
Abstract
Mesenchymal stem cells (MSCs) directly differentiate into neurons and endothelial cells after transplantation, and their secretome has considerable potential for treating brain injuries. Previous studies have suggested that the effects of MSCs priming with exposure to hypoxia, cytokines, growth factors, or chemical agents could optimize the paracrine potency and therapeutic potential of MSCs. Studies have suggested that thrombin-primed Wharton's Jelly-derived mesenchymal stem cells (Th.WJ-MSCs) significantly enhance the neuroprotective beneficial effects of naive MSCs in brain injury such as hypoxic-ischemic brain injury (HIE) and intraventricular hemorrhage (IVH). This study aimed to characterize WJ-MSCs in terms of stem cell markers, differentiation, cell proliferation, and paracrine factors by comparing naive and Th.WJ-MSCs. We demonstrated that compared with naive MSCs,
Introduction
Mesenchymal stem cells (MSCs) are fibroblastic spindle-like adherent cells isolated from adipose tissue, bone marrow, Wharton's jelly (WJ), and other tissue sources with a multipotent differentiation capacity into osteocytes, adipocytes, and chondrocytes in vitro [1 –4]. MSCs from WJ of the umbilical cord (WJ-MSCs) are multipotent stem cells with self-renewal and proliferation capacities that can differentiate into multiple mesenchymal lineages such as adipocytes, chondrocytes, osteoblasts, skeletal muscle cells, endothelial cells, cardiomyocyte-like cells, and neuron-like cells [5 –8].
Recent studies have suggested that the various protective effects (such as anti-inflammatory, antioxidative, and antiapoptotic) of transplanted MSCs are mediated predominantly by paracrine actions through the secretion of several biological factors, including immunomodulatory, angiogenesis, antiapoptosis, antioxidant, mitochondrial transfer factors, and exosomes, instead of by direct actions [9,10].
In addition to MSCs, which directly differentiate into neurons and endothelial cells after transplantation [11,12], their secretome has significant potential for treating brain damage, such as stroke, hypoxic-ischemic brain injury (HIE), and intraventricular hemorrhage (IVH), by inducing neuroprotection, neurogenesis, and angiogenesis [13 –17]. Several studies have demonstrated that MSC priming with exposure to hypoxia, cytokines, growth factors, and pharmacological or chemical agents can optimize their paracrine potency and therapeutic potential [9,18,19]. Thrombin plays a central role in coordinating blood coagulation, inflammation, and wound repair after tissue injury [20,21]. Interestingly, it also regulates several important cellular activities in the central nervous system (CNS) [22]. Thrombin has been demonstrated to protect rat primary hippocampal neurons and astrocytes from hypoglycemia, hypoxia, and growth supplement deprivation in vitro [23]. Thrombin priming has been shown to enhance the efficacy of WJ-derived MSCs in HIEs and IVHs in vitro and in vivo [16,17].
The present study aimed to investigate and compare transcriptome and secretome patterns, as well as paracrine neuroprotective effects between thrombin-primed (Th.) WJ-MSCs and naive WJ-MSCs. We also investigated whether thrombin priming enhances the therapeutic efficacy of human WJ-MSCs in vitro using oxygen-glucose deprivation (OGD) model. In addition, we identified a cytokine that mediates the neuroprotective effects of thrombin-primed Wharton's Jelly-derived mesenchymal stem cells (Th.WJ-MSCs) among various cytokines, hepatocyte growth factor (HGF).
Our results assisted in clarifying the characteristics of Th.WJ-MSCs, thereby facilitating better logical application of Th.WJ-MSCs in clinical settings.
Materials and Methods
Human WJ-MSCs isolation with enzymatic digestion
For WJ-MSC culture, informed written consent was obtained from each patient according to guidelines approved by the Institutional Review Board of Seoul National University Bundang Hospital (SNUBH; Seongnam, Republic of Korea; IRB file No. B-2005-613-302). The isolation process involved enzymatic digestion of WJ using a modified protocol [24]. Equal amounts of WJ segments were digested in Minimum Essential Medium alpha (MEM-α; Gibco, Waltham, MA) containing 0.1% collagenase type I (Gibco) for 60 min at 37°C. The enzymatic digestion was neutralized by adding 10 mL of MEM-α medium supplemented with 10% fetal bovine serum (FBS; Gibco) and 50 μg/mL gentamicin (Thermo Fisher Scientific, Sweden). The contents were centrifuged in 50 mL tubes, and the supernatant was discarded. A slurry containing ∼20 partially digested tissue pieces was transferred to a 75-cm2 culture flask (Corning, New York) with 15 mL of culture medium containing 10% FBS and 50 μg/mL gentamicin.
After a 3-week culture period, the primary (P0) cells were harvested. The culture medium was changed twice a week. P0 cells were detached using 0.05% trypsin-ethylenediaminetetraacetic acid (EDTA; Gibco), subcultured in new flasks, and labeled as passage number 1 (P1). Subsequent passaging was performed using T75-cm2 flasks every 3 days.
Preparation of conditioned media in WJ-MSCs
WJ-MSC-conditioned media (CM) was prepared as described previously, with some modifications [25,26]. In brief, WJ-MSCs at passage 5 were seeded at a density of 3 × 103 cells/cm2 and cultured to 80% confluence in 100 mm culture dishes (430167; Corning). Then MSCs were washed thrice with 10 mL of 1 × phosphate-buffered saline (PBS), and replaced with MEM-α with 10% FBS and 50 μg/mL gentamicin with or without 20 U/mL thrombin [Thrombine pow (5000iu); REYON PHARMACEUTICAL CO., LTD., Seoul, Republic of Korea]. After 3 h, the supernatant was removed and the samples were washed thrice with 1 × PBS and then replaced with MEM-α with 150 μg/mL gentamicin. Cells were maintained at 37°C with saturated humidity and 5% CO2.
After 48 h, the CM was harvested and designated as MSC-CM. After that, supernatants were collected, pooled and centrifuged at 1,000 g and filtered with a 0.2-μm filter (AP-4225; Pall Corporation, Port Washington, NY). Finally, the CM was lyophilized, stored at −80°C, and dissolved before used.
Immunofluorescence
The methods of immunofluorescence was performed as described previously [27]. Cells were treated with 4% paraformaldehyde (PFA) for 10 min, 0.1% Triton X-100 (X-100; Sigma-Aldrich, St. Louis, MO) +1% bovine serum albumin (BSA, A2153; Sigma-Aldrich) for 10 min at room temperature, and then primary antibody diluted in 0.1% Triton X-100 + 1% BSA at 4°C, overnight; mouse antimicrotubule-associated protein 2 (MAP2) (1:500, ab11267; Abcam), rabbit anti-MAP2 (1:500, ab32454; Abcam), mouse antineuronal nuclei (NeuN) (1:500, MAB377; EMD Millipore, Burlington, MA), rabbit anti-NeuN (1:500, ab177487; Abcam), neuron-specific class III beta-tubulin (Tuj1) (1:500, 801202; Biolegend, San Diego, CA), and glial fibrillary acidic protein (GFAP) (1:500, V2129; NSJ Bioreagents, San Diego, CA).
Terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) staining was performed using an in situ Cell Death Detection kit (12156792910; Roche) according to the TMR Red protocol. In brief, 50 μL of TUNEL reaction mixture was applied to each sample at 37°C for 1 h. Nuclei were visualized by treatment with 4′,6-diamidino-2-phenylindole solution at RT for 5 min. Immunocytochemistry products were observed in three randomly chosen areas by confocal laser microscopy. As no immunoreactivity was observed in the negative controls, signal-positive cells were counted using ImageJ software.
Primary culture of cortical neurons and OGD model
The primary culture method for cortical neurons and the OGD model have been described previously [27]. In brief, primary cortical neurons were isolated using a method described previously (aging us). Seven days after the primary culture of isolated cortical neurons, the culture media were replaced with Dulbecco's Modified Eagle Medium (DMEM) glucose-free neurobasal media, and culture plates were placed in a humidified incubator at 37°C in 5% CO2 and 1% O2 for 1 h to create the OGD model. Promptly after OGD, the culture medium was replaced with DMEM/F12 (incomplete medium), naive WJ-MSC-CM (naive MSC-CM), and thrombin-primed WJ-MSC-CM (Th. MSC-CM) or
3-(4,5-dimethylthiazolyl-2)-2,5-diphenyltetrazolium bromide assay
The methods of Ha et al. [27]. Cell viability was measured using the 3-(4,5-dimethylthiazolyl-2)-2,5-diphenyltetrazolium bromide MTT assay (298-93-1; Sigma-Aldrich). MTT solution (10 μL of MTT solution) was added to each well of a 96-well plate at 37°C for 3 h, and 100 μL of dimethyl sulfoxide ( D2650–100ML; Sigma-Aldrich) was added to each well at 37°C for 30 min. The absorbance was measured at 570 nm using an Auto X Mark microplate reader (10319; Bio-Rad, Hercules, CA). Relative cell viability (%) was calculated as the relative difference in absorbance compared with that of the control (without OGD): (absorbance/absorbance of control) × 100.
Western blot analysis
The method of the western blot analysis has been previously described [27]. In brief, cells were harvested and lysed in radioimmunoprecipitation assay lysis buffer (sc-24948; ChemCruz, Texas). The amount of protein in the samples was quantified using Bradford Protein Assay Dye (5000006; Bio-Rad). Equal amounts of protein were loaded onto a OneGel PAGE Kit Plus (MP014; BIOSOLUTION, Suwon, Republic of Korea). Equal amounts of proteins were separated using sodium dodecyl sulfate-polyacrylamide gel electrophoresis and analyzed using immunoblotting with the indicated antibodies.
The following antibodies were used: Bax (1:1,000, sc-7480; Santa Cruz Biotechnology), Bcl-2 (1:100, sc-7382; Santa Cruz Biotechnology), β-actin (1:1,000, sc-47778; Santa Cruz Biotechnology), Cleaved-caspase 3 (1:1,000, 9664; Cell Signaling), p-STAT3 (Tyr705)(1:1,000, 9145S; Cell Signaling), STAT3 (1:1,000, 30835S; Cell Signaling), Akt (1:1,000, 9272; Cell Signaling), p-Akt (1:1,000, 9271; Cell Signaling), p44/42 mitogen-activated protein kinase (MAPK) (Erk1/2) (1:1,000, 9102; Cell Signaling), and p-p44/42 MAPK (Erk1/2)(Thr202/Tyr204)(1:1,000, 4370; Cell Signaling). Bands were quantified using ImageJ software and normalized to β-actin levels.
Microarray and real-time quantitative reverse transcription-polymerase chain reaction
Microarray and quantitative reverse transcription-polymerase chain reaction (qRT-PCR) analyses were performed as described previously [27,28]. In brief, primary cortical neurons were harvested, and the pellet was analyzed by microarray according to the procedure described by Macrogen Co., (Seoul, Republic of Korea), and the study was generating cDNA from the same total RNA used in the SAGE protocol. cDNA was synthesized using the GeneChip Whole Transcript (WT) Amplification Kit (Thermo Fisher Scientific). The synthesized sense cDNA was fragmented and labeled with biotin using a GeneChip WT Terminal Labeling Kit (Thermo Fisher Scientific). Approximately 5.5 μg of labeled DNA target was hybridized to the GeneChip™ Rat Gene 2.0 ST Array (Thermo Fisher Scientific) at 45°C for 16 h. The hybridized arrays were washed, stained, and scanned. Data computation, CEL file generation, and normalization were performed using Affymetrix GeneChip Command Console software (AGCC) and the robust multiaverage method.
Differential gene expression analysis was conducted, identifying genes with a fold change ≥2 as differentially expressed genes (DEGs). Hierarchical cluster analysis was used for each DEG set. Gene enrichment and functional annotation analyses were performed using Gene Ontology (GO) and Database for Annotation, Visualization, and Integrated Discovery (DAVID). Data analysis and DEG visualization were performed using R 3.3.2. qRT-PCR was performed for confirmation. Total RNAs were extracted, reverse transcribed using an EasyScript™ cDNA Synthesis Kit, and subjected to qRT-PCR analysis using a Rotor-Gene Q real-time PCR detection system. Normalized gene expression was determined using Rotor-Gene Q software, with β-actin as the reference for normalization. Gene-specific primer sequences are listed in Supplementary Table S1. This methodology allowed for comprehensive gene expression analysis and validation using qRT-PCR.
Measurement of cytokine levels through enzyme-linked immunosorbent assay
Enzyme-linked immunosorbent assay (ELISA) was performed using commercial kits, according to the manufacturer's instructions. The following ELISA kits were used: Human (h)HGF Quantikine ELISA Kit (DHG00B; R&D Systems, Inc., Minneapolis, MN), Human TIMP-2 Quantikine ELISA Kit (DTM200, R&D Systems, Inc.) and Human IGFBP-4 DuoSet ELISA Kit (DY804; R&D Systems, Inc.). All kits included standard proteins; therefore, the protein counts were determined based on the standard curve from each kit.
In vitro differentiation
In vitro differentiation of MSC-CM was performed as previously described, with some modifications [26]. For adipocyte differentiation, WJ-MSCs were cultured in adipogenesis medium [MesenCult™ Adipogenic Differentiation Kit (Human); 05412; STEMCELL Technologies, Vancouver, Canada] for further 3 weeks by following manufacturer's instructions. In brief, the medium was changed every 3–4 days and the wells were stained with Oil Red O solution (LL-0052; Lifeline Cell Technology, Frederick, MD). For chondrocyte differentiation, WJ-MSCs were cultured in chondrogenic medium (MesenCult-ACF Chondrogenic Differentiation Kit; 05455; STEMCELL Technologies) for further 3 weeks by following manufacturer's instructions.
The medium was changed every 3–4 days, and the wells were stained with 1% Alcian Blue solution (LL-0051; Lifeline Cell Technology). For osteoblast differentiation, WJ-MSCs were cultured in osteogenic medium [MesenCult Osteogenic Differentiation Kit (Human), 05465; STEMCELL Technologies] for further 3 weeks by following manufacturer's instructions. The medium was changed every 3–4 days and the wells were stained with 2% Alizarin Red S (CM-0058; Lifeline Cell Technology).
Flow cytometric analysis
The Flow Cytometric Analysis method has been described previously [26]. WJ-MSCs were harvested using 0.25% trypsin-EDTA (25200-072; Gibco) in fluorescence-activated cell sorting (FACS) tubes (BD Bioscience, Franklin Lakes, NJ). When the cells were stained with surface marker, the monoclonal antibodies, CD44 (1:100, 559942; BD Bioscience), CD73 (1:100, 560847; BD Bioscience), CD90 (1:100, 555595; BD Bioscience), CD105 (1:100, 562408; BD Bioscience), CD166 (1:100, 559263), CD11b (1:100, 562793; BD Bioscience), CD14 (1:100, 555397; BD Bioscience), CD19 (1:100, 555415; BD Bioscience), CD34 (1:100, 555821; BD Bioscience), CD45 (1:100, 555482; BD Bioscience), and HLA-DR (1:100, 559866; BD Bioscience), were added in tube and incubated in the dark at RT.
After 20 min, the FACS buffer (BD Biosciences) was added to the tubes. This was followed by centrifugation at 500 g for 3 min and the cells were resuspended in Dulbecco's phosphate-buffered saline containing 4% PFA. Fluorescence intensity was determined using a FACSCalibur (Becton Dickinson, San Jose, CA). Data analysis was performed using the FLOWJO software (Tree Star, Inc., Ashland, OR).
Protein identification using nano liquid chromatography with tandem mass spectrometry analysis
Previously, a method for protein identification by nanoliquid chromatography with tandem mass spectrometry (LC-MS/MS) has been described [29], according to the procedure described by Ebiogen, Inc. (Seoul, Republic of Korea). Protein concentrations were determined using the Pierce BCA Protein Assay Kit (Thermo Fisher Scientific). Protein digestion was conducted using a Filter-Aided Sample Preparation technique on a Microcon 30 K centrifugal filter (Millipore). Each sample was treated by reducing with Tris(2-carboxyethyl) phosphine, followed by alkylation with iodoacetic acid under dark conditions. After washing with lysis buffer and 50 mM Ammonium Bicarbonate, trypsin digestion (enzyme-to-protein ratio 1:50; w/w) was performed at 37°C for 18 h. The resulting peptide mixtures were collected and trypsin inactivation was achieved by acidification with formic acid.
Digested peptides were desalted using C18 spin columns and eluted with a mixture of 80% acetonitrile and 0.1% formic acid in water. LC-MS/MS analysis was performed using a Q Exactive Orbitrap hybrid mass spectrometer equipped with an Ultimate 3000 system (Thermo Fisher Scientific). For proteome data analysis, Thermo MS/MS raw files were processed using Proteome Discoverer™ software (ver. 2.5). For data analysis, identified proteins were filtered to remove reverse hits and contaminants. We used the STRING database version 9.1 (
Statistical analysis
All data are presented as mean ± standard error of standard deviation. Statistical analyses were performed using the Prism 9 software (GraphPad Software, Inc., San Diego, CA). Two-tailed Student's t-tests assuming equal variance, one-way analysis of variance, and Tukey's post hoc analysis were used for comparisons between two samples and among multiple groups, respectively. Statistical significance was set as follows: *P < 0.05, **P < 0.01, and ***P < 0.001.
Results
Characterization and identification of WJ-MSCs
In the present study, three WJ-MSCs were successfully isolated and cultured three types of WJ-MSCs. Different WJ-MSCs maintained their typical fibroblastic spindle-like morphology (Fig. 1A). The expression pattern of the surface antigen was analyzed using FACS. WJ-MSCs were positive for CD44, CD74, CD90, CD105, and CD106, but negative for CD11b, CD14, CD19, CD34, CD45, and HLA-DR (Fig. 1B). The immunophenotype of the WJ-MSCs was derived from three sources. The expression pattern of the surface antigen was similar to that of the immunophenotype populations (Fig. 1C). Subsequently, to determine in vitro differentiation potential, three different WJ-MSCs were cultured to confluence, and the culture medium was changed to adipogenic, osteogenic, or chondrogenic media for 21 days. Oil Red O and Alizarin Red staining revealed lipid and calcium vacuole deposits, respectively. Alcian blue staining confirmed the chondrogenic differentiation (Fig. 1D).
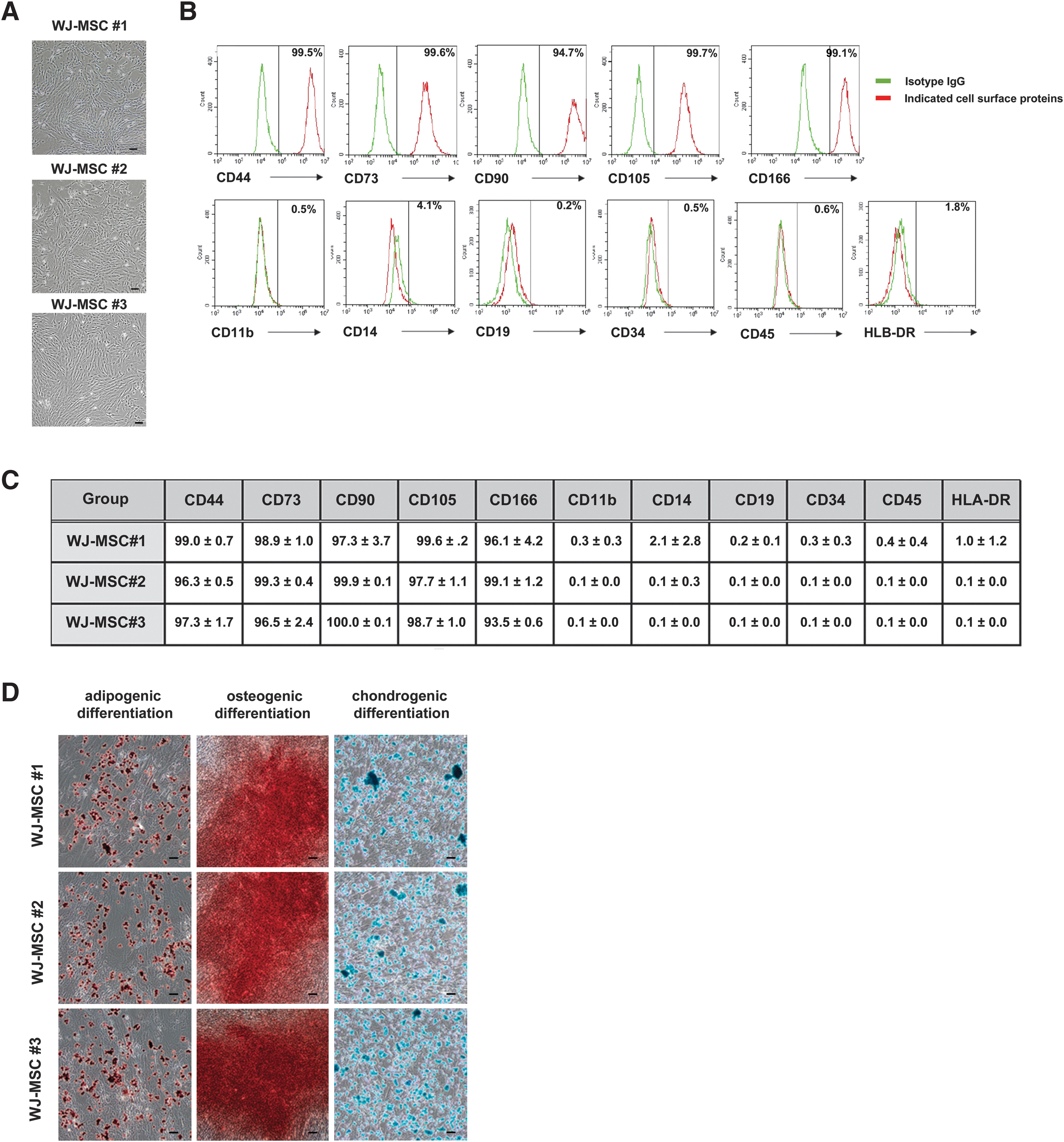
Primary isolation and characterization of three different WJ-MSCs. Primary isolated WJ-MSCs were cultured and characterized.
Paracrine neuroprotective effects of WJ-MSC CM
We examined whether the neuroprotective effects of WJ-MSCs were mediated by paracrine mediators in vitro. Primary cortical neurons were cultured from the brains of rat fetuses and were characterized. In primary cortical neurons, we observed the expression of neuron-specific markers, such as MAP2, Tuj1, and NeuN, but not the astrocyte-specific marker GFAP (Fig. 2A). To elucidate the neuroprotective effects of human WJ-MSCs on neurons, cerebral ischemic conditions were reproduced using an in vitro OGD model to mimic CNS insults in vivo. Previous studies have suggested that the Th.WJ-MSCs significantly enhance the neuroprotective effects of naive WJ-MSCs [17,30].

In vitro paracrine neuroprotective effects of thrombin primed WJ-MSC conditional media.
First, we examined the neuroprotective effects of Thrombin. Compared with naive WJ-MSCs, WJ-MSCs were significantly enhanced in vitro. Under OGD conditions, the viability of primary cortical neurons decreased significantly. However, treatment with CM from naive or Th.WJ-MSCs significantly recovered the damaged and dead primary cortical neurons. In addition, thrombin-preconditioned WJ-MSCs were significantly enhanced compared with naive WJ-MSCs in vitro (Fig. 2B).
Next, we examined whether the characteristics of naive WJ-MSCs were altered by thrombin priming. We tested the morphology, differentiation potency, chromosomal stability, and telomerase activity of naive WJ-MSCs and Th.WJ-MSCs. Thrombin-primed WJ-MSCs did not alter the characteristics of naive WJ-MSC such as morphology, differentiation potency, cell proliferation, chromosome stability, or telomerase activity (Supplementary Fig. S1). In addition, the three different CM of thrombin-preconditioned WJ-MSCs significantly enhanced the neuroprotective effects compared to the OGD control (Fig. 2C). The in vitro paracrine neuroprotective effects of CM from thrombin-preconditioned WJ-MSCs (Th. MSC-CM#1) was confirmed by TUNEL staining of primary cortical neurons under OGD conditions (Fig. 2D, E). These results indicated that the neuroprotective effects of WJ-MSCs were mediated by paracrine mediators. Paracrine neuroprotective effects of Th.WJ-MSCs were verified using western blot analysis for Bcl-2-associated X protein (Bax), B cell lymphoma 2 (Bcl-2), and cleaved caspase 3 (CC3).
Western blot analysis indicated a significant reduction in Bax and CC3 expression and a significant increase in Bcl-2 expression 24 h after Th.WJ-MSC-CM#1 under OGD conditions (Fig. 2F, G).
Relative pathways underlying neuroprotective activities of WJ-MSCs
We investigated the mechanisms mediating the paracrine neuroprotective activities of WJ-MSCs and the transcriptomes of primary cortical neurons treated with Th.WJ-MSC-CM was analyzed for 24 h under OGD conditions. The DEGs among primary cortical neurons in the normal condition (control), OGD condition (OGD), and those treated with Th.WJ-MSC-CM for 24 h under OGD (OGD+Th.WJ-MSC-CM) were filtered out and hierarchical clustering was performed (Fig. 3A). Genes with a fold change value ≥2 in the OGD+Thrombin. The WJ-MSC-CM group, compared with the OGD group, was classified as an upregulated gene. The 137 upregulated genes were further analyzed using the KEGG pathway clustering (Fig. 3B). KEGG pathway clustering identified the MAPK, Janus family tyrosine kinase (Jak)/signal transducers and activators of transcription (STAT), phosphatidylinositol 3-kinase (PI3K)/Akt, Hypoxia-inducible factor-1, and Tumor Necrosis Factor (TNF) signaling pathways, which matched well with the treatment mechanisms underlying the neuroprotective effects of WJ-MSCs in this study.
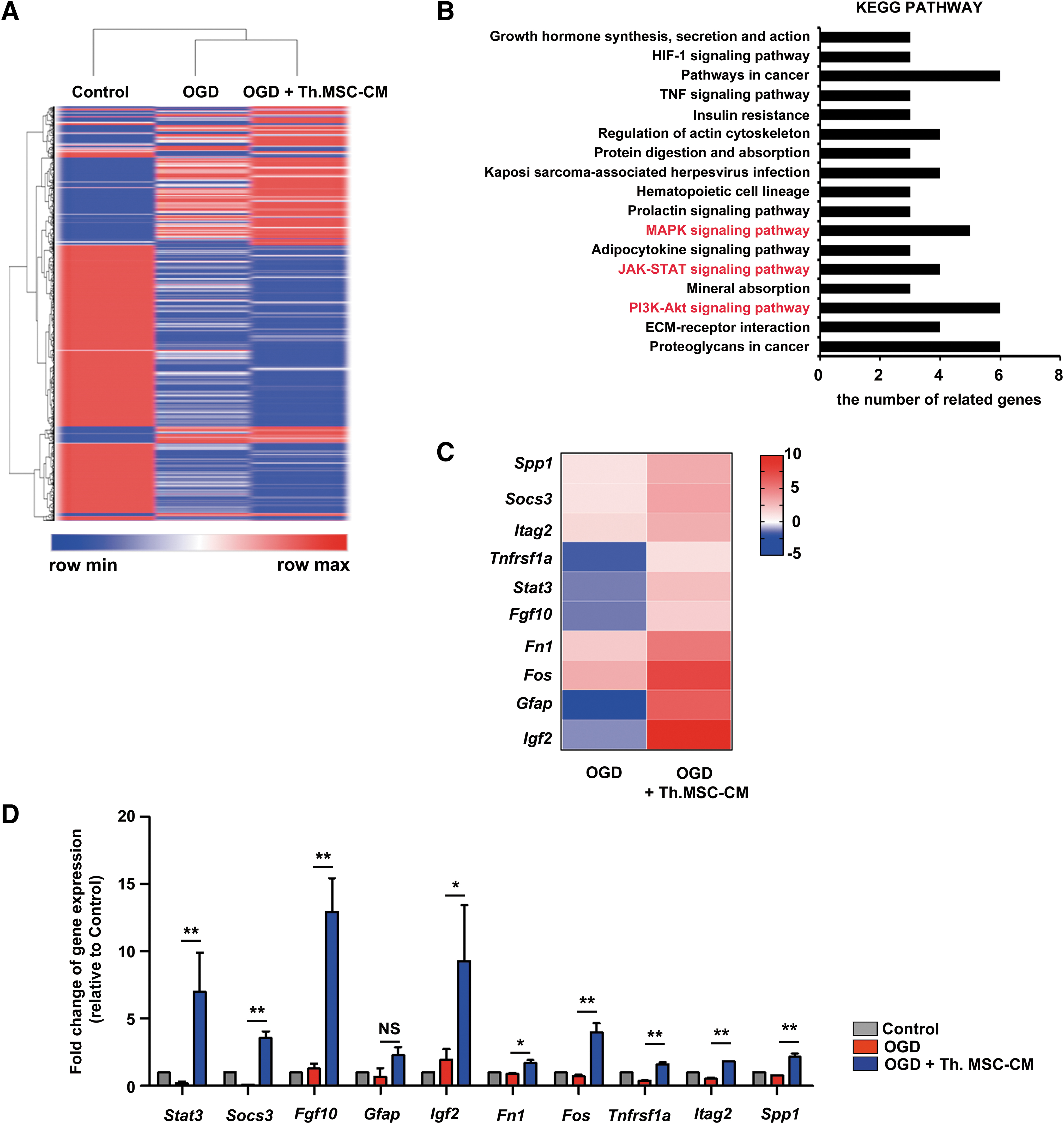
DEGs and biological processes associated with effects of thrombin primed WJ-MSCs.
High expression of the 10 most upregulated genes, Spp1, Socs3, Itag2, Tnfrsf1a, Stat3, Fgf10, Fn1, Fos, Gfap, and Igf2, in OGD+Thrombin in the WJ-MSC-CM group (Fig. 3C) was confirmed by qRT-PCR analysis (Fig. 3D). In the transcriptome analysis of primary cortical neurons, GFAP expression levels in OGD+Thrombin. The WJ-MSC-CM conditions did not change and the expression levels of other genes increased significantly (Fig. 3D). These results indicated that Th.WJ-MSC-CM activated the MAPK, JAK-STAT, and PI3K/Akt signaling pathways to promote neuroprotective effects.
Identification of secreted proteins in the Th.WJ-MSC-CM
Previous studies investigated MSCs in cell-based therapies involving paracrine actions for their anti-inflammatory, immunosuppressive, immunomodulatory, and regenerative mechanisms [31,32]. Paracrine actions depend on the MSC secretome, such as growth factors, cytokines, chemokines, and microvesicles/exosomes carrying proteins and/or miRNAs to the target cells [31 –33]. We identified differentially expressed proteins in naive WJ-MSC-CMs and Th.WJ-MSC-CM by LC-MS/MS-based analysis (Fig. 4A). In this study, the expression of secreted proteins in naive WJ-MSC-CMs (n = 550) and Th.WJ-MSC-CMs (n = 551) were analyzed using LC-MS/MS, and 578 proteins were identified (Fig. 4B). The 262 proteins [including HGF, TIMP2, IGFBP-4, leukemia inhibitory factor (LIF), and matrix metalloproteinase-1 (MMP1)] with a fold change value of ≥1.5 in the Th.WJ-MSC-CM group (n = 551), compared to the naive WJ-MSC-CM group (n = 550), was classified as an upregulated protein.
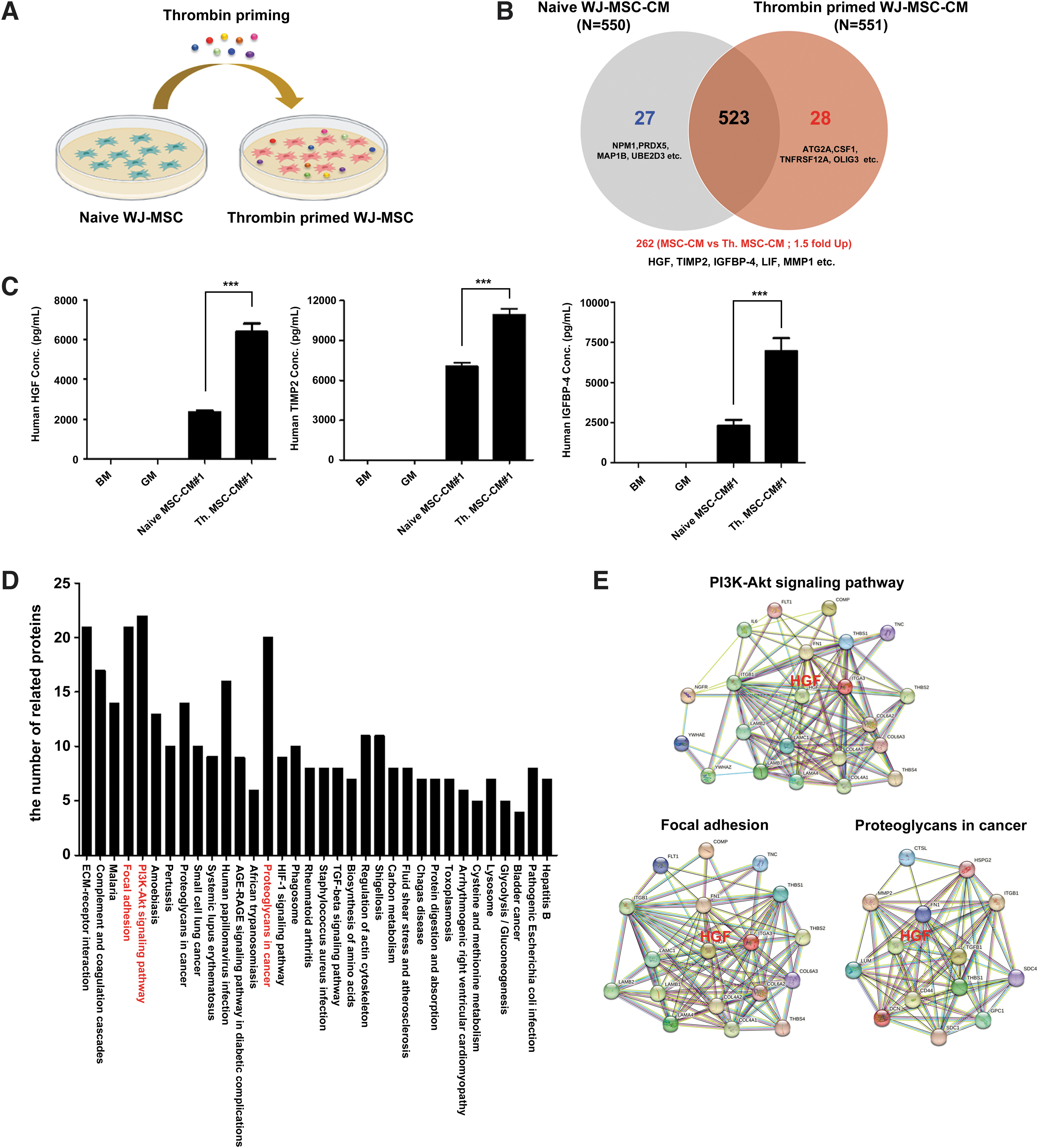
LC-MS/MS-based analysis of differentially expressed proteins in naive WJ-MSC-CM (naive MSC-CM) and thrombin primed WJ-MSC-CM (Th. MSC-CM).
We examined whether proteins with a fold change value of ≥1.5 (HGF, TIMP2, and IGFBP-4) were upregulated in the Th.WJ-MSC-CM group compared with the naive WJ-MSC-CM group using ELISA. We show the three proteins (HGF, TIMP2, and IGFBP-4) with significantly increased expression levels (with a fold change value ≥1.5) in the Th.WJ-MSC-CM group compared to the naive WJ-MSC-CM group (Fig. 4C). The 262 upregulated proteins were further analyzed using string network analysis of the KEGG pathways. KEGG pathway clustering revealed 34 pathways (Fig. 4D) and specific interactions of the top 3 KEGG pathways involving the PI3K/Akt signaling pathway, focal adhesion, and proteoglycans in cancer (Fig. 4E). Interestingly, HGF is also involved in these mechanisms. These results indicate that HGF can activate the PI3K/Akt signaling pathway, focal adhesions, and proteoglycans in cancer.
Role of HGF in the neuroprotection effects of WJ-MSCs
HGF and its tyrosine kinase transmembrane receptor, c-Met, have been identified in the mammalian brains [34,35], suggesting their role in the CNS. Other studies demonstrated that HGF promotes the survival and maturation of cultured neural cells obtained from the CNS and spinal cord motor neurons [36,37] suggesting that HGF regulates neuronal survival. To examine whether HGF has similar expression levels in the three different Th.WJ-MSC-CM conditions, HGF concentrations were analyzed using ELISA and ELISA revealed similar HGF expression levels in the three Th.WJ-MSC-CM conditions (Fig. 5A). To examine whether HGF induced neuroprotection, OGD-damaged cortical neurons were treated with different concentrations of recombinant (r)hHGF. Higher concentrations of recombinant human HGF (rhHGF) demonstrated increased protection of OGD-damaged cortical neurons (*P < 0.05, ***P < 0.01; Fig. 5B). OGD-damaged cortical neurons were treated with rhHGF or Th.WJ-MSC-CM. rhHGF and Th.WJ-MSC-CM increased the protection of OGD-damaged cortical neurons without affecting cell viability (Fig. 5C).
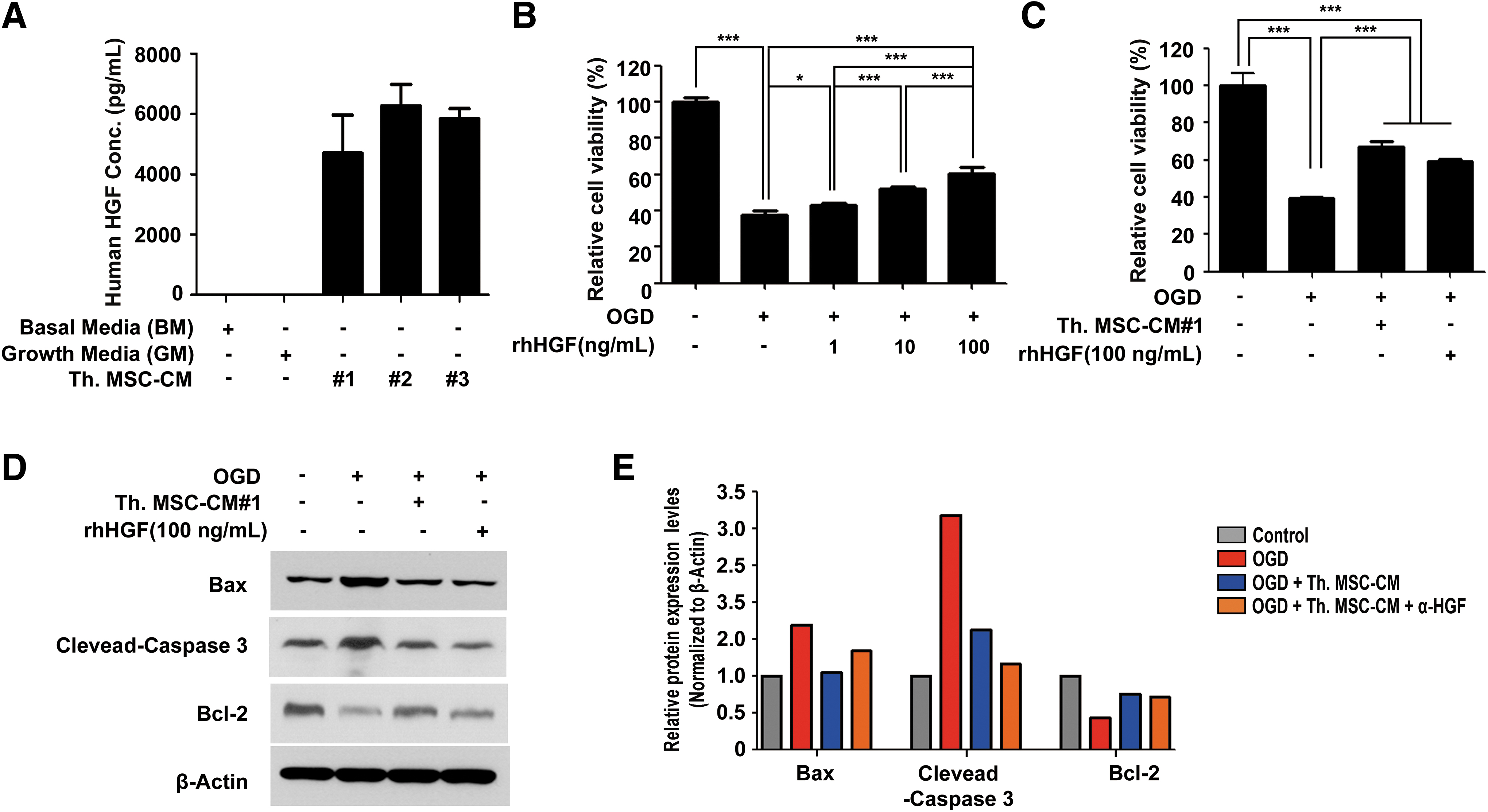
Neuroprotective effect of HGF in
Neuroprotective effects of rhHGF and Th.WJ-MSC-CM were verified using western blot analysis of Bax, Bcl-2, and CC3 expression. The results revealed that the expression levels of Bax and CC3 were significantly reduced, whereas that of Bcl-2 was significantly increased, 24 h after rhHGF or thrombin treatment. WJ-MSC-CM treatment was compared to the OGD group (Fig. 5D, E). These results suggest that HGF plays an important role in the neuroprotective effects on hypoxia-injured primary cortical neurons.
Activation of the Akt/STAT3 signaling pathway by HGF in Th.WJ-MSC-CM
To examine whether HGF depletion reduced these neuroprotective effects, OGD-damaged cortical neurons were treated with Th.WJ-MSC-CM and were blocked by a neutralizing antibody against HGF (α-HGF). HGF depletion abrogates the neuroprotective effects of Th.WJ-MSC-CM, whereas anti-IgG (Immunoglobulin G) showed no effect on cell survival (***P < 0.001; Fig. 6A). The in vitro neuroprotective effects of Th.WJ-MSC-CM was confirmed using TUNEL staining of primary cortical neurons under OGD conditions (Fig. 6B, C). These results indicate that the neuroprotective effects of WJ-MSCs were mediated by HGF. HGF/c-Met signaling activates multiple signal transduction pathways, including the PI3K/Akt, MAPK, and signal transducer and activator of transcription 3 (STAT3) pathways [38 –40]. The results of the nano LC-MS/MS analysis revealed that HGF is involved in the PI3K/Akt signaling pathway, focal adhesion, and proteoglycans in cancer (Fig. 4).
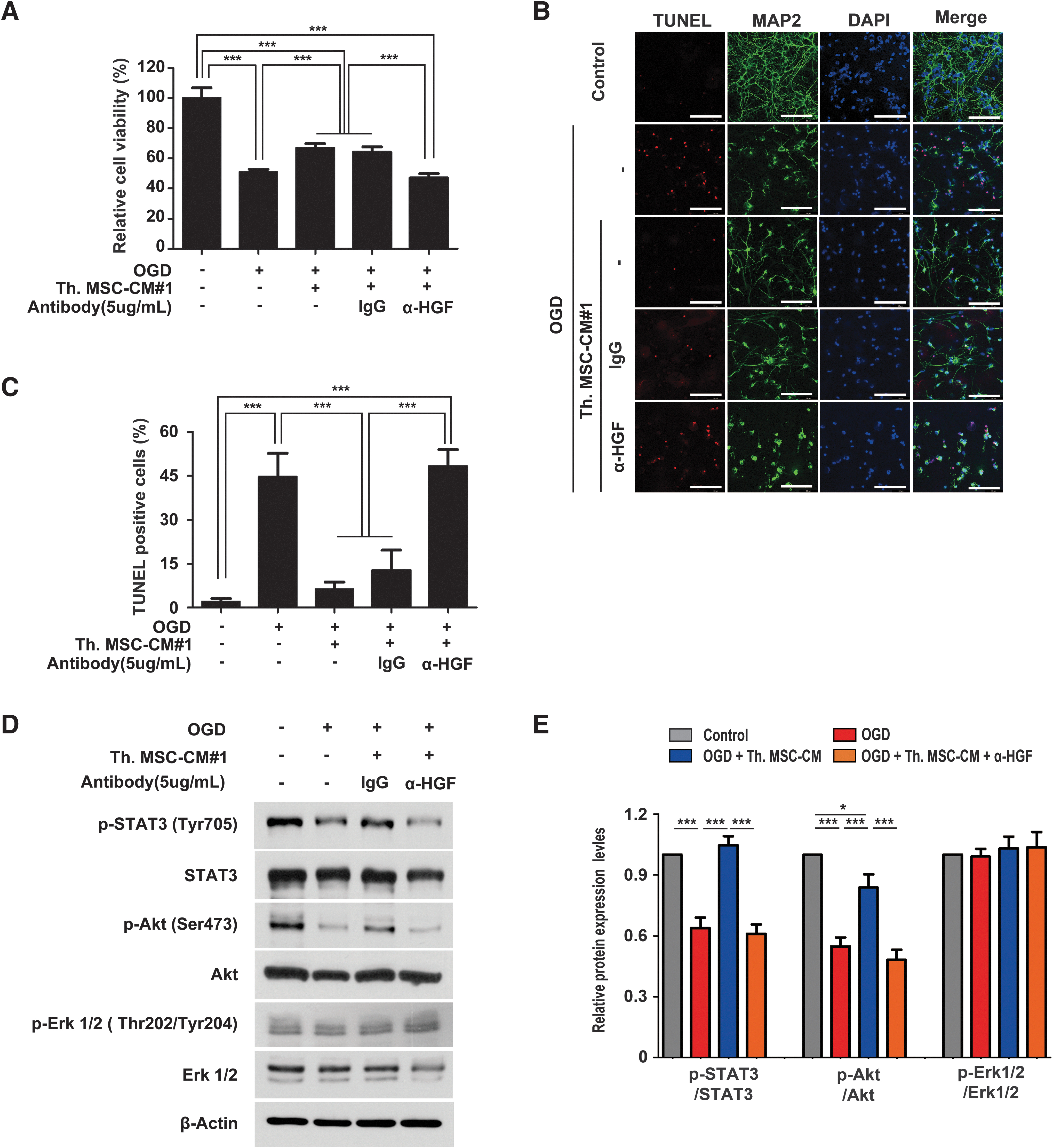
Depletion of HGF was significantly reduced the neuroprotection through AKT/STAT3 signaling pathways in primary cortical neurons.
We investigated whether HGF-mediated signal transduction has neuroprotective effects against Th.WJ-MSC-CM in the PI3K/Akt, MAPK, and STAT3 pathways and is involved in HGF/c-Met signaling. Western blot analysis revealed that the expression levels of phospho-STAT3 (Tyr705) and phospho-Akt (Ser473) were significantly reduced, whereas those of phospho-Erk1/2 were unchanged in the OGD+anti-HGF group compared to those in the OGD+IgG group (Fig. 6D, E). These results indicated that the neuroprotective effects of WJ-MSCs were mediated by the HGF/Akt/STAT3 signaling pathway.
PI3K/Akt, MAPK, and JAK-STAT signaling pathways in the neuroprotection effects of HGF
HGF activates diverse intracellular signaling pathways, including PI3K/Akt, MAPK, and JAK-STAT3 [41,42]. To determine whether the PI3K/Akt, MAPK, and JAK-STAT signaling pathways are involved in the neuroprotective effects of HGF, LY294002 (a PI3K inhibitor), AG490 (a JAK2/STAT3 inhibitor), and PD98059 (a non-ATP-competitive MEK inhibitor) were used. Treatment of rhHGF exhibited protective effects on OGD-damaged cortical neurons. However, when OGD-damaged cortical neurons were treated with a combination of rhHGF and three different inhibitors (LY294002, AG490, or PD98059), the protection effects were reduced significantly (Supplementary Fig. S2A). In addition, the phosphorylation of PI3K, Akt, ERK1/2, and STAT3 were also reduced in OGD-damaged cortical neurons by the three inhibitors (Supplementary Fig. S2B). These results indicate that the neuroprotective effect of HGF is associated with the PI3K/Akt, MAPK, and JAK-STAT signaling pathways.
Discussion
Studies have demonstrated that MSCs have cell-protective and anti-inflammatory effects in neonatal brain injuries such as stroke, HIE, and IVH [13 –17]. However, a novel strategy for enhancing the therapeutic effect of MSCs is required to maximize their therapeutic potential against various brain injuries. Various recent studies have reported that thrombin priming enhances the paracrine and therapeutic potentials of MSCs [9,18,19]. Thrombin plays an important role in various processes after tissue injury by secreting cytokines and growth factors [20,21,43,44]. It promotes neuroprotection of CNS cells against brain injury [23]. In vivo studies on the protective effects of thrombin in ischemic and hemorrhagic stroke have reported that intracerebral injection of a low dose of thrombin can reduce the subsequent brain damage caused by intracerebral hemorrhage or cerebral ischemia [45,46]. In contrast, the intracerebral infusion of a high concentration of thrombin induces brain edema and neuronal death [47 –49].
Recent studies have indicated that thrombin priming enhances the efficacy of WJ-derived MSCs in neonatal HIE and IVH in vitro and in vivo [16,17]. Therefore, we hypothesized that the neuroprotective efficacy of naive WJ-MSCs can be enhanced by thrombin priming [50] in preclinical models of IVH and HIE. In the present study, we observed that, compared with naive WJ-MSCs, Th.WJ-MSCs significantly enhanced antiapoptotic effects in an in vitro model of OGD-induced neuronal injury.
Considering the differentiation potential and paracrine factors of WJ-MSCs, they may exhibit therapeutic effects in IVH and HIE through the differentiation and secretion of neuroprotective paracrine factors [51 –53]. In a previous study, the presence of human DNA in MSCs transplanted into the lumbar spinal cord of superoxide dismutase 1 mice was observed ∼5 days after transplantation using RT-PCR with human-specific primers [54 –56]. The amount of human DNA gradually decreased at the transplantation [55,56].
Moreover, human DNA in WJ-MSCs transplanted into Sprague–Dawley rats was detected in the brain tissue ∼7 days after transplantation through RT-PCR using human-specific primers (unpublished data). As functional recovery through WJ-MSC transplantation in animal models was not reduced over time, the neuroprotective effects of WJ-MSCs in animal models of IVH or HIE may be primarily mediated by the paracrine factors of WJ-MSCs. Our study demonstrated that the CM of WJ-MSCs and transplantation of Th.WJ-MSCs inhibited neural cell apoptosis under oxidative stress and exhibited neuroprotective effects in vitro.
Secretory factors such as growth factors, cytokines, chemokines, extracellular matrix components, and metabolic products have been detected in the CM of MSCs and contribute to immune modulation, tissue remodeling, and cellular homeostasis during regeneration [57 –59]. In the present study, we identified differentially expressed secretory factors in Th.WJ-MSC-CM, compared with those in naive WJ-MSC-CM, by LC–MS/MS analysis and observed that HGF, TIMP2, IGFBP-4, LIF, and MMP1 had a fold change value of ≥1.5. Moreover, the expression levels of two secretory factors (TIMP2 and IGFBP-4) significantly increased in the CM of Th.WJ-MSCs compared to naive WJ-MSCs. Thus, IGFBP-4 released from MSCs may serve as a key neuronal survival factor in OGD-treated primary cortical neurons [60]. HGF levels were relatively higher in the CM of Th.WJ-MSCs compared to WJ-MSCs. HGF concentration was similar in the three independent batches of Th.WJ-MSC-CM, high HGF expression levels may be a key feature of WJ-MSCs.
HGF is a pleiotropic cytokine of mesenchymal origin that promotes mitogenic, motogenic, morphogenetic, and antiapoptotic activity in various cells, such as epithelial and endothelial cells [42,61]. After full ligand-dependent tyrosine phosphorylation, the Met receptor expressed on MSCs activates the PI3K/Akt, ERK1/2, and p38 MAPK pathways, which are the main transduction pathways activated in other HGF-responsive cells [42,61]. These pathways have been reported to contribute to different biological responses elicited by HGF. The PI3K/Akt pathway is mainly associated with motogenic and antiapoptotic effects, whereas the ERK1/2 and MAPK pathways are mostly associated with HGF-related mitogenic and morphogenic effects induced by the HGF/c-Met signaling pathways [61 –63]. Moreover, HGF activates STAT3 through phosphorylation after ischemic or traumatic injury. STAT3 activation has been linked to the neuroprotective roles of these factors following injury [40,64].
In addition, HGF and its tyrosine kinase transmembrane receptor, c-Met, have been identified in mammalian brains [34,35] suggesting their potentially important role in the CNS. Other studies reported that HGF promotes the survival and maturation of cultured neural cells obtained from the CNS and spinal cord motor neurons [36,37] indicating that it regulates neuronal survival. In the present study, we investigated the neuroprotective effects of thrombin. The effect of WJ-MSC-CM on the survival of rat primary cortical neurons in the OGD model was offset and reversed using an HGF-neutralizing antibody. In the present study, we demonstrated that thrombin priming promotes the neuroprotective effects of WJ-MSCs via the HGF/Akt/STAT3 signaling pathway. In addition, the neuroprotective effects of HGF are associated with the PI3K/Akt, MAPK, and JAK-STAT signaling pathways. Collectively, these results suggest that HGF is a major paracrine factor mediating the in vitro and in vivo neuroprotective effects of WJ-MSCs.
Conclusion
In conclusion, we investigated and compared the secretory factors and paracrine neuroprotective effects of Th.WJ-MSCs and naive WJ-MSCs. This study focused on the neuroprotective effects of an in vitro OGD model that mimicked CNS insults in vivo. In particular, the HGF released by Th.WJ-MSCs may play an important role in neuroprotective and antiapoptosis mechanisms in IVH and HIE models.
Footnotes
Acknowledgments
We acknowledge support by the Medical Research Center (MRC) and BIO R&D Projects (BIORP) of Sungkyunkwan University. We thank Editage (
Author Disclosure Statement
No competing financial interests exist.
Funding Information
This research was funded by the National Research Foundation (NRF-2016R1A5A2945889, NRF-2020R1F1A1073261 and 20172-MFDS-194) and Korea Basic Science Institute (2020R1A6C101A191).
Supplementary Material
Supplementary Data
Supplementary Table S1
Supplementary Figure S1
Supplementary Figure S2