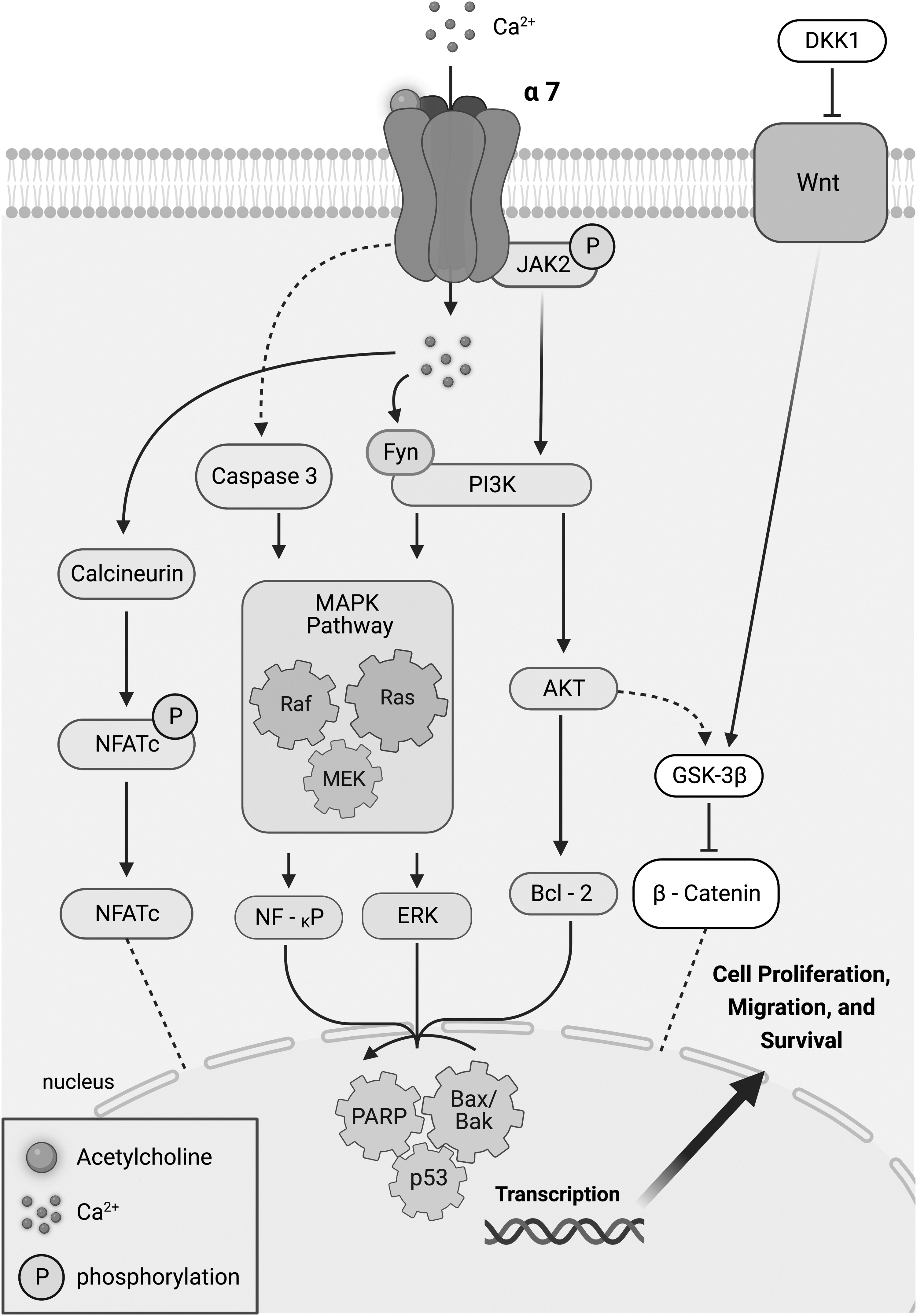
Diagram depicting the α7 nAChR signaling pathway. Upon stimulation with ACh, or a subunit-specific agonist, nAChRs increase cytosolic Ca2+ concentration, initiating several signaling cascades. The Ca2+ influx with activation of calcineurin can induce NFATc dephosphorylation and translocation back to the nucleus. This recruits transcription factors that govern cell differentiation. Similarly, the α7nAChR, through the Wnt/AKT pathway, can translocate β-catenin into the nucleus and subsequently activate expression of target genes that modulate differentiation. Common signaling pathways activated by nAChRs include the MAPK through the PI3K pathway. This can occur in Fyn-dependent manner or by means of upstream phosphorylation of JAK2. In addition, inhibition of JAK2 activates caspase 3, leading also to activation of the MAPK downstream signaling pathway. Later effectors such as NF-KP, ERK, and Bcl-2 signal nuclear transcription factors (e.g., PARP, Bax/BaK, and p53) were found to play a role in the downstream signaling of α7 nAChR activation. Depending on the downstream signaling pathways being activated, these pathways can impinge on transcription factors that control synthesis and repair of DNA or promote apoptosis; consequently, influencing cell proliferation, migration, and survival.
In most of the studies, this results in inhibition of MSC growth or even in initiation of apoptosis. While other studies show inhibition or suppression of MSC potential to differentiate as seen with chondrogenic differentiation of BM-MSCs [69] and osteogenic differentiation of PDL-MSCs [72].
AChRs as a potential target to regulate MSC function
The evidence suggests that AChRs may present as a promising therapeutic target to control the regenerative potential of MSCs. Indeed, the reviewed studies presented data for subtype-selective AChR agonist and antagonists in manipulating stem cell function. For example, the selective M2 agonist APE placed AD-MSCs in a quiescent state without affecting the viability of the cells [9]. This may be favorable during transplant and for directing in vivo regeneration. Migration, an important function of stem cell therapy during in vivo regeneration, can be controlled through AChRs. Indeed, the M2 selective agonist APE suppressed AD-MSC migration through the M2 mAChRs [9]. Another promising option is to block undesirable AChR function through selective and non-selective agonists. For instance, atropine the general mAChR antagonist was able to enhance FM-MSC viability by blocking the M1 receptor [57]. It is not unexpected that mAChRs have this central role in MSCs; these receptors were shown to be influential in many body systems, for example, nervous, cardiovascular, and muscular [80].
As mAChRs are GPCRs, they belong to the most successful therapeutic targeted family of proteins that continue to be the most prominent aim of biomedical research [24 –26].
Nonexcitable cells are now known to express a plethora of ion channels, and these are also interesting targets for pharmacological intervention. In this study, MSCs were shown to express several subunits of nAChRs, among which there are functional ones that are susceptible to pharmacological stimulation. Indeed, the selective α7 nAChR agonist ICH3 was shown to enhance MSC migration [70]. Likewise, α-BTX, the α7 nAChR selective antagonist, had the ability to block the detrimental effects of nicotine in several MSCs [67,71,72]. These data display an interesting potential for targeting ion channels that influence MSC function. It is also worth mentioning that other classical excitable ion channels are being investigated in the control of MSC phenotype.
Worthy of mention are the Piezo ion channels, which regulate osteogenesis of MSCs by regulating the expression of BMP2 [81] and migration through adenosine triphosphate release [82]. Likewise, of interest are the transient receptor potential ion channels that can influence MSC differentiation [83], cell cycles [84], and survival [81].
ACh signaling, mediated by mAChRs and nAChRs, is indeed involved in regulating MSC function. Targeting both mAChRs and nAChRs with pharmacological agents may therefore reveal novel mechanisms to tune MSC function and thus their regenerative output. Indeed, there is a plethora of AChR agonists and antagonists licensed for the treatment of a variety of diseases [25,80,85,86] and these are yet to be explored with regard to their effects on the regenerative potential of MSCs and the possibility of their repurposing into the regenerative medicine arena. Furthermore, there is more to learn about the metabotropic downstream signaling of mAChRs in MSCs and indeed even if they do play a naturally occurring role in MSC homeostasis or differentiation. It is therefore imperative that more studies are undertaken to investigate the precise functions of AChRs in MSCs.
Footnotes
Acknowledgment
Figures were created with BioRender.com
Author Disclosure Statement
The authors have no relevant financial or nonfinancial interests to disclose.
Funding Information
M.C.B. was funded by GlaxoSmithKline Consumer Health care and the BBSRC Industrial GlaxoSmithKline CASE PhD studentship (BB/V509541/1).