
Abstract
Prolonged tissue ischemia and inflammation lead to organ deterioration and are often accompanied by microvasculature rarefaction, fibrosis, and elevated systemic Activin A (ActA), the level of which frequently correlates with disease severity. Mesenchymal stromal cells are prevalent in the perivascular niche and are likely involved in tissue homeostasis and pathology. This study investigated the effects of inflammatory cells on modulation of phenotype of adipose mesenchymal stromal cells (ASC) and the role of ActA in this process. Peripheral blood mononuclear cells were activated with lipopolysaccharide (activated peripheral blood mononuclear cells [aPBMC]) and presented to ASC. Expression of smooth muscle/myofibroblast markers, ActA, transforming growth factors beta 1-3 (TGFβ1–3), and connective tissue growth factor (CTGF) was assessed in ASC. Silencing approaches were used to dissect the signaling cascade of aPBMC-induced acquisition of myofibroblast phenotype by ASC. ASC cocultured with aPBMC or exposed to the secretome of aPBMC upregulated smooth muscle cell markers alpha smooth muscle actin (αSMA), SM22α, and Calponin I; increased contractility; and initiated expression of ActA. Interleukin (IL)-1β was sufficient to replicate this response, whereas blocking IL-1β eliminated aPBMC effects. ASC-derived ActA stimulated CTGF and αSMA expression in ASC; the latter independent of CTGF. Induction of αSMA in ASC by IL-1β or ActA-enriched media relied on extracellular enzymatic activity. ActA upregulated mRNA levels of several extracellular matrix proteins in ASC, albeit to a lesser degree than TGFβ1, and marginally increased cell contractility. In conclusion, the study suggests that aPBMC induce myofibroblast phenotype with weak fibrotic activity in perivascular progenitors, such as ASC, through the IL-1β-ActA signaling axis, which also promotes CTGF secretion, and these effects require ActA extracellular enzymatic processing.
Introduction
Vascular rarefaction and fibrosis are the primary contributors to organ dysfunction in many chronic ischemic and inflammatory conditions and with aging. Such pathologies are driven by interactions between resident, as well as circulating inflammatory cells and tissue-specialized cells, stromal cells, and vasculature. Uncovering the signaling pathways of these interactions will guide development of therapies to mitigate fibrosis and decline in tissue perfusion.
Studies over the past 15 years have provided strong evidence that tissues of adult organisms have high prevalence of mesenchymal stromal cells (MSC). 1 –3 Explorations of MSC niches revealed that MSC have vascular localizations, either periendothelial in smaller vessels 4,5 or in media adventitia in larger caliber vessels, 6 and expansion of the vascular networks during organ growth correlates with an increase of MSC in the organ. 7 The high prevalence of MSC in all tissues of adult organisms suggests that together with tissue-resident fibroblasts and macrophages, MSC, in response to stressors such as hypoxia, inflammation, reactive oxygen species, and toxins, may promote tissue recovery. However, if stress persists, MSC most likely contribute to the manifestation of pathology. Although much work has been done in evaluating the contribution of fibroblasts and macrophages to tissue homeostasis, 8,9 the role of MSC in these processes is understudied.
In addition to their endogenous activity, MSC, when extracted from their natural niche and used for therapeutic applications in animal models and clinical trials, effectively mitigate acute and chronic pathologies associated with inflammation and hypoxia. MSC improve perfusion of ischemic tissues, 10,11 decrease tissue loss (heart, brain), slow disease progression in chronic pathological conditions (heart), and even promote recovery of organ function (kidney, 12 pancreas 13 ). These effects are attributed to paracrine activities of MSC, which promote survival and proliferation of host cells (endothelial cells, 10 pancreatic beta-cells, 14,15 keratinocytes 16 ). Furthermore, MSC promote vasculogenic program of endothelial cells 17 –19 and vessel stability and remodeling, 4,20,21 modulate deposition of extracellular matrix, 22 and contribute to immune homeostasis. 23,24 Effects of MSC on inflammatory cells have been assessed by many groups, including the ability of MSC to promote pro- and anti-inflammatory phenotypes of macrophages and T-cells, depending on the level and composition of local inflammatory factors. 25 Although the effects of MSC on resident cells have received a lot of attention, the rapid clearance of MSC after administration 26 limits the extent to which effects of hypoxia and inflammation on donor MSC fate (postinfusion) can be explored.
Many pathologies with inflammatory and hypoxic components are accompanied by elevated systemic Activin A (ActA), a member of the TGFβ family of pleiotropic factors. Whether ActA promotes tissue recovery or exacerbates pathology is tissue dependent. Elevated ActA in the brain after insult 27 or traumatic injury 28 promotes organ recovery, 14 but its upregulation in the kidney after acute ischemic insult is detrimental. 29 Patients with heart failure show increased systemic ActA, which correlates with the severity of the condition. 30 Myocardial infarction is also associated with increased ActA, 31 but its contribution to disease progression and resolution is controversial. 31,32 Recent discovery that expression of ActA in endothelial cells is under hypoxia-inducible factor-1 (HIF-1α) regulation 33 suggests that ActA represents an early marker of local ischemia. 32,34 Furthermore, ActA may limit compensatory angiogenesis in ischemic tissues by virtue of its angiostatic activity 11,35 –37 and contribute to fibrosis. 38 Contribution of ActA to inflammation is multifaceted. Some studies suggest that ActA initiates the inflammatory cascade, 39,40 but other studies show it has anti-inflammatory activities. 41,42 Taken together, this suggests that there is much left to understand about effects of ActA, in particular in ischemia and during inflammation, and what controls its tissue-dependent and time-dependent actions.
It has been established that MSC and fibroblasts express ActA. 35,43,44 Furthermore, we have recently reported that activated immune cells, and interleukin (IL)-1β particularly, significantly upregulate ActA in adipose MSC. 37 At the same time, inflammation is known to be a key driver of fibrosis, and contribution of ActA to this process has been proposed. Although fibroblasts are recognized as the primary contributors to fibrosis, MSC may also play role in this pathology. 45 –47 In this study, interactions between adipose MSC and activated peripheral blood mononuclear cells (aPBMC) have been explored with the focus on MSC transition to myofibroblasts and role of ActA in these interactions. This study expands current knowledge on modulation of tissue-resident MSC by inflammatory cells.
Materials and Methods
Cell preparations
Adipose stromal cells (ASC), isolated and expanded as previously described, 4,48 were used at passages 3–6. Normal human cardiac fibroblasts (NHCF) and coronary artery smooth muscle cells (CASMC) were purchased from Lonza, expanded in FGM-3 or EGM-2MV (Lonza), and used at passages 3–6. Procedures for blood collection from healthy donors (ages 25–35, both sexes) were approved by the University of Florida Institutional Review Board. Blood was drawn into CPT tubes prefilled with Ficoll Hypaque solution (BD Biosciences), and PBMC were isolated by density gradient centrifugation. Isolated PBMC were resuspended in EBM-2/5% fetal bovine serum (FBS) and were used directly or activated with either 100 ng/mL lipopolysaccharide 49 (LPS, Sigma) (15 mins at room temperature, then washed to remove LPS) or with T-Cell Activation/Expansion Kit (Miltenyi Biotech, #130-091-441; beads coated with CD2, CD3, and CD28 IgGs). PBMC were also cryopreserved for future experiments.
ASC monocultures and ASC–PBMC cocultures
ASC were plated at 6 × 104 cells/cm2 and incubated in EBM-2/5% FBS. The next day, either nonactivated or activated PBMC (at quantities defined in the Results section) were applied to the ASC monolayer and incubated for up to 5 days in EBM-2/5% FBS alone or with (1) neutralizing ActA immunoglobulin G (IgG) (10 μg/mL); or (2) neutralizing IL-1β IgG (10 μg/mL); or (3) matching isotype control IgG (all from R&D Systems); or (4) SB431542 (10 μM, Sigma), an ALK4/5/7 receptor inhibitor; or (5) vehicle, 0.08% dimethyl sulfoxide (DMSO) (Sigma). ASC monocultures were also exposed to (1) IL-1β (0.01–10 ng/mL, GeminiBio) alone or with Aprotinin (FisherScientific, 400U/ml); (2) secretome of nonactivated PBMC (PBMC-S) diluted to 50% in control media; (3) secretome of activated PBMC (aPBMC-S; undiluted or diluted to 2%–50% in control media), alone or in the presence of neutralizing ActA IgG (10 μg/mL), or IL-1β IgG (10 μg/mL), or isotype control IgG.
To produce secretomes, PBMC or aPBMC were resuspended in EBM-2/5% FBS at 106 cells/mL and incubated for 24 h, then media were collected, centrifuged at 500g to remove cells and debris, and immediately used in experiments or cryopreserved until further use.
The concentrations of ActA, connective tissue growth factor (CTGF), follistatin (FST), and TGFβ1 in media conditioned by PBMC, ASC mono- and ASC+PBMC cocultures were measured by enzyme-linked immunosorbent assays (R&D Systems).
EC proliferation test
Cord blood–derived endothelial cells were plated at 5 × 103 cells/cm2 in EBM-2/5% FBS. The next day, cells in a subset of wells were stained with Hoechst 33342 to establish a “baseline” cell number, whereas the remaining wells were treated with fresh EBM-2/5% FBS alone or supplemented with secretomes of ASC, or ASC+PBMC, or ASC+aPBMC cocultures (in ratio of fresh media:secretome 1:1) for 4 days. Wells were then fixed, stained with 4′,6-diamidino-2-phenylindole (DAPI), imaged, and processed using Image J software for cell counts.
Cell transfection with small interfering RNA constructs
ASC were plated at 5 × 104 cells/cm2 in EBM-2/5% FBS and transfected with Inhibin Ba (INHBA, Origene) or CTGF (CCN2, ThermoFisher Scientific [TFS]) silencing RNA or corresponding scrambled RNA, using Lipofectamine RNAiMAX (TFS). Cells were used for experiments 24 h after transfection.
RNA isolation and reverse transcription-quantitative polymerase chain reaction
RNA was isolated from cells using NucleoSpin RNA II kit (Macherey-Nagel) and reverse-transcribed to cDNA using iScript Reverse Transcription Supermix (Bio-Rad). Quantitative polymerase chain reaction was performed with TaqMan Fast Advanced Master mix and TaqMan probes (TFS) on a QuantStudio 3 Real-Time PCR System (Applied Biosystems). Expression of β-actin was used for normalization. Gene expression was analyzed using QuantStudio™ Design & Analysis Software.
SDS-PAGE and immunoblotting analysis
Cells were lysed with Radio-Immunoprecipitation Assay (RIPA) buffer containing proteinase and phosphatase inhibitor cocktail (TFS). Lysates were clarified, and protein concentration was determined by Bradford assay. Samples were resolved on 12% Mini-PROTEAN TGX Precast Protein Gels (Bio-Rad) and transferred onto 0.2 μm PVDF membranes (Bio-Rad). Membranes, blocked in 5% bovine serum albumin (BSA)/Tris-buffered saline (TBS), were probed for alpha smooth muscle actin (αSMA, Sigma A2547, 1:1,400), Calponin 1 (CST, #17819, 1:1,000), SM22α (CST, #40471, 1:1,000), SMAD2,3 (CST, #8685, 1:1,000), pSMAD2 (CST, #3108, 1:500), β-actin (CST, #3700, 1:5,000), and β-tubulin (CST, #2128, 1:1,000). Protein bands were visualized with IRDye 800CW or IRDye 680RD secondary antibodies, imaged with an Odyssey CLx and quantitatively analyzed using Image Studio software (all LI-COR). Alternatively, membranes were incubated with Horseradish peroxidase (HRP)-conjugated secondary IgG, and bands were revealed with SuperSignal West Femto Maximum Sensitivity Substrate (TFS) and imaged with a ChemiDoc Imaging system (Bio-Rad) and quantitatively analyzed using ImageJ. Expression of β-actin or β-tubulin was used as loading controls.
Immunofluorescence
Cultures were fixed with methanol (−20°C, 5 min), blocked with 2% BSA/phosphate-buffered saline (PBS) (30 min), probed with αSMA IgG (Sigma) or mouse isotype control IgG for 1 h, washed in 0.05% Tween-20/PBS, and incubated with anti-mouse Alexa Fluor 594 IgG (Invitrogen) for 30 min. Nuclei were revealed with DAPI (Sigma). Images were captured using a fluorescent microscope (Nikon Ti with camera and NIS Elements Software) or scanned for quantitative analysis of fluorescence signal intensity using a CLARIOstar plate-reader (BMG Labtech). Alternatively, cells were incubated with αSMA IgG, followed by anti-mouse IRDye 680RD IgG for 1 h and then imaged and analyzed on an Odyssey CLx using Image Studio software (LI-COR).
Statistical analysis
Data analysis was performed using Prism 10 (GraphPad) and expressed as mean ± standard error of mean. An unpaired nonparametric Mann–Whitney test was used for statistical analysis of the data that included only two experimental groups. When one group was used for normalization and expressed as “1” or “100%,” one sample t-test was performed. For the data that had three or more groups, Shapiro–Wilk test was performed to confirm normal distribution, followed by one-way analysis of variance (ANOVA) with Tukey’s multiple comparisons. Kruskal–Wallis test was used for data that did not pass normal distribution test. Two-way ANOVA with Dunnett’s multiple comparisons test was used for longitudinal studies.
Results
Inflammatory cells induce a myofibroblast/smooth muscle cell phenotype in ASC
ASC at baseline express low levels of αSMA, a marker of smooth muscle cells/myofibroblasts, and its level was not changed by contact of ASC with naïve PBMC but was substantially upregulated when in contact with LPS-activated PBMC (aPBMC) (Fig. 1A). Expression of other markers of smooth muscle differentiation, such as SM22α and Calponin 1, was also upregulated in ASC when cocultured with aPBMC (Fig. 1B); however, expression of MYH11, a key marker of mature smooth muscle cells, was not detected (not shown). Upregulation of αSMA in ASC was also observed through treating cells with cell-free aPBMC secretome (aPBMC-S; media conditioned by aPBMC for 24 h) (Fig. 1C). Importantly, exposure of ASC to aPBMC-S for merely 24 h was sufficient to cause a profound upregulation of αSMA when assessed 4 days later. Extending treatment to 48 h produced an even stronger expression of αSMA (Fig. 1C). To evaluate the extent to which ASC acquire functional characteristics of myofibroblasts, a collagen matrix contraction assay was performed. ASC exposed to media alone or nonactivated PBMC for 5 days contracted collagen gels to 57.9 ± 4.6% and 56.6 ± 3.5% of their original size, respectively, over the first 24 h after cell embedding. aPBMC-treated ASC increased collagen matrix contraction by 10 percentage points to 46.7 ± 2.9% of their original size (Fig. 1D). Interestingly, mRNA expression of extracellular matrix proteins, such as collagens (Col1, 3, 5), nidogen (NID1, NID2), and fibronectin (FN), which are known to be associated with fibrosis, was unaffected in ASC exposed to aPBMC for 5 days (Fig. 1E). These findings suggest either an incomplete transition of ASC to myofibroblast phenotype at this duration of aPBMC exposure or their differentiation into a previously undefined cell type.
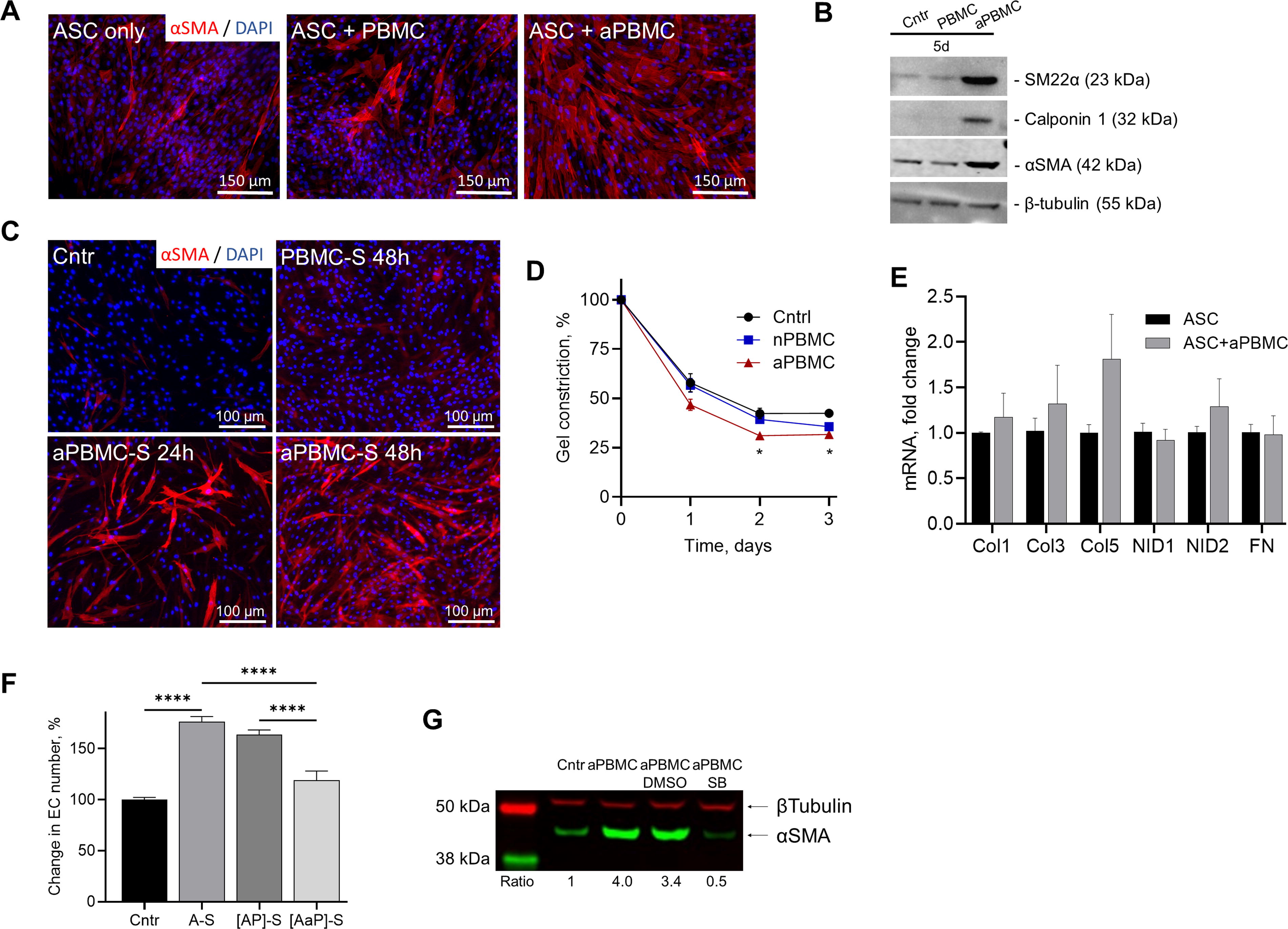
Activated PBMC upregulate markers of myofibroblast in ASC.
To test whether aPBMC affected the angiogenic secretory activity of ASC, EC were exposed to secretomes produced by ASC monocultures (A-S), or by ASC cocultured with either PBMC ([AP]-S) or aPBMC ([AaP]-S). EC exposed to A-S or [AP]-S showed a 75% and 60% increase in cell enumeration, respectively, whereas [AaP]-S was completely ineffective (Fig. 1F). This suggests that either aPBMC cause the release of factor(s) from ASC that converts their activity from angiogenic (promoting EC proliferation) to angiostatic (inhibiting EC proliferation) or aPBMC themselves release an angiostatic factor.
It has been established that both ActA and TGFβ stimulate myofibroblast phenotype acquisition by ASC. 35,50 Incubation of ASC+aPBMC cocultures in the presence of SB431542, which silences canonical ActA and TGFβ signaling pathways, prevented upregulation of αSMA in ASC (Fig. 1G), verifying that upregulation of αSMA by aPBMC was also attributable to the activities of either ActA or TGFβ.
Inflammatory cells upregulate expression of ActA in perivascular cells and fibroblasts
We have previously shown that aPBMC upregulate ActA in ASC. 37 Here, as an extension of that prior observation, a positive correlation between the number of aPBMC cocultured with ASC and the amount of ActA in incubation media was observed (Fig. 2A). Although aPBMC or ASC in monocultures did not produce ActA (in 48 h), it was detected in incubation media of all cocultures, even those with a low aPBMC number. Daily sampling of media from ASC+aPBMC cocultures revealed an initial substantial induction of ActA secretion in the first 24 h, followed by a steady rate of ActA accumulation over the next 6 days (the length of the experiment) (Fig. 2B).
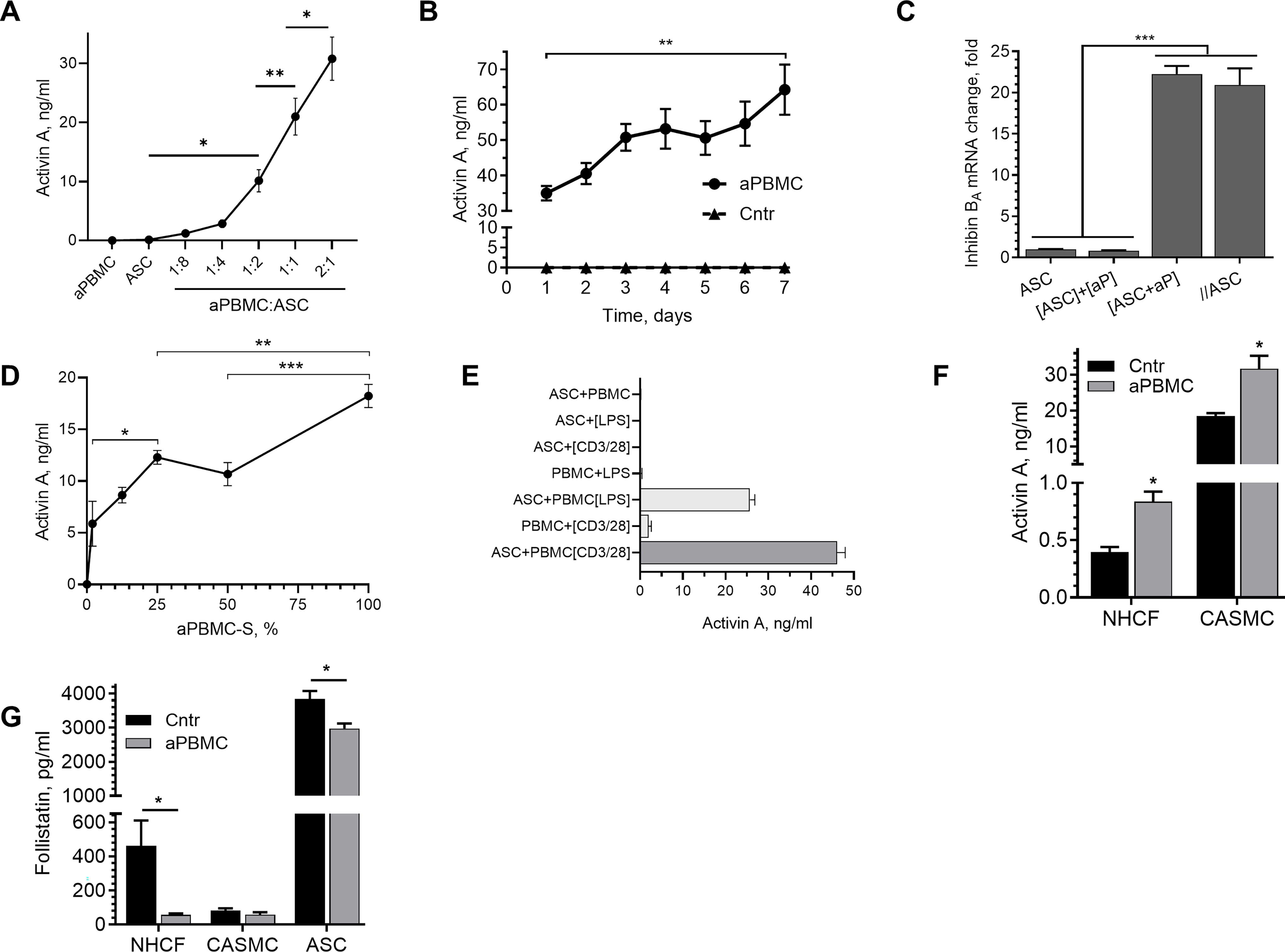
aPBMC induce Activin A (ActA) in perivascular cells.
To confirm that ASC were the source of ActA in ASC+aPBMC cocultures, ASC and aPBMC were incubated in the same well but separated by a membrane with 0.4 µm pore size for 24 h, and Inhibin Ba (homodimer of ActA) mRNA expression was analyzed in each cell type. Inhibin Ba expression was upregulated in ASC, separated from aPBMC, to the same extent as in the ASC+aPBMC cocultures with cell contact (Fig. 2C). This suggested that induction of ActA in ASC was attributable to paracrine activity of aPBMC. Importantly, isolation of mRNA from aPBMC samples, corresponding to aPBMC numbers is ASC+aPBMC cocultures, did not produce sufficient material to conduct downstream tests, suggesting negligible contribution of PBMC mRNA to the overall changes in mRNA in cocultures.
Analysis of the dose-response curve of various dilutions of aPBMC-S on ActA secretion by ASC revealed a positive correlation with no sign of reaching a plateau even with undiluted secretome (100%) (Fig. 2D).
Activation of either subset of PBMC with LPS (monocyte/macrophage) or anti-CD2/CD3/CD28 IgGs (lymphocyte) resulted in similar levels of ActA secretion by ASC subjected to aPBMC (Fig. 2E).
To assess whether upregulation of ActA expression in response to inflammatory stimuli is a particular feature of ASC or a common response of other perivascular cells, CASMC and NHCF were exposed to aPBMC. Unlike ASC, these cells secreted ActA at baseline, whereas aPBMC stimulation further increased its secretion (Fig. 2F). In parallel, a decrease in secretion of follistatin, an endogenous inhibitor of ActA, was observed in cocultures of ASC and NHCF with aPBMC but unchanged in CASMC+aPBMC cocultures, the latter of which may be attributed to its already low level of expression at baseline (Fig. 2G).
ActA mediates aPBMC-induced ASC differentiation and decrease in ASC mitogenic effect on EC
To test whether ActA mediates aPBMC-induced phenotypic switch in ASC, cells were exposed to aPBMC-S in the presence of neutralizing ActA IgG. Although strong upregulation of αSMA was observed in ASC exposed to aPBMC-S alone or premixed with isotype control IgG, ActA IgG blocked this effect (Fig. 3A). Also, based on semi-quantitative western blotting, no upregulation of αSMA was observed in ASC exposed to aPBMC in the presence of ActA IgG (0.8 of control) (Fig. 3B) or when aPBMC were introduced to ASC transfected with Inhibin Ba silencing RNA (0.7 of control) (Fig. 3C) versus control ASC cocultured with aPBMC (2.7-fold) and scrambled RNA-transfected ASC cocultured with aPBMC (1.7-fold).

Activin A mediates aPBMC-induced acquisition of myofibroblast phenotype by ASC.
It is well established that factors secreted by ASC promote EC proliferation, 3,36 whereas factors secreted by aPBMC cause cell loss. 37 However, whether aPBMC affects ASC paracrine angiogenic activity has not been established. As we have shown previously and confirmed in Fig. 3D, exposure of EC to ASC-S stimulated EC proliferation (75% increase in cell enumeration), but here we also show that medium conditioned by cocultures of ASC with aPBMC was ineffective. However, if this medium was pretreated with ActA neutralizing IgG, a complete restoration of EC mitogenic response was observed (Fig. 3D).
Concomitant with increased ActA, analysis of TGFB expression in ASC in response to aPBMC revealed an increase in mRNA expression for all three TGFB forms (Fig. 3E). In parallel, ASC treated with ActA (25 ng/mL) for 24 h show no change in TGFB1 expression, a decrease in TGFB2 mRNA by 60% and a fourfold increase in TGFB3 mRNA (Fig. 3F).
As TGFβ induces fibrosis, the ability of ActA to affect well-established characteristics of myofibroblasts, for example, expression of extracellular matrix proteins, in aPBMC-treated ASC was also assessed. Exposure of ASC to ActA did not change the expression of collagens 1, 3, 4, and 5, which, except for collagen 4, was opposite to the effect TGFβ1 had (Fig. 3G). However, ASC treated with ActA show a slight increase in collagen matrix contraction (Fig. 3H).
aPBMC-induced ActA expression in ASC is mediated by IL-1β
We have previously observed that IL-1β induces ActA in ASC. 37 ActA was detectable in media conditioned by ASC treated with 0.1 ng/mL IL-1β and dramatically increased in response to 1 ng/mL IL-1β, with no further change with the next 10-fold increase in IL-1β concentration (Fig. 4A). Accumulation of ActA in ASC incubation media was detectable 4 h after ASC was exposed to 10 ng/mL IL-1β (1.1 ± 0.1 ng/mL) and linearly increased over the next 8 h (23.8 ± 7.3 ng/mL) after which it reached a plateau (Fig. 4B). Exposure of ASC to IL-1β (10 ng/mL) for 5 days substantially increased αSMA expression (Fig. 4C), but this effect was eliminated if incubation medium was admixed with either ActA IgGs or SB431542, an inhibitor of the ActA receptor (Fig. 4D). Furthermore, only a minor upregulation of αSMA was detected in ASC treated with aPBMC-S in the presence of IL-1β neutralizing IgG (Fig. 4E). Collectively, these data suggest that aPBMC-secreted IL-1β is necessary and sufficient to upregulate both ActA and αSMA in ASC.
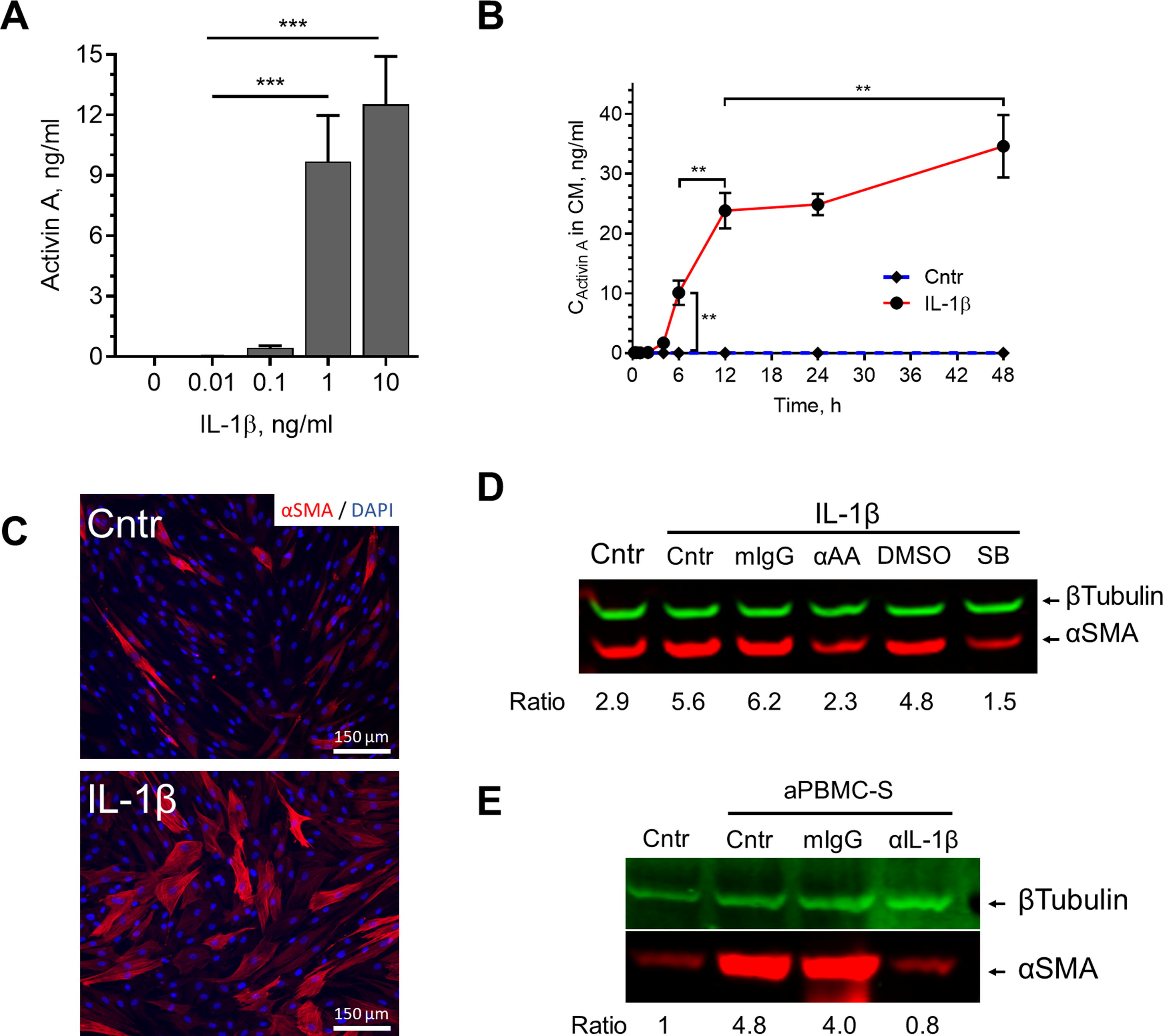
IL-1β mediates aPBMC-induced expression of ActA in ASC.
ActA mediates aPBMC-induced CTGF expression in ASC
CTGF is a known profibrotic factor 51,52 that is highly expressed in ischemic and inflammatory conditions. 53 –55 A twofold increase in CCN2 (CTGF) mRNA was observed in ASC after 24 h of exposure to aPBMC (Fig. 5A). Interestingly, CTGF protein was not detectable until 72 h of exposure (Fig. 5B). As TGFβ and ActA have similar actions on mural cells and fibroblasts, and it is well established that CTGF mediates the profibrotic effects of TGFβ, 56,57 the contribution of CTGF to the effects of ActA was evaluated. A 5.5-fold increase in CCN2 mRNA was detected in ASC after 24 h of stimulation with ActA (25 ng/mL) (Fig. 5C), which was accompanied by a threefold increase in CTGF protein in incubation media (Fig. 5D). To assess whether ActA was the sole mediator of induction of CTGF by aPBMC, aPBMC were incubated with either unmodified ASC or cells transfected with Inhibin Ba small interfering RNA (siRNA) or scrambled RNA (>80% silencing efficiency was confirmed at mRNA level; not shown). Intact ASC and ASC transfected with scRNA, but not with Inhibin Ba siRNA, upregulated CTGF secretion in response to aPBMC, suggesting that ActA expression was necessary for upregulation of CTGF secretion (Fig. 5E).
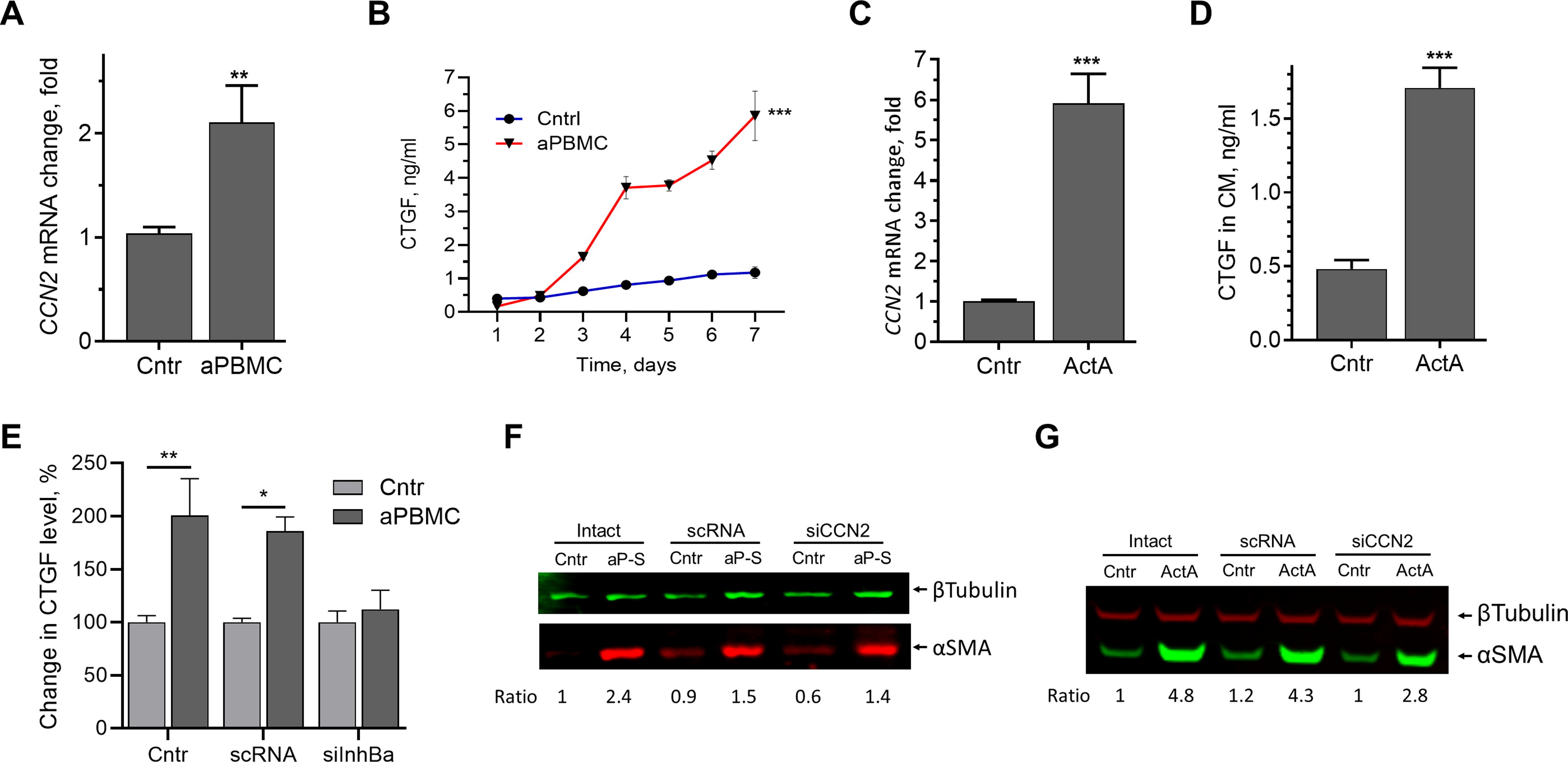
ActA mediates aPBMC-induced CTGF expression in ASC.
CTGF does not mediate ActA-induced αSMA expression in ASC
To define the role of CTGF in aPBMC-induced expression of αSMA in ASC, cells transfected with CCN2 siRNA were exposed to aPBMC secretome (aP-S) for 5 days (>90% silencing efficiency was confirmed at mRNA level; not shown). Expression of αSMA was similar in control and CCN2-silenced ASC (Fig. 5F) and 5 days of exposure to ActA (25 ng/mL) upregulated αSMA expression in CCN2-silenced ASC to the level just 35% below of its level in scRNA-transfected ASC (Fig. 5G).
ActA requires extracellular enzymatic activation to display its activity
ActA is produced as a proprotein that requires enzymatic activation to attain full biological activity. 58,59 Proprotein convertase furin is considered the primary candidate for activation of the proprotein of ActA; however, whether this occurs intra- or extracellularly is not well-defined. 60 To test whether ActA secreted by ASC requires extracellular proteolytic activation, ASC were treated with either IL-1β alone or aprotinin, an inhibitor of multiple serine proteases, including plasmin, trypsin, kallikrein, and furin. 61 Although IL-1β induced a 2.2-fold increase in αSMA, it had only a minor effect (37% increase) in the presence of aprotinin (Fig. 6A, B). In parallel, naïve ASC exposed to ASC-secretome enriched with ActA (by exposure of ASC to IL-1β followed by secretome collection) strongly upregulated αSMA, but aprotinin inhibited this effect (Fig. 6C, D). Finally, aprotinin decreased the secretion of ActA by IL-1β-stimulated ASC by 40% (Fig. 6E), which could be attributed to the positive autocrine loop regulating ActA expression by ASC. Specifically, treatment of ASC with ActA led to upregulation of Inhibin Ba mRNA (Fig. 6F).
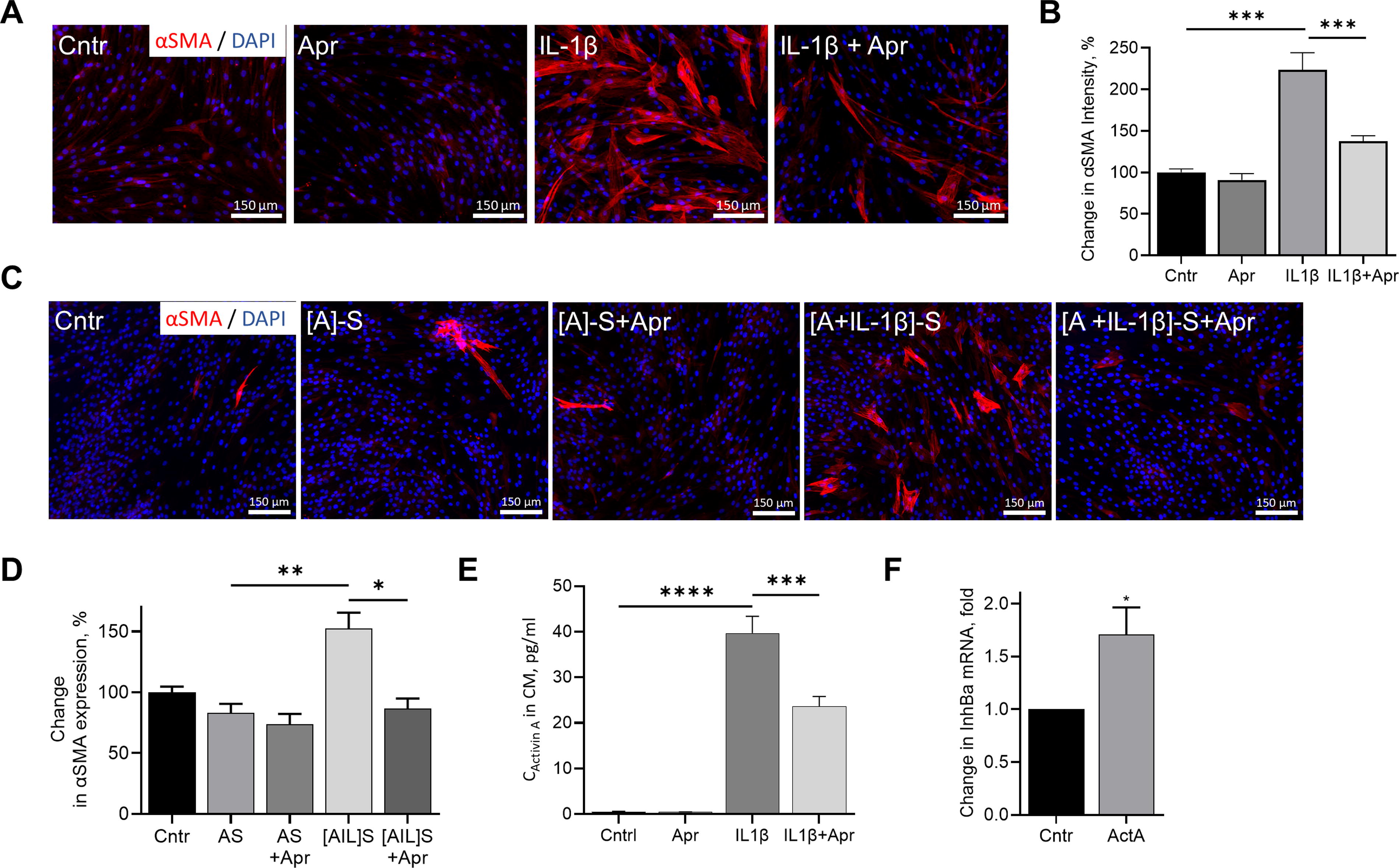
ActA activity depends on extracellular enzymatic processing.
Discussion
Inflammation promotes myofibroblast phenotype in ASC through IL-1β–ActA signaling axes
Persistent and severe hypoxia and chronic inflammation are major drivers of many pathologies, including heart failure, metabolic syndrome, or chronic kidney disease, often manifesting as decline in tissue perfusion (vessel rarefaction) and fibrosis, and eventually loss of function. Although tissue-resident fibroblasts and epithelial-to-mesenchymal transition are considered to be the primary contributors to fibrosis, the role of mesenchymal progenitors, despite their high prevalence in tissue stroma, is not well defined in this process. In this study, mesenchymal cells from adipose tissue were used as an example of tissue-resident perivascular/stromal cells. Most of the experiments were conducted with immune cells pretreated with LPS, which activates monocytes, macrophages, dendritic cells, 62 neutrophils, 63 and B cells. 64 It could be argued that such an approach models the immune response to bacterial infection, which might be represented by a specific profile of secreted cytokines and might not properly capture the inflammatory milieu of sterile inflammations of many pathologies associated with fibrosis. Although we have confirmed some of our observations with activated T cells (PBMC subjected to CD2/CD3/CD28 IgG), 65 further studies are needed to explore whether immune cells activated by other stressors induce myofibroblast phenotype in MSC and whether it is mediated by IL-1β–ActA signaling pathway.
ASC, when exposed to activated immune cells or their secretome, acquire myofibroblast markers, including αSMA, SM22α, and Calponin, and show increased contractility. Although activated immune cells secrete a wide variety of cytokines, we have identified IL-1β as the primary factor both necessary and sufficient for upregulation of myofibroblast markers in ASC from healthy donors (Fig. 1B). The general role of IL-1β in myofibroblast formation has been controversial. Conversely, several groups have reported that IL-1β blocks TGFβ-induced production of αSMA, calponin, collagen I, and fibronectin in human lung and dermal fibroblasts. 66,67 An explanation of such contradictory results is not yet clear, suggesting the need for further studies delineating the effects of IL-1β on (myo)fibroblasts.
It is important to recognize that even short (24 h) exposure to inflammatory stimuli was sufficient to prompt conversion of ASC into myofibroblasts (Fig. 1C). The fact that such brief stimulation was sufficient to set into motion a several-day long program of ASC acquisition of myofibroblast phenotype suggested that ASC respond to inflammatory stimuli by secreting factor(s) which perpetuates differentiation in autocrine fashion. Whether this causes a terminal or temporary and reversible transition of ASC into myofibroblasts requires further evaluation.
This study revealed that IL-1β-induced acquisition of myofibroblast phenotype by ASC was mediated by ActA (Fig. 4A–D). Importantly, ASC continued to secrete ActA as long as IL-1β was present, suggesting that prolonged inflammation will be accompanied by a persistently increased level of ActA and promotion of a myofibroblast program.
While intact ASC contract collagen gels, their differentiation into myofibroblasts led to a slight, but significant, increase in contractile activity. However, no dramatic increases in several tested extracellular matrix proteins were observed. These findings further support the notion that ASC, while sharing many phenotypic and functional characteristics with fibroblasts, 68,69 have distinct responses to similar stimuli. We hypothesize that exposure of ASC to inflammatory environment does not result in generation of myofibroblasts with strong fibrotic activity.
Although aPBMC upregulated TGFB1-3 in ASC (Fig. 3E), aPBMC-induced upregulation of αSMA was completely eliminated by blocking ActA. This suggests that TGFβ1–3 have limited, if any, contribution to αSMA upregulation in ASC. Although this observation challenges the prior dogma defining TGFβ as a primary driver of myofibroblast, 70 this study complements our prior work, showing that ActA is a primary factor responsible for acquisition of smooth muscle cells (SMC) phenotype by human ASC within vascular cords. 35 This may further support the notion that ASC differ from fibroblasts in regard to mechanism of their transition to myofibroblasts by relying primarily on ActA rather than on TGFβ; however, side-by-side studies are needed to clarify signaling pathways responsible for induction of myofibroblast phenotype in both cell types in response to activated immune cells.
In addition to their endogenous role, isolated and in vitro expanded ASC possess strong therapeutic effects per local or systemic delivery. Multiple studies have characterized ASC secretome as proangiogenic and used this finding to explain their therapeutic effects in ischemic conditions. However, it is important to recognize that most findings have been made with ASC in monoculture and contribution of other cells to ASC secretome has been disregarded. This study has revealed that although ASC produced angiogenic secretome in monocultures, 4,10 exposure to inflammatory cells caused loss of angiogenic properties of secretome via a pathway requiring IL-1β and ActA. This finding should be considered when planning to use ASC to stimulate angiogenesis in the tissues that manifest inflammation and perhaps patients with ischemia will have better recovery if treated with ASC secretome produced in optimal in vitro conditions rather than when treated with ASC directly.
ActA does not require CTGF to induce SMC/myofibroblast phenotype in ASC
TGFβ, as well as thrombin 71 and angiotensin, 72,73 possesses profibrotic activities, which are mediated by CTGF. 56,74,75 These factors also induce ActA, which, as established in this and other studies, 76 –78 upregulates αSMA in stromal cells. A prior study by Kusama et al. suggested that induction of αSMA in endometrial stromal cells by ActA was dependent on CTGF 76 ; however, studies with silencing CTGF have not been performed to confirm this hypothesis. The current study extends these previous observations by showing that ActA induces CTGF in ASC (Fig. 5C, D), but contrary to the prior study, it shows that ActA-induced αSMA expression in ASC was CTGF-independent (Fig. 5G). Although it is established that TGFβ-induced fibroblast-to-myofibroblast transition is CTGF dependent, additional studies are required to define to what extent ActA effects are CTGF-dependent and whether ActA is necessary for TGFβ effects, as suggested by Wada et al. 78
ActA extracellular activation
ActA, like many other members of TGFβ family, is produced as a proprotein and undergoes proteolytical cleavage to form an active protein. Previous reports have suggested that activation occurs in Golgi apparatus and is mediated by endoprotease (proprotein convertase) with furin as a primary candidate for such activity. 58,79 ActA is a homodimer of Inhibin Ba, with the apparent MW of each propeptide being approximately 47 kDa, with the resultant inactive homodimer at 92 kDa. 58 The premature protein gets cleaved to two prodomains (∼33 kDa each) and one mature dipeptide (24 kDa). 58 It has been reported that in addition to cytoplasmic activity, furin is active at the cell membrane, suggesting a possibility of extracellular activation of ActA. 60,80 Previous analysis of secretion and activities of intact versus cleaved forms of ActA has been limited to their effects on secretion of follicular-stimulating hormone and the results are contradictory. As such, only active form of ActA induced follicular stimulating hormone release from rat pituitary cells in in vitro test, 81 whereas systemic infusion of either form led to release of follicular stimulating hormone. 82
The current study revealed that aprotinin, an inhibitor of furin, prevented expression of αSMA in ASC stimulated with IL-1β or [ASC+IL-1β]-S, a media enriched with ActA (Fig. 6A–D). Lower level of αSMA in ASC treated with IL-1β+aprotinin correlated with a 40% lower level of ActA in incubation media (Fig. 6E), suggesting that interference with ActA activation disrupts a positive feedback loop important for sustained ActA secretion. In parallel, diminished upregulation of αSMA in ASC treated with [ASC+IL-1β]-S supports the significance of ongoing extracellular activation of ActA.
Footnotes
Data Availability
All data produced during this study are included in the article. Raw data are available on request.
Author Disclosure Statement
All authors declare no conflict of interest. No competing financial interests exist.
Funding Information
This work was supported by Veterans Administration Merit Review [1I01BX003888-01] and the UF Center for Regenerative Medicine and Gatorade Foundation.