
Abstract
As the body ages, it experiences a gradual decline in the functioning of cells, tissues, and systems, which eventually leads to dysfunction and increased susceptibility to disease. At the cellular level, a reduction in the activity or number of stem cells is an important feature of cell senescence, and such changes may also directly drive the aging of the organism. Thus, finding ways to prevent or even reverse stem cell senescence holds promise for the development of aging therapies in tissues and organisms. This review discusses the relationship between changes in stem cell senescence, tissues aging, and related diseases, focusing on four categories of tissue stem cells: hematopoietic stem cells, mesenchymal stromal/stem cells (MSCs), intestinal stem cells, and muscle stem cells.
Introduction
Cellular senescence and organismal aging are interconnected processes that mutually influence each other. The aging of organisms leads to changes in the internal environment, which in turn activate and promote cellular senescence. Simultaneously, the accumulation and dysfunction of senescent cells contribute to the aging phenotype observed in the organism [1]. This relationship is also reflected in the context of stem cells. In adults, stem cells play a vital role in maintaining tissue regeneration during normal conditions, and elaborate repair mechanisms during stressful conditions. Therefore, any dysfunction or depletion of these cells due to aging can subsequently contribute to a range of tissue and organism dysfunctions, ultimately leading to aging-related diseases [2]. The senescence of stem cells, which are predominantly concentrated within specific tissues (Fig. 1), can impede tissue renewal and impair its overall functionality [3,4].
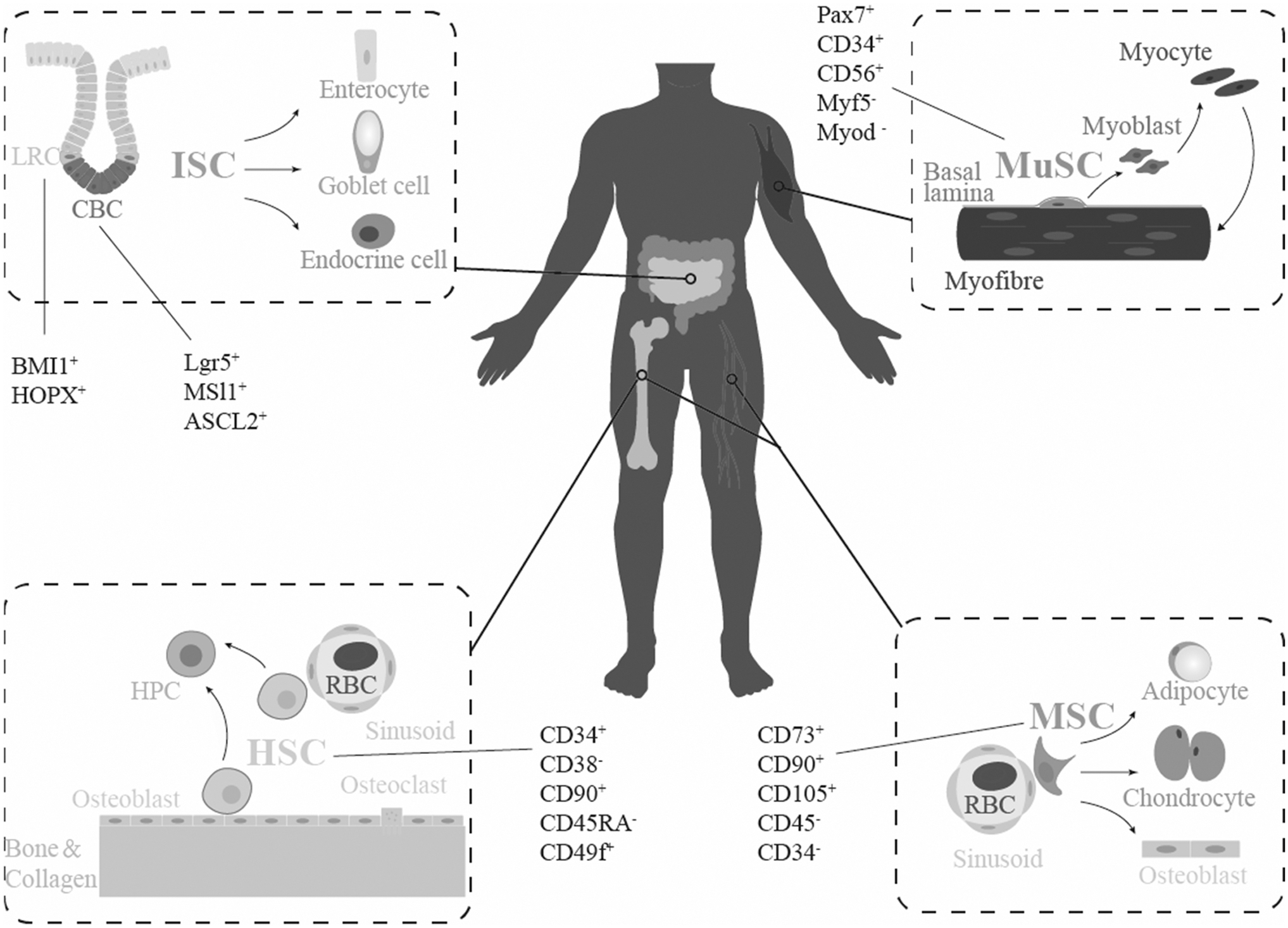
Schematic diagram of the main distribution of HSCs, MSCs, ISCs, and MuSCs in human body, direction of cell differentiation, and major molecular markers. CBC, crypt-base columnar cell; HSCs, hematopoietic stem cells; HPC, hematopoietic progenitor cell; ISCs, intestinal stem cells; LRC, labeled lagging cell; MSCs, mesenchymal stromal/stem cells; MuSCs, muscle stem cells; RBC, red blood cell.
The mechanisms by which stem cell senescence leads to stem cell dysfunction in different tissues are diverse, but cellular alterations share many commonalities, including the disruption of genomic stability due to the accumulation of DNA damages, metabolic imbalances resulting from excessive or inadequate autophagy, and the damage to macromolecules due to elevated levels of reactive oxygen species (ROS). For instance, ionizing radiation causes DNA damage that activates Ataxia telangiectasia mutated (which is often activated by DNA double-strand breaks) and Ataxia telangiectasia and Rad3-related protein (which are activated by different types of DNA damage), both of which phosphorylate downstream p53 to prevent ubiquitination degradation and activate downstream pathways. p53 can activate genes that cause apoptosis, like p53 up-regulated modulator of apoptosis (PUMA) and BCL2-Associated X Protein, which causes apoptosis. In addition, the activation of p21 by p53 induces cell cycle arrest into cell senescence [5,6].
Certainly, the sensitivity of different types of stem cells to various risk factors can be influenced by their unique microenvironments and external stimuli patterns. Consequently, the effectiveness of treatment strategy may vary among stem cells due to these individual mechanisms and sensitivities.
It is crucial to consider these variations to optimize treatment outcomes. To better understand the impacts of senescence on various stem cells, this review will discuss the aging changes connected to stem cells from various tissues, the intrinsic mechanisms, and the associated pathway of aging salvage. Hematopoietic stem cells (HSCs), mesenchymal stromal/stem cells (MSCs), intestinal stem cells (ISCs), and muscle stem cells (MuSCs) are the four primary categories of adult stem cells that we primarily covered.
Senescent HSCs and Age-Related Diseases
HSCs are a type of adult stem cells that possess the ability to self-renew and undergo differentiation into various types of blood cells (erythrocytes, myeloid leukocytes, lymphocytes, platelets). The gradual differentiation of HSCs is typically categorized into three stages of transformation: pluripotent HSCs, directed hematopoietic progenitor cells, and various types of blood cells, with each progression to the subsequent stage. There is a gradual decline in their proliferative potential [7]. Throughout the step-by-step differentiation process, HSCs manifest a variation in their characteristics, displaying heterogeneity and expressing distinct molecular markers. These changes occur as a result of alterations in the niche environment, coupled with genetic and epigenetic modifications prompted by various molecular and cellular stimuli. Molecular biomarkers are valuable tools for categorizing HSCs into distinct groups. Due to species differences, relevant markers between humans and mice may vary, but with similarities. In mice, the most pivotal molecular markers for identifying HSCs are linage−, sca1+, and c-kit+, commonly known as LSK.
The lineage markers encompass CD3e, CD4, CD8, B220, Gr1, Ter119, CD11b, which serve as indicators for HSC differentiation and maturation. Other markers of undifferentiated HSCs are CD150+ CD48− flk2− [8,9]. In humans, HSC is represented by linage− CD34+ CD38− CD90+ CD45RA− CD49f+ as biomarkers [10].
HSCs are accountable for the generation of blood cells in the body. During this process, a single HSC will produce two offspring HSC through unequal division: one to replenish the HSC pool, and the other with a tendency for differentiation that will eventually complete the formation of fully differentiated blood cells [11]. Although new offspring HSCs are formed to fill the parental gap, this process is accompanied by a decline in their ability to differentiate. As the body ages and experiences blood loss due to diseases, the proliferation of HSCs is consistently activated, leading to the accumulation of DNA replication and mitochondrial ROS. In addition to these adverse factors, HSCs undergo senescence and ultimately become depleted, alongside epigenetic modifications and telomere attrition [12 –14].
The senescence of HSCs is strongly linked to the disruption of cell homeostasis, including diminished cell polarity, compromised cell plasticity, and dysregulation of cell metabolism, all of which ultimately impede their capacity for differentiation and self-renewal. The senescence and depletion of HSCs result in the development of anemia, myelodysplastic syndromes, clonal hematopoiesis, and other hematological diseases [15,16].
Furthermore, the senescence of HSCs will serve as the underlying cause of immune system aging disorder [17], and the related dysfunction is manifested as follows: (1) The overall decrease in white blood cell levels, caused by the depletion of HSCs, weakens the immune system and elevates the susceptibility to infectious diseases [18]. (2) The decline in the proportion of lymphoid and B cells further diminishes the level of adaptive immunity, while the increase in myeloid cells proportion raises the expression of inflammatory factors and overall inflammation within the body, consequently leading to inflammatory senescence [19]. (3) Immune system disorders can potentially result in the development of myeloid and lymphoid malignancies, increasing the risk of age-related myeloma, acute myeloid leukemia, and chronic lymphocytic leukemia [20 –22].
The mechanisms responsible for senescence-related dysfunction in HSCs encompass the accumulation of DNA damages, impaired autophagy, gene expression changes, reduced proteasome degradation, and alterations in the bone marrow (BM) niche (Fig. 2). These changes detrimentally affect the differentiation potential and lineage determination of HSCs to varying extents, ultimately leading to HSC dysfunction and the onset of the aforementioned diseases. Although the determinant of HSC senescence has not been well clarified, the adverse environmental stimuli are usually the initial factors of HSC senescence, such as DNA and macromolecular damage caused by ionizing radiation, rapid proliferation of HSC caused by blood loss, and the interference of senescence-associated secretory phenotype (SASP) molecules in niche, which leads to changes in relevant pathways, resulting in the dysfunction of HSC metabolism and homeostasis [8,23,24]. First off, senescent cells develop as a result of DNA damages brought on by environmental stressors such as exogenous toxins and endogenous replication errors over time [25].
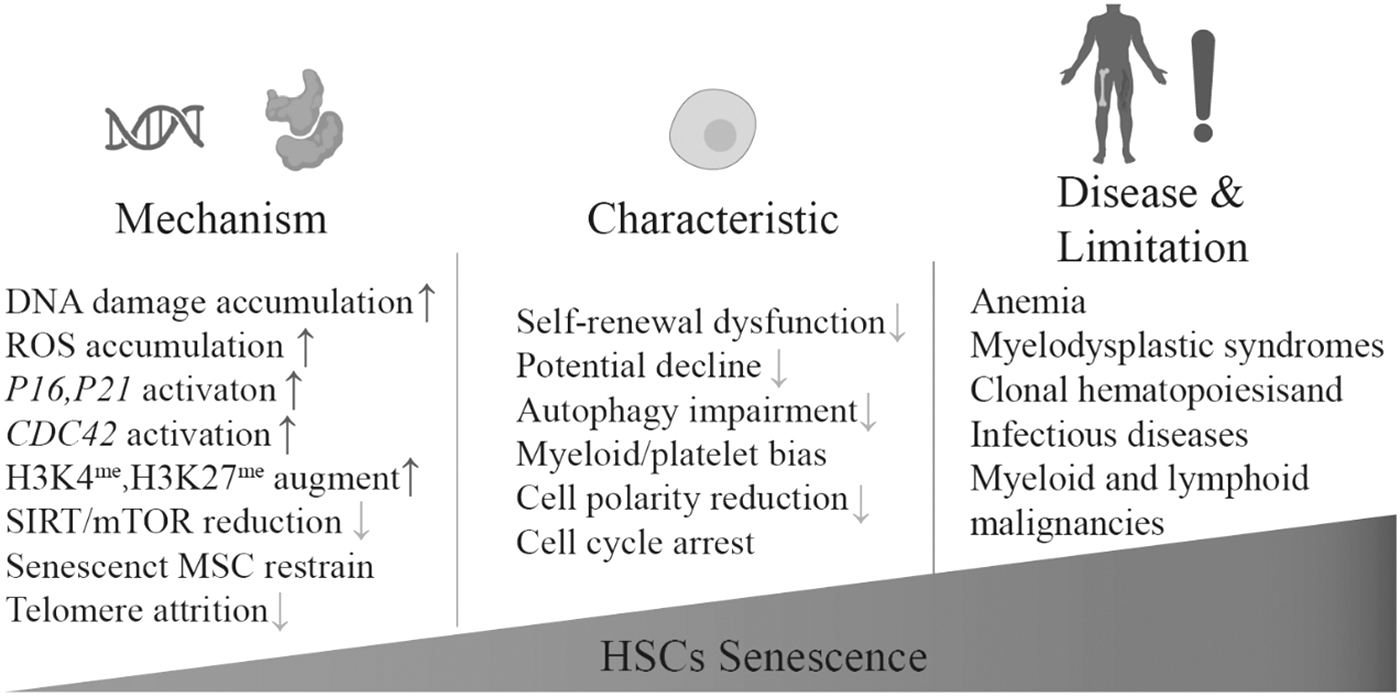
Aging and senescence lead to dysfunction of HSCs, resulting in disorders of the hematopoietic system and immune system, and further leading to age-related diseases like anemia and increased risk of infection. mTOR, mammalian target of rapamycin; ROS, reactive oxygen species.
The occurrence of DNA damage triggers DNA damage response (DDR) and cell cycle arrest through three key pathways: p53–p21 mediates G0/G1 arrest, p16 activation mediates G0/G1 arrest, and p53-PUMA mediates cell apoptosis [8]. In addition, a significant feature of senescent HSCs compared with normal HSCs is the change in the distribution of specific proteins, resulting in a reduction of cell polarity [26]. At the molecular level, Cdc42-Borg4-Septin7 axis has emerged as a crucial factor in governing the polarity and functionality of HSCs. Specifically, the activation of Cdc42 activation is widely recognized as a significant contributor to the occurrence of myeloid/platelet bias and subsequent depletion within HSCs [27].
Subsequently, autophagy maintains normal cell material turnover and organelle renewal, which has a significant impact on maintaining cell function. However, autophagy defects occur in senescent HSCs, leading to the accumulation of damaged organelles represented by mitochondria, resulting in high concentration of ROS damage and metabolic stress [26,28]. Mammalian target of rapamycin (mTOR) pathway is considered a crucial regulatory pathway of autophagy [29].
Studies have shown that the mTOR pathway is activated in senescent HSCs, leading to impaired autophagy in these HSCs. Therefore, maintaining mitochondrial function and inhibiting the mTOR signal are potential strategies for promoting the recovery of HSCs function from the perspective of autophagy [12,30].
Additionally, senescence is characterized by changes in gene expression, including epigenetic modifications, which also occur in HSCs. Senescent HSCs exhibit decreased expression of Sirt1, Sirt3, and Sirt7 genes, which are closely associated with aging-related dysfunction. Furthermore, restoring the expression of these genes can reverse the state of cell senescence state, suggesting that they can be used as indicators of cellular senescence to some extent [31]. In terms of epigenetic changes, it has been observed that compared with young mice, HSCs in older mice display a significant increase in global methylation levels, particularly in H3K4me and H3K21me modifications of genes related to cell cycle regulation and lineage determination [32]. Specifically, the prevalent alterations observed in elderly individuals with myelodysplasia and acute myeloid leukemia involve enzymes, such as DNA-methyltransferase 3A (DNMT3A), enhancer of zeste homolog 2, tet methylcytosine dioxygenase 2, and SET domain bifurcated 1, all of which are closely associated with DNA methylation [33,34].
Importantly, the BM niche shift has a considerable effect on the senescence and exhaustion of HSCs. Relevant studies have found that HSCs from old mice restored their original potential after transplantation into BM of young mice, which illustrates how HSC niche are required for HSCs to play a normal function [35]. As HSCs enter senescence state, the cellular components of niche, mesenchymal cells and endothelial cells, also undergo notable alterations, which further leads to the changes of cytokines in niche, including diminished levels of stem cell factor, Cxcl12, IL7, and Notch ligands [36,37]. At the same time, the inflammatory changes associated with senescence result in increased secretion of inflammatory factors, including C-C motif chemokine ligand 5, interleukin-1β, tumor necrosis factor ligand superfamily member 2, interferon-γ, Wnt5, and transforming growth factor-β (TGF-β). These factors further facilitate the bias of myeloid cells and contribute to the dysfunction of HSC [38].
Senescent MSCs and Age-Related Diseases
MSCs are pluripotent stem cells originating from the mesoderm. Since the discovery of MSCs in the BM in 1968, their application and research have become a hot topic in the field of stem cells [39]. Accompanied by blood vessels, MSCs exist in nearly all tissues, and have differentiation potential for a variety of tissues [40]. Due to their immunosuppression of T cells, low immunogenicity, and lower tumorigenicity, MSCs hold great potential in the field of stem cell-based therapies. In human, MSCs are stably labeled with CD73, CD90, and CD105, as well as a lack of CD45, CD34, CD14, CD19, CD11β, and CD79α [41].
As being senescent, MSCs may exhibit age-related dysfunction, characterized by limitations in their ability to proliferate and a decline in their stemness properties [42] (Fig. 3). In their normal state, MSCs have the remarkable ability to differentiate into various cell types, including osteoblasts (involved in bone formation), adipocytes (involved in fat storage), and chondrocytes (involved in cartilage formation). However, as MSCs enter senescence state, their osteogenic capacity is significantly reduced. Long-term in vitro MSC culture experiments indicated that senescent MSCs decreased adipogenic ability and loss of osteogenic ability, but show differentiation bias of adipogenic ability on the basis of proliferation and differentiation disorders [43,44]. As a result of the reduced osteogenic ability of senescent MSCs, the elderly population is more prone to developing osteoporosis and experiencing difficulties in the healing process after bone fracture. Moreover, this dysfunction also poses limitation of MSCs-based stem cell therapy.
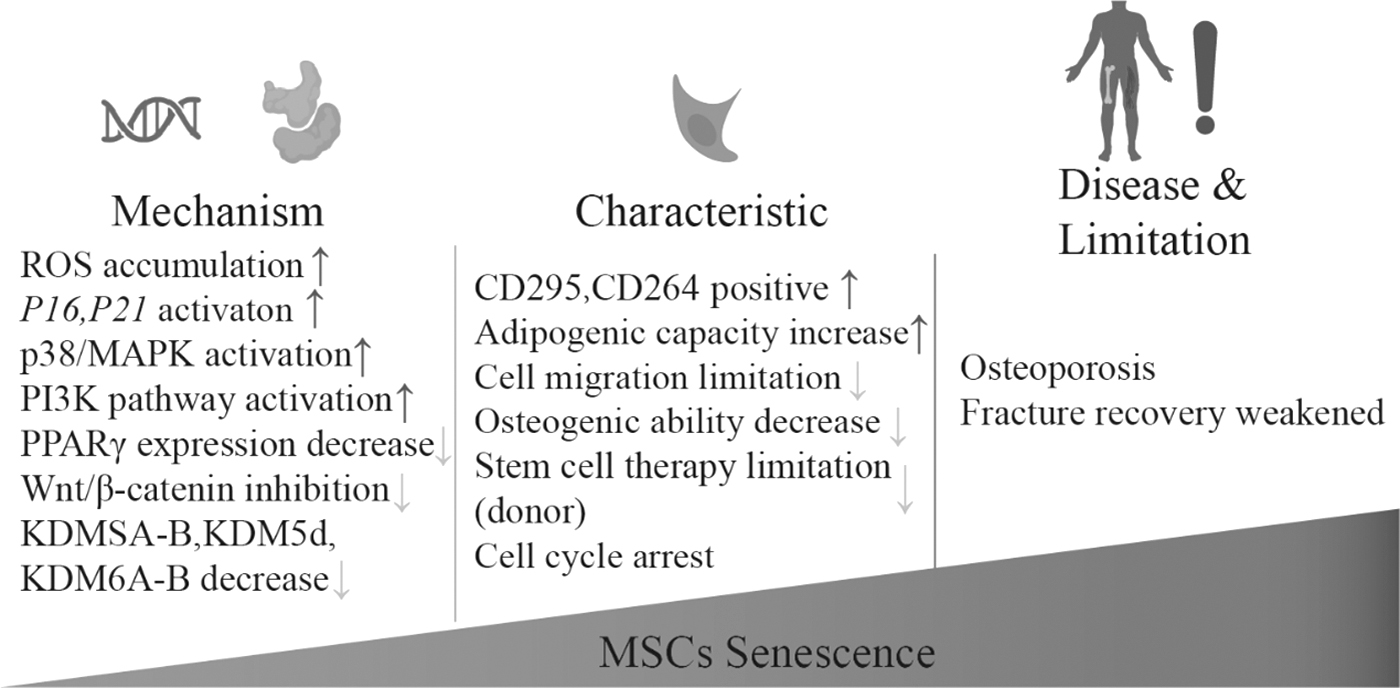
Aging and cellular senescence cause MSC dysfunction, and deficiencies in normal functioning result in age-related diseases such as osteoporosis.
Additionally, MSCs play a crucial role as a component of the HSC niche. When MSCs enter the senescence state, they can activate the inflammatory transcriptome of HSCs through SASP. The activation of SASP can lead to impairment in the function of HSCs, limiting their stemness and inducing premature senescence. Consequently, the impaired hematopoietic system and immune system can be adversely affected by this process [45,46].
In addition to the changes in cell morphology, there are various mechanisms involved in the senescence of MSCs. Senescent MSCs often exhibit flattened cells, nuclear pyknosis (shrinkage), and cytoplasmic granulation. Moreover, there is an increase in the abundance of actin stress fibers and elevated activity of galactosidase (SA-β-Gal) in senescent MSCs. Other molecular marker expression included increased CD295 and CD264 [47].
As MSCs undergo senescence, the expressions of certain surface markers, including CD146 and CD106, are downregulated. CD146, also known as melanoma cell adhesion molecule, is involved in cell adhesion and migration [48]. Its downregulation in senescent MSCs reflects a decline in their ability to migrate and potentially contribute to their reduced regenerative potential. CD106, also known as vascular cell adhesion molecule 1, is involved in immune regulation and the recruitment of cells to inflamed tissues [49]. The downregulation of CD106 in senescent MSCs suggests a decline in their immune regulatory function and impaired ability to attract immune cells to inflammatory sites for tissue repair.
These changes in surface marker expression further contribute to the functional alterations associated with the senescence of MSCs. In senescent MSCs, there is a noticeable increase in the expression of peroxisome proliferator-activated receptor-γ (PPARγ). PPARγ is a significant controller of the formation of adipocytes. This increase in PPARγ expression suggests that MSCs undergo a shift toward an adipogenic capacity [50]. In contrast, the osteogenic ability of MSCs decreased. The osteogenic activity of MSCs is related to various signaling pathways, notably the inhibition of Wnt/β-catenin signaling pathway and the activation of TGFβ/Smad3 signaling pathway. Intriguingly, overactivation of Wnt signaling pathway, which is downregulated with aging also leads to senescence in MSCs [50,51].
In addition, the senescence of HSCs leads to myeloid bias in differentiation, which increases the activity of osteoclasts from the mononuclear and macrophage system, increases the efficiency of osteolysis, and leads to osteoclast–osteogenesis imbalance, which is also an important cause of the decline of osteogenesis in older populations [52].
DDR is an important mechanism of MSC senescence. MSCs enter cell cycle arrest through p21 and p16 due to accumulation of genome damage caused by replication or oncogene mutations [53]. These two signaling pathways are initiated by DDR and then inhibit the phosphorylation of Rb through the interaction with cyclin-dependent kinase (CDK), which prolong the G0/G1 phase of MSCs and significantly reduce the S phase. Telomere attrition is also a classic factor in DNA damage. However, it should be noted that telomere length alone cannot be relied upon as a definitive indicator to accurately predict the degree and state of cell senescence, mainly because there are variations in telomere length between individuals. Nonetheless, this does not diminish the importance of understanding the role telomeres play in the aging process.
ROS production and accumulation and mitochondrial dysfunction are also important causes of MSC senescence. ROS can directly or indirectly activate p38/MAPK signaling pathway, promote phosphorylation of p53, and cause cell cycle arrest [54]. ROS can also activate PI3K to catalyze the synthesis of phosphatidylinositol 3, 4, 5-triphosphate, which then activates the downstream-related pathways, which ultimately lead to MSC senescence [50].
Elevated levels of ROS contribute to the damage of mitochondrial genome. Additionally, damaged mitochondria further produce increased levels of ROS, establishing a detrimental positive feedback loop. In this intricate cycle, mitochondria play an indispensable role [55].
In terms of epigenetic changes, the inhibition of DNA methyltransferases DNMT1 and DNMT3a, 3b in senescent MSCs can directly demethylate CDKN1A and CDKN2A to induce senescence [56]. On the other hand, the expression of specific tyrosine demethylases, KDM3A-B, KDM5d, and KDM6A-B, also decreased, which led to the silencing and inactivation of genes important for the potentiality of MSCs and limited the function of MSCs [57]. Finally, aging-related alterations in niche also significantly affect the normal function of MSCs. Adipocytes are the main source of proinflammatory factors in the niche and their composition in the niche increases with age, limiting the osteogenic capacity of MSCs. Senescent MSCs have the ability to induce chemotaxis in macrophages toward a specific microenvironment, which subsequently leads to intercellular interactions through the release of the SASP factor. Macrophages also regulate MSCs through the secretion of regulatory factors such as oncostatin M, prostaglandin E2, and bone morphogenetic protein-2 [58].
Macrophages produce oncostatin M (OSM) through cox-pge2 regulatory circuits, then OSM further induces osteoblast differentiation by activating the transcription factor STAT3, leading to bone formation [59]. The role of extracellular matrix (ECM) cannot be ignored either, as ECM provides support for MSCs; ECM (sinusoidal or arteriolar noncellular structural proteins, adhesive proteins, and colloidal substances), possess the capacity to harbor MSC and promote its stemness. The increased level of SASP in aging tissues makes ECM present a persistent and active inflammatory state, and ECM produced by senescent MSCs in turn promotes cell proliferation inhibition in MSCs [60].
Senescent ISCs and Age-Related Diseases
The small intestinal epithelium undergoes a rapid renewed process every 3–6 days, which necessitates the rapid proliferation and differentiation of a specific cell population to sustain the proper number of cells within the intestinal epithelium. The task of rapid proliferation and differentiation is taken up by the ISCs at the base of the crypt within the crypt–villus structure.
There are about 4–6 ISCs in each crypt, and existing theories generally divide them into two categories according to the molecular surface markers and their properties: one is crypt-base columnar cells (CBCs), which are intercalated and arranged between the Paneth cells at the base of the crypt, with molecular markers, Lgr5, MSl1, and ASCL2, relatively active in the physiological state and sensitive to radiation damage; the other category is the labeled lagging cells (LRCs) located at the +4 position of the crypt, with molecular markers, including bmi1 polycomb ring finger oncogene and homeodomain-only protein homeobox, which remain quiescent in the physiological state and are resistant to radiation damage. Their relative physiological properties suggest that LRCs may be a reservoir for CBCs. The interconversion of the two classes is also within the scope of ISC plasticity [61,62].
ISCs require a high renewal rate to maintain intestinal epithelial cell (EC) populations, and aging-related metabolic disorders and signaling abnormalities are major causes of abnormal ISC plasticity and imbalance in gastrointestinal barrier homeostasis [63]. The senescence of the ISCs, an important role of the intestine that is linked to multiple systems, not only directly impairs the body's protection against chemical, biological, and physical damage from the gastrointestinal tract, but also creates varying degrees of impairment in the transmission of immune signals, communication with the intestinal flora, and response to physiological and pathological stimuli [64]. These alterations can lead to a range of local or systemic pathologies, including obesity, inflammatory bowel disease, and metabolic disorders. Also, disturbances in epithelial homeostasis may lead to chemogenic lesions in the gastrointestinal tract, which are usually precursors to cancer, which exhibit some correlation with age. Some researches indicate that senescence of ISCs (gut adult stem cell, to be precise) is a direct cause of colorectal cancer [65,66].
It is worth noting that ISCs in the usual state have a short lifespan, while senescent ISCs just meet the initiation conditions of long-lived tumors with high mutation (eg, drosophila mothers against decapentaplegic protein 4, kirsten rat sarcoma viral oncogene). Moreover, changes in aging-related inflammatory factors in ISC niche and intestinal flora can also directly or indirectly initiate tumorigenesis [67 –69].
The genes involved in maintaining the gastrointestinal barrier and intestinal epithelial homeostasis in both mouse and Drosophila exhibit a high degree of homology with the human genome. These genes also share conserved signaling pathway activities, including the regulation of self-renewal and lineage differentiation by Notch pathway, as well as others such as Wnt, epidermal growth factor (EGF), bone morphogenetic protein, and Hippo [70]. As a result, mice and Drosophila have emerged as the most extensively researched models for studying ISCs.
When it comes to senescent mechanisms, ISCs face greater replication pressure compared with other types of stem cells due to their high renewal and replication needs. Age-related impairments lead to a decline in the regenerative capacity of intestinal tissue and an imbalance in the proportion of differentiated progenitor cells derived from ISCs. Disruptions of the tight junctions between ECs in small intestine can occur due to the inability of cells to renew in a timely manner, and lead to increased intestinal permeability and the development of intestinal inflammatory bowel disease, which in turn lead to the development of leaky gut syndrome [71].
In addition, changes in mitochondrial activity and protein homeostasis are also important manifestations of senescence in ISCs. Thus, increasing mitochondrial activity, decreasing endoplasmic reticulum stress levels, and maintaining protein homeostasis in ISCs can repair ISC function and maintain the gastrointestinal barrier homeostasis, thereby delaying aging and increasing lifespan. For example, overexpression of Drosophila PPARγ coactivator 1 homologs can increase the lifespan of Drosophila from the perspective of increased mitochondrial activity [64]. The normal state of cellular autophagy is also important for the function of ISCs and the delay of the senescence. Autophagy maintains the renewal of organelles and a normal energy state. The abnormal consequences of autophagy are the accumulation of damaged DNA and mitochondria, leading to an increase in the number of functionally abnormal ISCs, resulting in abnormal intestinal epithelial proliferation and differentiation of stem cells [72] (Fig. 4).

Aging and cellular senescence result in ISC dysfunction, which exacerbates digestive issues and age-related disorders such as leaky gut syndrome and inflammatory bowel disease.
On the contrary, the preservation and induction of autophagy have a restorative impact on senescence. In experiments involving Drosophila, it was discovered that activating the adenosine 5′-monophosphate activated protein kinase (AMPK) pathway or autophagy-associated gene 1 pathway during senescence can enhance autophagy levels, decelerate the progress of senescence, and extend lifespan. Likewise, the administration of mTOR inhibitors, which impede the mTOR pathway to trigger autophagy, yields similar outcomes [73]. In the aging mouse model, the observed rise in Cdc42 activity in ISC coincided with declining regenerative capacity, and the reduction of Cdc42 activity by pharmacological means can improve the regeneration of elderly intestinal ECs, indicating the mechanistic role of Cdc42 in senescent ISC [74].
The aging-induced changes in the niche of ISCs also contribute to the senescence of ISCs; however, it is important to note that there are differences in the cellular composition and interactions within the ISC niche of mouse and Drosophila ISC niche. In the mouse ISC niche, regulatory factors such as Wnt, Notch, and EGF are predominantly secreted by Paneth cells, which in turn regulate the proliferation and differentiation of ISCs. In contrast, the structure of Drosophila small intestine differs from that of mouse, and in the ISC niche of Drosophila, the relevant regulatory factors are secreted by visceral muscle cells, enteroblasts, and intestinal ECs [75].
The instability of heterochromatin in Drosophila's ECs, which is associated with aging, can trigger abnormal signaling pathways like the JNK pathway. This can potentially result in the death of ECs and indirectly contribute to the senescence of ISCs. In aged mice, the ISC niche reveals an increase in the number of Paneth cells and goblet cells. Moreover, senescent Paneth cells in the aging niche exhibit an elevated secretion of Notum, an inhibitor of Wnt signaling. This leads to a reduced Wnt signaling in the aging niche, thereby limiting the functionality of ISCs [76].
Senescent MuSCs and Age-Related Diseases
Skeletal muscle possesses a remarkable ability to regenerate, primarily due to the presence of a specialized type of stem cells called satellite cells. These satellite cells are situated on the surface of the muscle fibers and beneath the basement membrane, commonly referred to MuSCs [77]. Pax7 is a widely used marker to identify MuSCs as it is expressed in any state of MuSC development. Other biomarkers like CD34 and CD56 express on MuSC surface in humans. Myogenic regulatory factors of MuSCs—MRFs (Myf5, Myod, Myogenin, Mrf4), are not expressed in a quiescence state. Skeletal muscle enters the cell cycle after injury and progressively expresses MRFs, which differentiates into myoblast to undertake muscle regeneration and tissue repair, after which the myoblast cells cease to proliferate and gradually fuse to form muscle fibers to complete tissue repair and regeneration [78,79].
After muscle injury, inflammatory infiltrating cells (first neutrophils and then macrophages) will release a variety of inflammation-related signaling molecules to activate the rapid expression of MRFs within MuSCs, and eosinophils can also indirectly activate MuSCs by interacting with fibroadipogenic precursors within the niche of MuSCs [80] (Fig. 5).
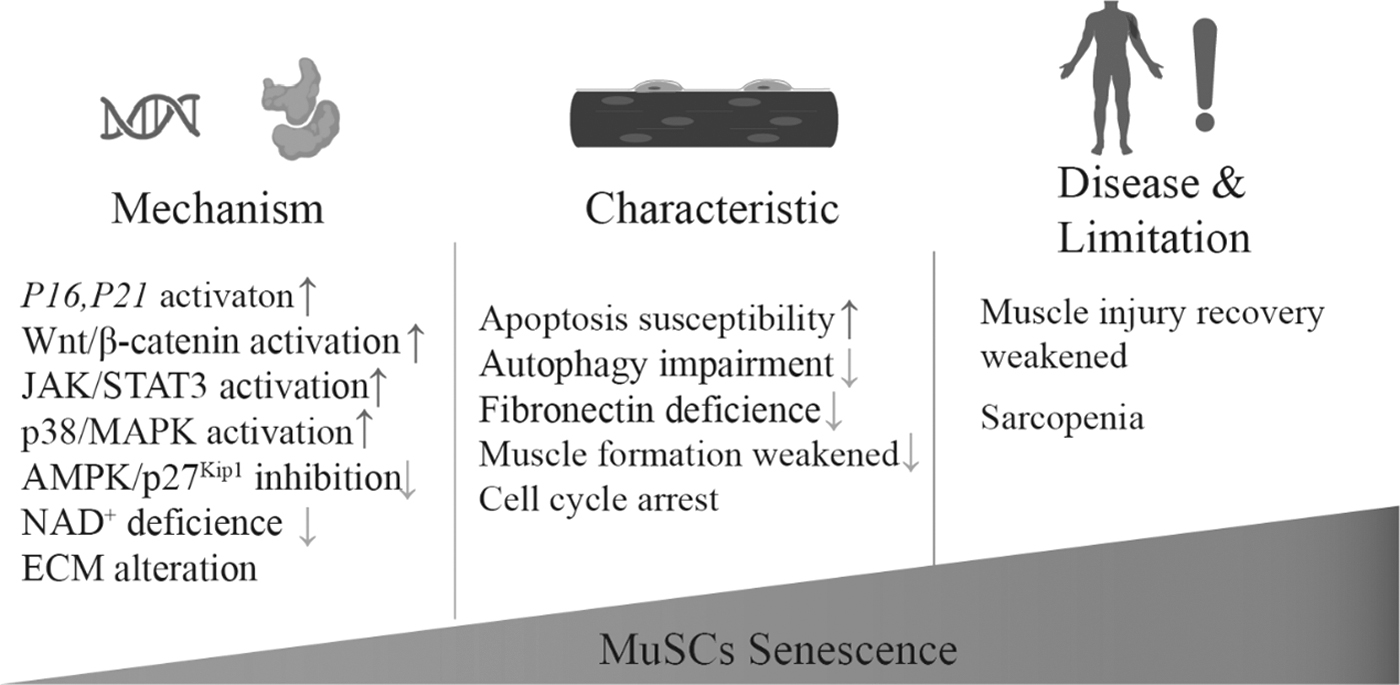
Aging and cellular senescence lead to dysfunction of MuSCs, cause poor skeletal muscle status, and further cause age-related diseases such as muscle fibrosis and atrophy. ECM, extracellular matrix.
The ability of muscles to recover after injury is greatly reduced in the elderly, and the occurrence of sarcopenia, osteoporosis, and fractures all increase with age, suggesting that MuSCs exhibit dysfunction with aging [81]. Moreover, chronic diseases such as cancer, chronic infectious diseases, diabetes, or related therapies have a higher probability of causing muscle atrophy, and all these processes have a complex relationship with MuSC senescence. However, several studies have shown that age-related depletion of MuSCs only increases age-related muscle fibrosis and does not affect sarcopenia [82]. While there is still ongoing debate surrounding this topic, the significance of this indicates that MuSCs play an essential role in maintaining muscle function under normal physiological conditions.
The niche of MuSCs has been found to be intricately linked to the senescent changes of MuSCs. In a study dating back to 2005, it was discovered that the transplantation of MuSCs from aged mice into young mice resulted in the restoration of MuSC activity. This finding strongly suggests a crucial role of the niche in regulating the stemness of MuSCs [83]. MuSCs engage in interactions with various cellular components, including myofibers, endothelial cells, macrophages, and fibro-adipogenic progenitors, as well as the extensive protein network known as the ECM. The aging process impacts both the components within the niche and the activity of signaling pathways involving MuSCs. Consequently, the musculogenic potential and regenerative capacity of MuSCs decline.
The onset of differentiation has been reported to be a process of conversion of Notch to Wnt signaling, with elevated levels of Wnt signaling within the aged muscle. The attachment of MuSCs to fibronectin in the ECM within the niche requires a role for the syndecan-4 and Wnt receptor frizzled-7 coreceptor complexes, and thus this process is also regulated by the Wnt signaling pathway, both suggesting an important regulatory role of Wnt signaling in MuSC senescence [84].
Senescent MuSCs are more prone to undergoing apoptosis, resulting in a reduction in the population of these cells within aging mice [80]. Additionally, there is still controversy as to whether the number of MuSCs in mice decreases significantly with aging. MuSC also shows the similar shift in differentiation profile with aging as HSCs, a tendency for fiber-derived spectral differentiation and less expression of myogenic spectral determinants, with alterations in Wnt and TGFβ signaling pathways [85]. As individuals age, several cell signaling pathways in MuSCs, including fibroblast growth factor 2, TGF-β, Wnt, JAK/STAT, p16, and p38 undergo changes. These alterations can lead to impaired self-renewal and differentiation abilities, ultimately resulting in the dysfunction of MuSCs [80]. In particular, activation of the p38/MAPK signaling pathway is an important endogenous cause of cellular self-renewal impairment.
Elsewhere, MuSCs are known to have active autophagy under normal physiological conditions, and autophagy level decreases with aging, which is related to the fact that senescence MuSCs exhibit inhibition of the AMPK/p27Kip1 pathway, less autophagic, and more prone to apoptosis [86].
Metabolically, aging MuSCs showed a significant decrease in NAD+ levels, and the use of nicotinamide riboside (NAD+ precursor) could save the senescence phenotype of MuSC. This is a result of the NAD+ salvage-induced response of the mitochondrial unfolded protein and the synthesis of the proline protein. This suggests that NAD+ metabolism and NAD+-related pathways play an important role in MuSC cell homeostasis [87]. The discovery of alterations in these pathways opens up the possibility of reversing the senescence of MuSCs through gene therapy or pharmacological interventions.
Overview and Conclusions
Stem cells, as undertaker of tissue regeneration, exist in embryo (embryonic stem cells), adult organism (tissue-specific stem cells), and tumor (tumor stem/tissue-specific stem cells). Embryonic stem cells are totipotent, but later, as they become specialized, the stem cell's potential gradually decreases to unipotent. Stem cells continuously generate new tissue cells in the body to renew tissues and maintain a pool of stem cells for self-renewal.
Over time, the accumulation of damage factors leads to the eventual cell cycle arrest and senescence, mirroring the natural aging process. Severe damage factors, such as ionizing radiation, toxic chemicals, or genetic disorders, can trigger an accelerated aging process in stem cells, severely hindering tissue renewal and repair following injury. These detrimental effects can also result in systemic changes due to alterations in aging phenotypes, ultimately paving the way for the development of various diseases. Hence, it is of utmost importance to establish a connection between cellular senescence, particularly the senescence of stem cells, and the onset of diseases. This could shed light on the relevant disease mechanisms from a stem cell perspective and provide a better basis for stem cell-based therapies.
The study of the senescence of tumor stem cells can be used to achieve the purpose of inhibiting tumor growth by inducing cell senescence, and at the same time, it can achieve better therapeutic effect by the use of senolytic therapy [88,89]. Advancements in single-cell histology techniques, imaging technologies, and biological model systems have significantly deepened our comprehension of stem cell senescence on a molecular level [90]. The identification and isolation of molecular markers specific to distinct cell types and their states, along with the establishment of animal models like mice and Drosophila, have revolutionized our ability to investigate diverse adult stem cells and unravel their aging-associated phenomena [91].
The progress in the analysis of stem cell senescence-related pathways, the anatomical analysis of various adult stem cell niche, and the determination of their composition and interactions will allow us to better understand the process of physiological and pathological aging of the body and to clarify the mechanisms of senescence related to stem cells. This will allow us to develop drugs and therapies to slow down and reverse stem cell senescence and body aging, as well as providing guidance and reference for the application of stem cell therapies.
So, to what extent stem cells, as a strong correlate of lifespan, affect the aging and health of the organism needs to be investigated further. While there are well-established cellular and animal models, more in-depth experiments are needed to determine whether the findings will work the same way in humans.
At the same time, in the study of cellular senescence, special attention should be paid to the distinction between cells with the Hayflick limit and senescent cells in aged tissues. The Hayflick limit represents the maximum number of replication cycles that occur under relatively stable conditions. [92], while stem cells in aging tissues need to account for the full interference of other adverse factors inside and outside the cell. The fundamental problem with both cases is the mechanism by which telomere and telomerase activity changes. Experimental conditions for aging should be distinguished and excluded [93]. Moreover, does the reversal of stem cell senescence really lead to a delay and reversal of aging in humans, and does the short-term acute recovery have long-term effects? These are currently controversial issues in stem cell research. What is certain, however, is that the link between stem cells senescence and aging is becoming clearer.
The application of stem cell therapy to slow down aging would be an intervention in the future. Stem cells are different from organs, but this inevitably raises a question of donor origin, biosecurity, and related ethical issues [94]. With advancements in technology and the establishment of ethical frameworks, there is a hopeful anticipation that the new era will witness increased and improved utilization of stem cells to enhance the health and extend the lifespan of the individuals.
Footnotes
Acknowledgments
The authors thank the Tianjin Key Laboratory of Radiation Medicine and Molecular Nuclear Medicine, Institute of Radiation Medicine, Chinese Academy of Medical Science & Peking Union Medical College.
Author Disclosure Statement
No competing financial interests exist.
Funding Information
This research and the APC were funded by the National Nature Science Foundation of China (Grant No. 81972975), CAMS Medicine and Health Technology Innovation Project (Grant No. 2021-I2M-1-060, 2021-RC310-010), and the Applied Basic Research Key Program of Tianjin (Grant No. 22JCZDJC00430).