
Abstract
Neural organoids derived from human-induced pluripotent stem cells (iPSCs) provide a model to study the earliest stages of human brain development, including neurogenesis, neural differentiation, and synaptogenesis. However, neural organoids lack supportive tissues and some non-neural cell types that are key regulators of brain development. Neural organoids have instead been cocultured with non-neural structures and cell types to promote their maturation and model interactions with neuronal cells. One component of the brain that does not form de novo in neural organoids is the meninges, a trilayered structure that surrounds the central nervous system and secretes key signaling molecules required for mammalian brain development. Most studies of meninges-brain signaling have been performed in mice or using two-dimensional cultures of human cells, which do not accurately recapitulate the architecture and cellular diversity of the tissue. To overcome this, we developed a coculture system of neural organoids generated from human iPSCs fused with fetal leptomeninges (LPM) from mice with fluorescently labeled meninges (Col1a1-GFP), which we call leptomeningeal neural organoid (LMNO) fusions. This proof-of-concept study tests the stability of the different cell types in the LPM (fibroblasts and macrophages) and the fused neural organoid (progenitors and neurons), as well as the interface between the organoid and meningeal tissue. We test the longevity of the fusion pieces after 30 and 60 days in culture, describe best practices for preparing the meninges sample before fusion, and examine the feasibility of single or multiple meninges pieces fused to a single organoid. We discuss potential uses of the current version of the LMNO fusion model and opportunities to improve the system.
Introduction
The development of the human central nervous system (CNS) is a complex process requiring the precise coordination of signaling molecules and various cell types. Our understanding of human CNS development is vastly derived from rodent and nonhuman primate models, along with the investigation of postmortem human brain tissue. Neural organoids derived from human pluripotent stem cells, both embryonic stem cells and induced pluripotent stem cells (iPSCs), have become useful tools for understanding and mimicking various in vivo processes of human brain development. Neural organoids model aspects of neurogenesis and synaptogenesis, and produce a heterogenous mixture of cell types. 1 –3
Neural organoids can form distinct layers, reminiscent of cortical layers found in vivo, and bear an organizational and transcriptomic resemblance to the gestational human cortex. 3 –5 Additionally, small molecules and other morphogens can be used to pattern the organoids into specific cell fates or encourage the patterning into different developmental regions, such as telencephalic or cerebellar-like neural organoids. 2,6,7 Thus, neural organoids have become central tools for studying early human CNS developmental processes and for translational applications.
One major drawback of current human neural organoid platforms is their lack of support structures that are required for proper CNS development and function in vivo. 2,8,9 These are structures or cell types that are not derived from the neuroectoderm that give rise to CNS structures including vasculature, microglia, and meninges. Some strides have been made to incorporate microglia and vasculature into neural organoids. 8,9 While these systems have improved our understanding of interactions between neural and non-neuronal cell types within neural organoid models of human CNS development, these platforms still lack many of the characteristics and spatial organization of human brain morphology. The meninges is a trilayer structure composed of fibroblasts, blood vessels, and immune cells that encase the entire CNS and is key for proper formation and maintenance of the CNS in vivo. It has been well established that the meninges play an essential role in brain development by the secretion of important factors: meningeal retinoic acid is required for cortical development in mice and humans 10 –12 meningeal CXCL12 directs Cajal-Retzius cell migration, 13 and the pia layer of the meninges forms the basement membrane interface, which is required for radial glial cell attachment. 14 Despite this, few attempts have been made to incorporate meningeal tissue into neural organoid models, and much of the work to understand interactions between the meninges and neuronal cell types during development has been conducted in rodent models or meningeal cell lines.
Overall, there is a need for human neural organoid systems that incorporate the meninges as a key component to achieve higher complexity. Here, we describe leptomeningeal neural organoid (LMNO) fusions, a three-dimensional coculture system generated from iPSC-derived human neural organoids fused with embryonic leptomeninges (LPM) from Col1a1-GFP mice. We show that the meningeal compartment of LMNOs retains key characteristics of LPM in vivo, such as layer-specific fibroblast markers and resident immune cells, up to 60 days in culture. We also find that neurons in the LMNO fusions undergo less cell death compared to neurons in nonfused organoids. Finally, we find evidence of iPSC-derived human brain organoid Reelin+ Cajal-Retzius-like neuronal cells localizing proximal to the meningeal compartment of LMNOs. This proof-of-concept study provides an important initial framework for the incorporation of meninges into neural organoid platforms.
Materials and Methods
Animals
All mice were housed in specific-pathogen-free facilities approved by AALAC and were handled in accordance with protocols approved by the IACUC committee on animal research at the University of Colorado, Anschutz Medical Campus. The following mouse line was used in this study: Col1a1-GFP 15 on a C57/BLK6J background. GFP+ embryos were identified by fluorescence detection using a stereoscope equipped with a fluorescent light source.
Bioinformatic analysis
Published human embryonic and fetal brain single nuclei datasets were generously provided by Dr. Aparna Shah 16 and Dr. Alexandro Trevino 17 as R objects (Dr. Shah) or were available as raw single-cell data from the NeMO Repository at (https://assets.nemoarchive.org/dat-0rsydy7) and GEO: GSE162170. From individual datasets containing nuclei isolated from Carnegie Stage 12–22 (corresponding to gestational weeks or GW 6–10) cortex/telencephalon, hindbrain, midbrain, or thalamus, clusters with expression of putative meningeal mesenchymal cell-enriched genes (COL1A1, COL1A2, LUM, S100A6, CRABP2, TBX18, FOXC1, DCN, NNAT, ALCAM, and BGN), endothelial genes (CLDN5, 321 PECAM1, SOX17), mural cell genes (RGS5, ABCC9, KCNJ8), or macrophages/monocytes/microglia (MRC1, CD74, AIF1, P2YR12) were subset, integrated, and reclustered. This dataset was further subsetted to remove a cluster of putative neural cells and reclustered at 0.8 resolution, leaving clusters of putative mesenchymal (combined meningeal mesenchymal and pericyte markers), endothelial, and macrophages/monocytes/microglia. From a dataset containing nuclei isolated from GW16-22 human cortex, previously annotated clusters 16 (“microglia”), 19 (“pericyte”), 20 (“endothelial cell”), and 22 (“vascular and leptomeningeal cell”) were subset, and cluster analysis was performed. Analysis of putative meningeal mesenchyme, pericyte/mural cell, monocyte/macrophage/microglia, and endothelial markers in the dataset showed the presence of clusters with neural identity. This dataset was further subsetted to remove a cluster of putative neural cells and reclustered at 0.8 resolution, leaving clusters of meningeal, pericyte/mural cell, monocyte/macrophage/microglia, and endothelial cells.
Human iPSCs
The WTC11 iPSC line used in this study was maintained in E8 medium in plates coated with Matrigel (Corning, cat. # 354277) at 37°C with 5% CO2. The culture medium was changed daily. Cells were checked daily for differentiation and were passaged every 3–4 days using Gentle Cell Dissociation solution (STEMCELL Technologies, cat. # 07174). All experiments were performed under the supervision of the Vanderbilt Institutional Human Pluripotent Cell Research Oversight Committee. Cells were checked for contamination periodically.
Neural organoids
Cerebral organoids were generated as previously described 1,2,18 with some modifications. Briefly, organoids were generated using the STEMdiff™ Cerebral Organoid Kit (STEMCELL Technologies; cat. # 08571, 08570), supplemented with dual SMAD inhibitors. iPSCs were dissociated into single cells using Gentle Cell Dissociation Reagent (STEMCELL Technologies, cat. # 07174) for 8 min at 37°C. Homogeneous and reproducible EBs were generated by using a 24-well plate AggreWell™ 800 (STEMCELL Technologies, cat. # 34815). On day 7, high-quality EBs were embedded in Matrigel (Corning, cat. # 354277). On day 10, the Matrigel coat was broken by pipetting up and down, and the healthy organoids were transferred to a 60 mm low-attachment culture plate (Eppendorf, cat. # 003070119). The plates were then moved to a 37°C incubator and a Celltron benchtop shaker for CO2 incubators (Infors USA, cat. # I69222) set at 85 rpm. Full media changes were performed every 3–4 days for 15 days.
Isolation of LPM
Embryos were collected from Col1a1-GFP pregnant dams at E16. Leptomeningeal tissue was dissected using previously established methods. 14 Briefly, a single lateral cut is made from the back of the head toward the eye, and the skin and calvarium are lifted dorsally, exposing the brain surface and GFP+ LPM. Whole pieces of LPM were then peeled off the forebrain (telencephalon) using a dissecting scope and then stored in ion-free HBSS on ice until placed into the culture system.
Generation of LMNO fusions and LPM aggregates
LPM from mice at E16 of embryonic development were isolated (described above) and seeded together with neural organoids (for LMNO fusions) or alone (for LPM aggregates) in a round-bottom tube placed within a modified 6-well plate. For LMNO fusions, the LPM fused to forebrain neural organoids within 6 days and were kept in culture for 30–60 days, with complete media changes and imaging using a fluorescent stereoscope every other day. For LPM aggregates, the LPM self-aggregated and were kept in culture for 14–28 days, with complete media changes and stereoscopic imaging every other day.
Immunohistochemistry and RNAscope
LMNO fusions and LPM aggregates were collected and sectioned prior to immunohistochemistry and detection of mRNA using RNAScope (days 30 and 60 for LMNOs, days 14 and 28 for LPM aggregates). LMNO fusions were fixed in 4% paraformaldehyde for 15 min at 4°C, washed in PBS, and then incubated in a 20% sucrose gradient at 4°C prior to embedding and flash-freezing in a solution of 7.5% gelatin/10% sucrose. LPM aggregates were fixed using the same methods and flash-frozen in Tissue-Tek OCT (Sakura, 4585). LMNO fusions and LPM aggregates were sectioned on a cryostat (Leica CM1950) at a thickness of 15 μm. For detection of leptomeningeal components and assaying for cell death, the following antibodies were used: CD206 (goat, R&D Systems, AF2535), Lyve1 (rat, R&D Systems, MAB2125), E-cadherin (ECAD) (mouse, BD Biosciences, 610181), Claudin-11 (rabbit, Invitrogen, 36-4500), CRABP2 (mouse, Sigma-Aldrich, MAB5488), and Cleaved Caspase-3 (rabbit, Cell Signaling Technology, 9661). For immunolabeling of Cajal-Retzius cells, the following antibodies were used: Reelin (rabbit, Proteintech, 20689-1-AP) and mitochondria (mouse, Abcam, ab92824). All primary antibodies were used at a dilution of 1:100. For detection of Cxcl12 transcript, the RNAScope Manual Detection Kit v.2.0 from ACDBio (Cat. No. 323100) and probe against mouse Cxcl12 (Cat. No. 422711-C3) was used.
Image acquisition
Confocal images were acquired on a Zeiss LSM 900 or Nikon Ti2 inverted light microscope equipped with a Yokogawa CSU-X1 spinning disk head, with 488, 561, and 647 nm excitation LASERs, 384, and Photometrics Prime 95B sCMOS camera.
Results
Cellular composition of neural organoids and leptomeningeal tissue
Prior to fusing, day 15 human neural organoids have begun to develop ventricular-like zones, which recapitulate the developing cytoarchitecture of the human cortex (Fig. 1A). The cells surrounding these ventricular-like zones express the early neural progenitor markers, PAX6 and SOX2 (Fig. 1A, B). By day 15, a few MAP2+ neurons start to emerge from the ventricular zone. This organization in neural organoids is intrinsic and develops prior to fusion with LPM (Fig. 1A, B). Absence of labeling for CTIP2+ deep-layer neurons show that at this time point, these cells have not yet emerged.
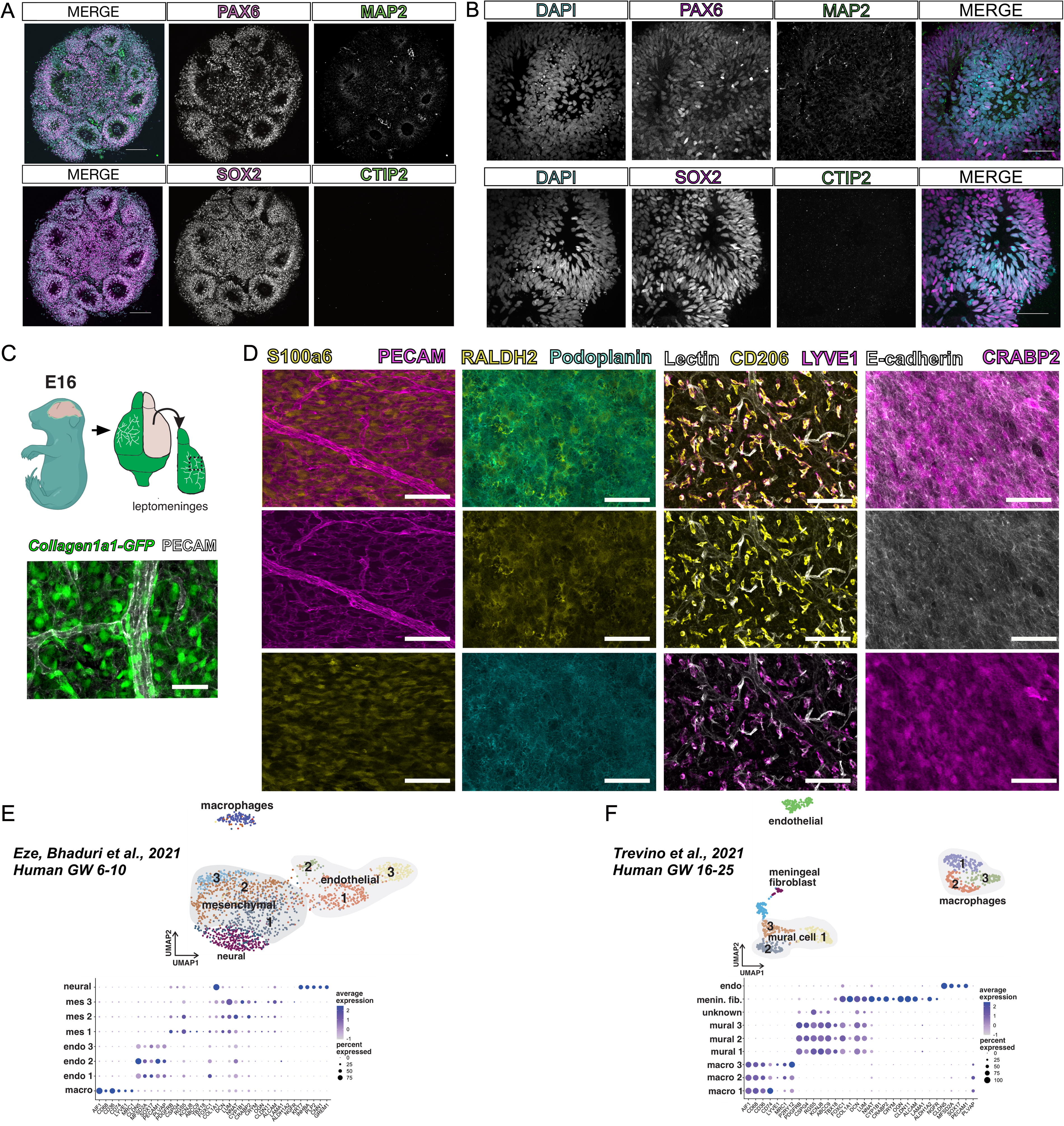
Characterization of neural organoids and mouse fetal meninges prior to fusion.
The LPM is comprised of the inner two layers of the meninges: the pia and the arachnoid. It contains pial and arachnoid fibroblasts that produce extracellular matrix (ECM), retinoic acid, CXCL12, and bone morphogenetic proteins (BMPs) vital for brain development. The mouse LPM can be isolated as a separate structure from the brain and the outer layer of the meninges, the dura. We isolated meninges from the Collagen1a1-GFP (Col1a1-GFP) reporter mouse line in which all fibroblasts are labeled with GFP (Fig. 1C, schematic); using previously described whole-mounting techniques 14 combined with immunofluorescent labeling, we can visualize Col1a1-GFP+ fibroblasts and the associated meningeal vasculature (Fig. 1C). Single-cell profiling studies of the meninges reveal unique layer-specific fibroblast subtypes and tissue-resident macrophages called border-associated macrophages (BAMs). 15 These cell populations can be visualized in the dissected LPM using specific markers: pia layer fibroblasts labeled by S100a6, arachnoid fibroblasts labeled with RALDH2 and PDPN, pseudoepithelial arachnoid barrier cells labeled by ECAD and CRABP2, and BAMs labeled by CD206 and LYVE1 (Fig. 1D).
We previously showed that some meningeal fibroblast layer-specific markers (e.g., S100a6, CRABP2) are shared between fetal mouse and human meninges. 15 To further test this, we used published single nuclear RNA sequencing datasets from early embryonic (human gestational weeks 6–10) and later fetal (gestational weeks 16–25) human brain development to identify cell clusters that are representative of putative meningeal mesenchymal/fibroblast, meningeal macrophage, and vascular cells (Fig. 1D, E). At early embryonic stages, three mesenchymal cell clusters were identified, with one of the clusters (mes 3) showing enriched gene expression of previously identified mouse fetal pia (NGFR, LAMA1, CYP1B1, LUM) and arachnoid cells (CLDN11, ALCAM) (Fig. 1E). The analysis of clusters at later stages of fetal human brain development identified a meningeal fibroblast-enriched cluster, likely representing a mixture of arachnoid and pial cells (Fig. 1F), as first identified in mouse fetal meninges. At both stages, we identified endothelial cell clusters with CNS-specific gene expression profiles (low PLVAP, high CLDN5, SOX17) and macrophage clusters with MRC1 and LYVE1 (Fig. 1E, F), consistent with studies on vascular and macrophage development in mice. This indicates that murine LPM contain meningeal fibroblasts, macrophages, and vasculature populations that are transcriptionally similar to embryonic and fetal human meninges. Further, these data provide supportive evidence that mouse LPM are a reasonable proxy for human-derived tissue.
Generation of LMNO fusions
To develop LMNO fusions, neural organoids were first generated from iPSCs as previously described 3,16 and grown to day 15 in culture (Fig. 2A). Then, LPM were isolated from embryonic day (E)16 Col1a1-GFP+ mice, in which meningeal fibroblasts express GFP under the Col1a1 promoter. 17 Prior to isolation of the meninges, embryos were perfused with ion-free HBSS on ice to remove blood contents (Fig. 2A). Then, the brains were removed, and the LPM were dissected from each forebrain hemisphere (Fig. 2A), as previously described. 19 Isolated meninges were collected in sterile, ion-free HBSS on ice until ready to be transferred into culture with neural organoids (Fig. 2A). One neural organoid was transferred along with 2–4 pieces of Col1a1-GFP+ forebrain LPM into one well of a static culture system consisting of a round-bottom Eppendorf tube secured with hot glue to the bottom of each well of a 6-well plate (Fig. 2A). Each well was filled with ∼300–500 µL of organoid differentiation media and given complete media changes every other day (Fig. 2A). The progression of the neural organoid-meninges fusion was monitored by capturing bright-field images every other day (Fig. 2A, B). On day 3 in culture, the organoid and meninges components remained mostly separate (data not shown), but by day 6, we observed fusion between the neural organoid and meninges, thus generating LMNOs (Fig. 2B). LMNOs remained fused throughout the course of the experiment, including after collection for staining at day 30 or 60 (Fig. 2A, B).

Leptomeningeal neural organoid (LMNO) fusion culture establishment and growth.
To assess the structural integrity and cellular composition of the LPM independent from neural organoids in the culture system, we generated and maintained LPM aggregates using methods identical to the derivation of LMNOs, with the exception of adding a neural organoid to the culture (Supplementary Fig. S1). By 3 days in culture, the isolated LPM collapses in on itself and forms aggregates (Supplementary Fig. S1). Aggregates were kept in culture until collection for staining on day 14 or 28 (Supplementary Fig. S1).
Characterization of specialized meningeal cell types within LMNO fusions
The meninges are a complex tissue consisting of a diverse array of cell types, including specialized fibroblasts and tissue-resident immune cells. Thus, we next sought to understand the cellular composition of the meningeal tissue compartment of the LMNO fusions. We have shown previously that mouse and human meningeal fibroblasts express layer-specific markers, 20 including RALDH2 and CRABP2, which mark arachnoid fibroblasts, and S100A6, which marks pial fibroblasts. We conducted fluorescent immunolabeling for RALDH2, CRABP2, and S100A6 on sections from LMNO fusions at days 30 and 60 and observed RALDH2 at day 30 (Fig. 3A) and CRABP2 and S100A6 at days 30 and 60 (Fig. 3B) within the meningeal compartment of LMNOs. Expression of CRABP2 and S100A6 is retained through day 60 in LMNOs (Fig. 3B), suggesting the fibroblasts located within the meningeal compartment retain their layer-specific identity throughout the duration of culture. We also observed widespread expression of these markers in LPM aggregates at days 14 and 28 in culture (Supplementary Fig. S1). Expression of these layer-specific markers is highest in LPM aggregates at day 14, with a marked decrease in labeling by day 28 (Supplementary Fig. S1); this is in contrast to robust expression of these markers by the meningeal compartment of LMNOs at day 30 (Fig. 3A, B), suggesting the neural organoid may secrete factors to promote arachnoid and pial fibroblast identity.
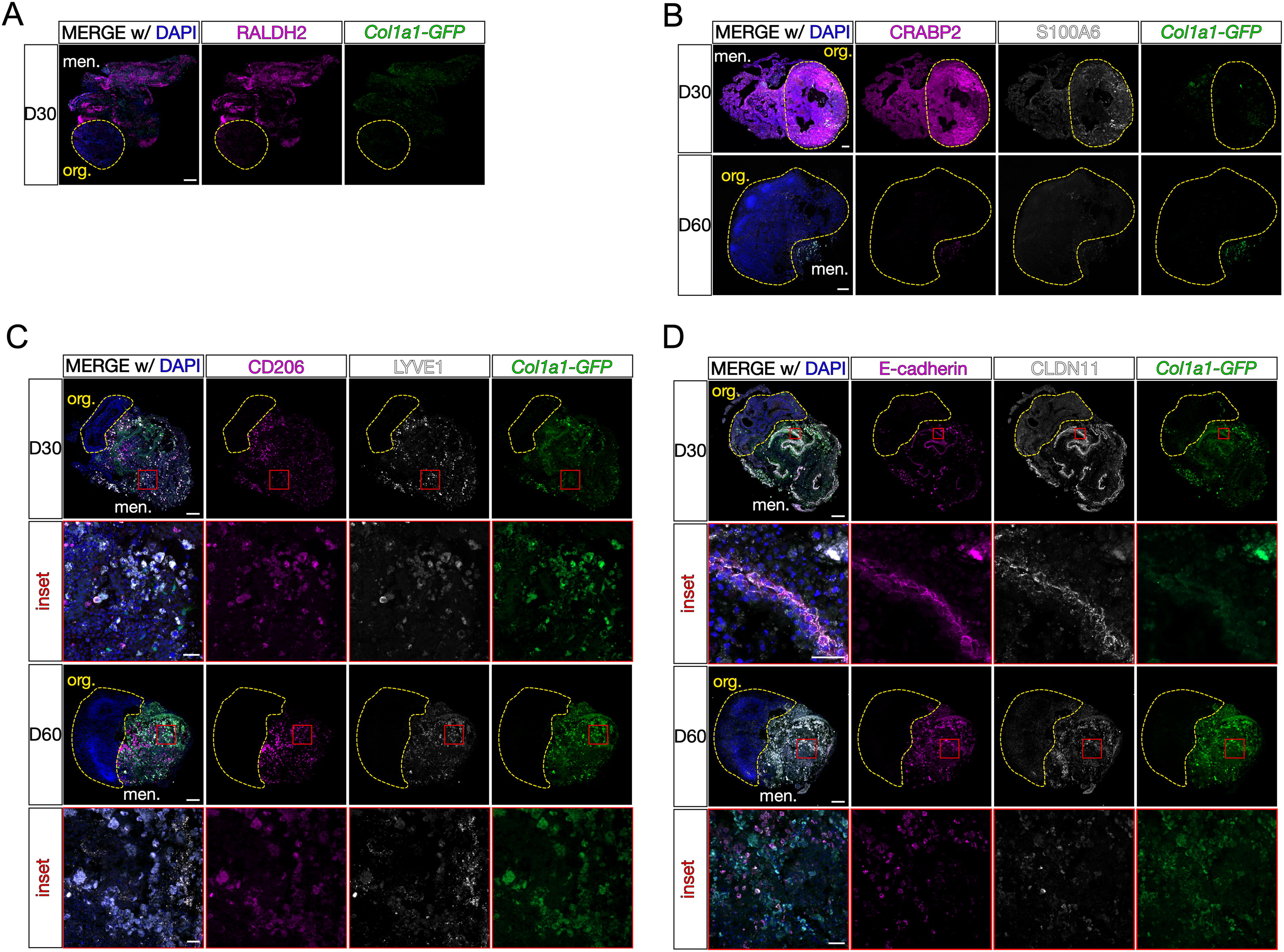
Characterization of meningeal components in LMNOs after 30 and 60 days in culture. Images showing LMNOs after 30 and 60 days in culture, labeled with markers for meningeal cell types:
The meninges contain tissue-resident yolk-sac-derived macrophages called BAMs that play an important role in immunological surveillance and response to infection and injury. 21 –23 BAMs appear in the meninges embryonically, persist through adulthood, 23 and are identified by the markers CD206 [also known as mannose receptor C-176 type 1 (MRC1)] and lymphatic vessel endothelial hyaluronan receptor 1 (LYVE1). 24 We conducted fluorescent immunolabeling for CD206 and LYVE1 on sections from LMNO fusions at day 30 and day 60 and found an abundance of CD206 and LYVE1 labeling of BAMs in the meningeal compartment of the LMNOs at both time points (Fig. 3C). The CD206 and LYVE1 labeling remained within the meningeal compartment, and no labeling was observed in the organoid compartment, suggesting that any BAMs that persist in the LMNO fusions stay confined to the meningeal compartment. On day 60, we observed that much of the Col1a1-GFP+ signal colocalized with CD206 and/or LYVE1 (Fig. 3C), suggesting that BAMs may be phagocytosing cells within the LMNO fusions. LYVE1- and CD206-positive cells are also abundant in LPM aggregates (Supplementary Fig. S1).
The meninges also contain specialized fibroblasts called arachnoid barrier (AB) cells that are critical for maintaining the barrier properties of the meninges. 25 AB cells are specified, and form elaborate cell–cell adhesions utilizing the molecules ECAD and claudin 11 (CLDN11) during embryonic development. 25 We conducted fluorescent immunolabeling for AB cell junctional proteins ECAD and CLDN11 on sections from LMNO fusions at days 30 and 60 and saw labeling for these markers within the meningeal compartment at both time points (Fig. 3D). On day 30, we observed “ribbons” of colocalized ECAD/CLDN11 labeling reminiscent of the AB layer in vivo (Fig. 3D, D30 inset). By day 60, these structures were mostly absent, and most of the ECAD/CLDN11 labeling was seen in ameboid-like structures (Fig. 3D, D60 inset). Given the abundance of BAMs, we suspect that any AB structures that remain in the meningeal compartment of the LMNO fusions are phagocytosed by this time point. A similar observation was made in the LPM aggregates, with characteristic ECAD and CLDN11 expression seen at day 14 and breaking down by day 28 (Supplementary Fig. S1). Taken together, these data show that specialized meningeal cell types (arachnoid and pial fibroblasts, BAMs, and AB cells) are able to persist in LMNO fusion cultures at days 30 and 60 and retain functional and structural characteristics.
Neural organoids in the LMNO fusion model display characteristic progenitor populations and reduced activated caspase-3
The neural organoid in the LMNO fusions contains the typical populations of PAX6-expressing progenitors with the potential of neuronal differentiation as shown by the expression of MAP2+ neurons (Fig. 4). The LPM retain the characteristic meningeal cell types (Col1a1-GFP+ fibroblasts, AB cells, and BAMs) 30–60 days into the culture, though we observe a significant amount of cell fragmentation and what appears to be phagocytosed cellular components by resident BAMs by day 60. Thus, we next quantified the relative frequency of apoptotic cell death as a readout for the overall health of the culture system. To do so, we conducted immunofluorescent labeling for the apoptosis pathway component activated Caspase-3, along with CRABP2, which labels both meningeal fibroblasts and neurons, in LMNO fusions at days 30 and 60 (Fig. 5A, LMNO). We also conducted the same staining in neural organoids not fused to meninges but kept in the same culture conditions as the LMNO fusions (Fig. 5A, org. only). Positive and negative staining controls were conducted to assess levels of background (Supplementary Fig. S2). We then measured the percentage of the area labeled with Caspase-3 of each LMNO compartment (organoid, meninges) (Fig. 5B) as a readout of apoptosis. At day 30, we find that the organoid compartment of LMNOs contains significantly less Caspase-3 labeling compared to the meninges compartment, and the organoid compartment had significantly less Caspase-3 labeling compared to organoids lacking meninges (Fig. 5B). This effect was only observed at day 30; at day 60, we find that the amount of Caspase-3 labeling is not significantly different between the organoid compartment of LMNO fusions and organoids alone (Fig. 5B). From these results, we speculate the meninges component of LMNOs are protective against cell death within the organoid compartment, potentially via secretion of pro-survival signals.

Characterization of neural progenitor populations in LMNOs.
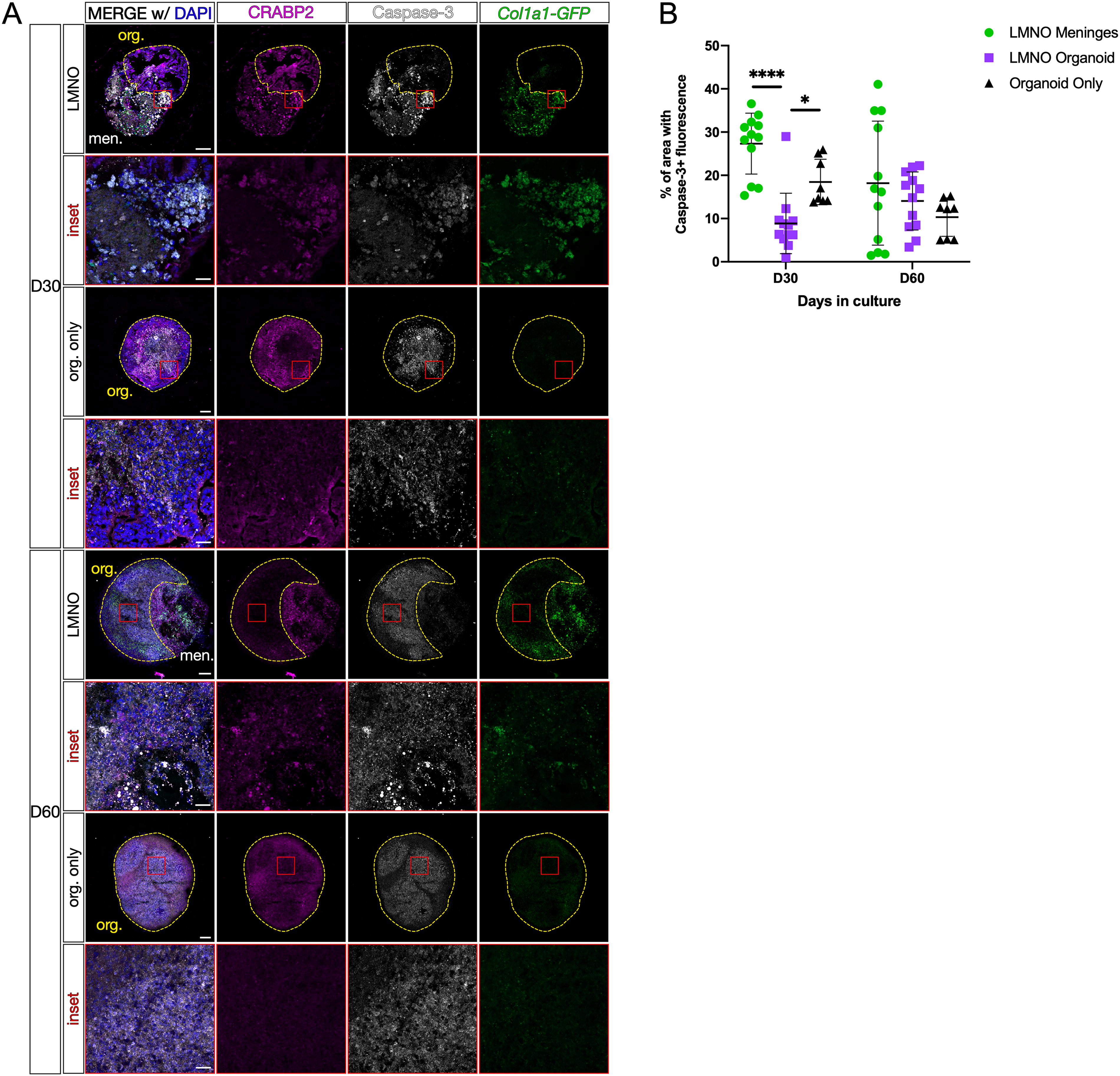
Neural organoids in LMNO fusions show reduced levels of apoptosis.
Human REELIN+ neurons are positioned proximal to meningeal tissue in LMNO fusion model
Given the evidence for neuronal cell migration in other iPSC-derived organoid models, 26 we examined whether any human cells migrated toward the meningeal compartment of the LMNO fusion. In vivo, Reelin-expressing Cajal-Retzius interneurons migrate and localize proximal to the LPM (Fig. 6A, inset/arrows). In LMNO fusions at both days 30 and 60, we observe Reelin-positive cells localized along the border of the organoid and meningeal compartments (Fig. 6B), reminiscent of what is observed in vivo. Using an antibody against human mitochondria to mark only human-derived cells, we show the Reelin-expressing cells located at the border between the organoid and meninges compartments in LMNOs appear human organoid-derived (Fig. 6B, Supplementary Fig. S3). Though we occasionally observed Reelin-positive cells located within the meningeal compartment of LMNOs (data not shown), they do not contain human mitochondria and could be attributed to Reelin-positive cells isolated during leptomeningeal dissection (Supplementary Fig. S3).
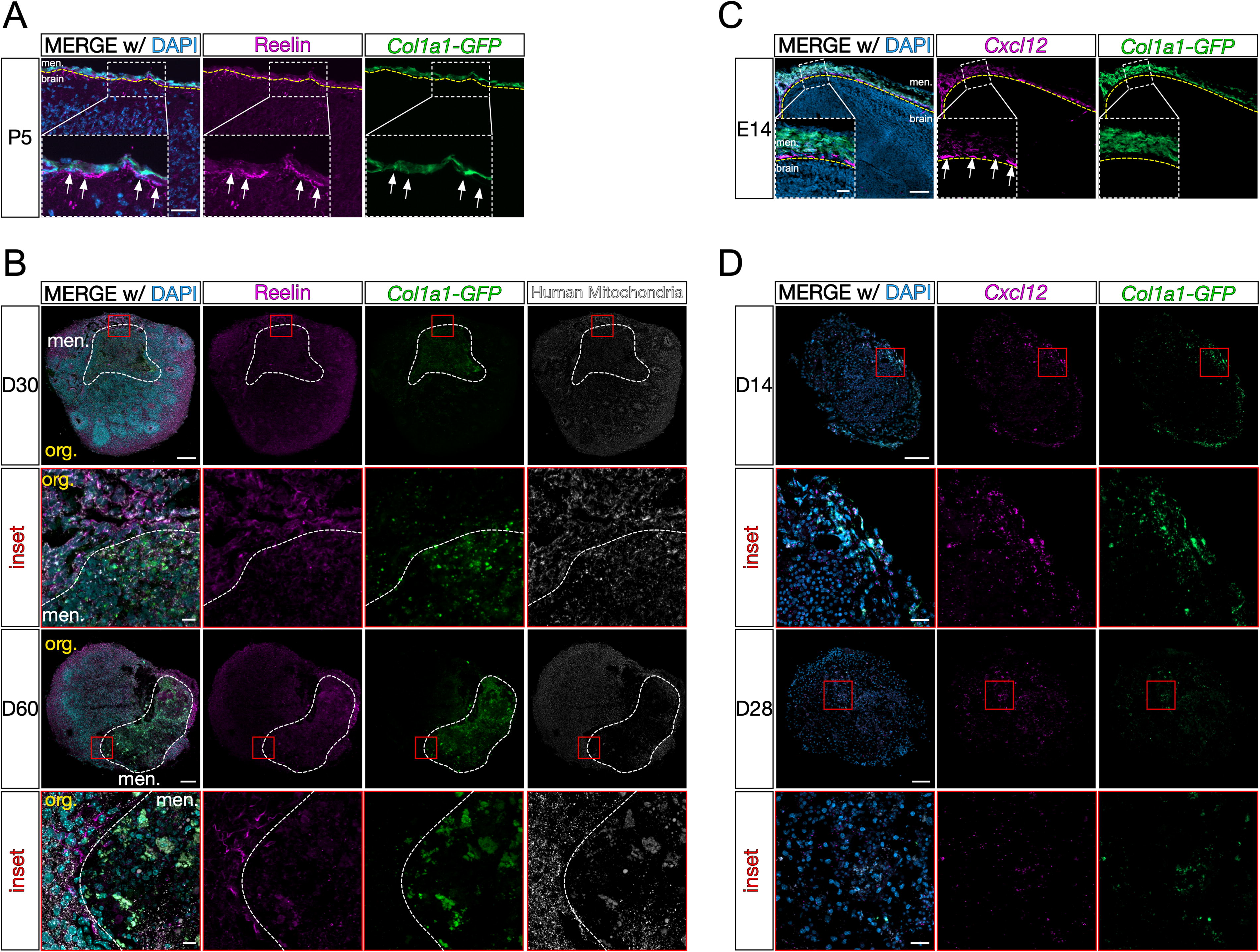
LMNO fusions recapitulate in vivo Cxcl12-dependent Reelin+ Cajal-Retzius cell positioning.
It is well established that meningeal-derived factors are essential for proper cortical development. One essential factor, CXCL12, is secreted by meningeal fibroblasts embryonically and is required for Cajal-Retzius cell migration in vivo. 13 Using RNAScope in situ hybridization, we confirm embryonic expression of Cxcl12 mRNA by Col1a1-GFP-expressing meningeal fibroblasts (Fig. 6C, inset/arrows). Given that we observe organoid-derived Reelin-positive Cajal-Retzius-like cells localized near the meningeal compartment of LMNOs, we next sought to determine if the meningeal compartment acts as a source for CXCL12 ligand. To do so, we conducted RNAscope in situ hybridization for Cxcl12 mRNA on LPM aggregates on days 14 and 28. At both time points, we observe Cxcl12 signal within Col1a1-GFP+ fibroblasts within LPM aggregates (Fig. 6D). This suggests that a CXCL12-dependent mechanism is driving Reelin-positive cell migration toward the meningeal compartment of LMNO fusions.
Discussion
We present a method for coculturing human iPSC-derived neural organoids with embryonic mouse meninges, referred to as LMNO fusions. In this system, embryonic mouse LPM cultured with neural organoids undergo fusion in 6 days and can remain in culture for 60 days. We find that the leptomeningeal compartment of LMNO fusions retains key characteristics of the meninges in vivo, with tissue-resident macrophages and specialized AB cells persisting in culture through 30 days. Furthermore, the cell types known to be present in neural organoids were present after fusion, suggesting that fusion did not disrupt neural organoid development. We also find that the LPM are potentially neuroprotective in the LMNO organoid compartment within the first 30 days in culture. Finally, we show that the LMNO organoid compartment recapitulates characteristics of cortical development in vivo, including producing migratory Reelin+ Cajal-Retzius-like cells that potentially respond to meningeal fibroblast-derived CXCL12.
Cells of the LPM cannot be derived de novo from human iPSC neural organoids (they are all non-neural lineages), and there are no protocols yet to derive human leptomeningeal fibroblast cell types from iPSCs in vitro for mixing with human neural organoids, as has been done for vascular or myeloid cell-derived microglia. Mouse LPM, which contain all the relevant cell types, are therefore a reasonable and easy-to-obtain tissue type to perform feasibility and functional studies fusing mouse and human neural organoids. The conservation of tissue types in the LPM between mouse and human and the expression of similar signaling factors, including CXCL12, support that the LMNO fusion model permits the study of the interaction of human neural organoids and mouse leptomeningeal tissue in vitro, which may give insight into key signaling pathways essential to deriving these tissues. Furthermore, this model provides evidence that meningeal tissue can be cultured in vitro in conditions that support organoid growth and retain the cellular diversity of the tissue. As protocols are developed to derive leptomeningeal cells from human iPSCs, information gained from mouse–human LMNO will accelerate the development of all human LMNO systems for modeling brain development in vitro and brain-meninges crosstalk.
Within the meningeal component of LMNO fusions, we observed the survival of meningeal tissue-resident macrophages, BAMs, marked by expression of CD206 and LYVE1. The BAMs appeared to remain confined to the meninges and were not observed in the organoid compartment of LMNO fusions, mirroring the homeostatic localization of BAMs in vivo. We also observed GFP+ fluorescence within the CD206+/LYVE1+ cells, suggesting that the macrophages present in the LMNO fusions may actively phagocytose fragments of dead fibroblasts. We also detect cells expressing the junctional proteins ECAD and CLDN11, which are expressed by AB cells in vivo. These AB-like structures are seen on day 30 but appear to break down by day 60 in culture. It has been established that the AB layer begins establishing cell–cell junctions along with its barrier function embryonically 25 ; however, it is unknown what signals contribute to maintenance of the AB layer in vivo. Potentially, other meningeal fibroblasts present in the LMNO meninges compartment produce pro-survival signals, or the BAMs act to reduce cytotoxicity by phagocytosing dead cells.
During development, the meninges secrete factors (CXCL12, retinoic acid, BMPs) that are essential for proper cortical development. In the LMNO fusion system, we observed organoid-derived Reelin-positive Cajal-Retzius-like cells are positioned proximal to the meningeal compartment. We also show fibroblasts in LPM aggregates express Cxcl12, which in vivo is necessary for driving Cajal-Retzius cell migration. These data suggest LMNOs may recapitulate key features of meninges-brain signaling during cortical development. A major advantage of this system is the ability to detect the discrete contributions from the meningeal and neural organoid components using species-specific antibodies; in this study, we leverage the use of an antibody specific to human mitochondria to detect the origin of Reelin-positive cells. We also found evidence that the meninges promote cell survival, as measured by apoptosis marker activated Caspase-3, in the organoid of the LMNO fusion through day 30 in culture. Potentially, other meningeal-derived cues promote neuronal survival in LMNO fusions. The current LMNO fusion platform is a suitable system for studying short-term (<30 days) meninges-neural organoid interactions.
Developmental apoptosis is an evolutionarily conserved pathway critical for neurodevelopment, 27 that is actively regulated during the generation of neural organoids. 28 However, increased cell death can also arise from long-term culturing conditions. At day 30, LMNO fusion organoids show less caspase activation than neural organoids alone, suggesting that the meningeal tissue secretes factors that could prevent cell death during the static conditions of the LMNO fusion protocol and that the factors generated from mouse tissue affect human cells.
The LMNO fusion system may benefit from experimentation with other aspects of the fusion process. For example, both the time postdifferentiation of the organoid and the developmental stage of the mice can be manipulated to interrogate different aspects of development. Fusing leptomeningeal tissue from an earlier time in development with a more developed neural organoid may be used to examine how signals from the developing brain impact leptomeningeal maturation. Further, fusion starting with an older neural organoid may allow for the examination of interactions between human iPSC-derived glia and leptomeningeal tissue. Overall, the LMNO fusion model shows that human neural organoids can functionally integrate into mouse meningeal tissue and that this platform can be used to reveal the intricate meninges-brain signaling networks that underlie early brain development.
Limitations of this study and future directions
Our method provides a novel framework for the coculturing of neural organoids with leptomeningeal tissue. In the LMNO system, whole meninges are placed into culture with neural organoids and undergo spontaneous fusion. Although the meninges in LMNOs do retain key cell types (Fig. 3), they do not retain their overall organization or form key structures at the interface between the meningeal and organoid components. In vivo, the meninges consist of three layers—the pia, arachnoid, and dura—with the pia layer residing closest to the brain surface, forming the glia limitans, and acting as a crucial attachment point for radial glial cells. 14 Future meninges-neural organoid platforms should aim to retain this organization, via scaffolding and/or methods to self-organize the meninges into the proper layer order around organoids. A dissociated meningeal cell line has been used to coat neural organoids with meningeal fibroblasts, 29 future systems could employ similar methods with primary embryonic meningeal cells to maintain cellular diversity.
The system reported in our study involves a fusion of murine LPM and human neural organoids. The different species used in the LMNO fusion model, due to the difficulty in obtaining human fetal meninges, limit to some extent the interpretation of feasibility. However, there are currently no protocols available for generating leptomeningeal fibroblast subtypes from human iPSCs. Our proof-of-concept studies lay the groundwork for more advanced approaches using human-derived leptomeningeal cell types.
In LMNOs, we observed colocalization of the Col1a1-GFP+ fibroblast marker with CD206 and LYVE1 BAM markers, leading us to speculate that BAMs present in the leptomeningeal compartment phagocytose dead fibroblasts. We did not examine the extent or timing of this phagocytic activity. Future experiments assaying for phagocytosis at multiple time points in LMNOs should be conducted. It is also unclear whether the presence of BAMs is helpful or harmful to the health of the LMNO system. We hypothesized that BAMs improve the health of the system by engulfing dying or dead fibroblasts. However, fibroblasts are the major source of secreted developmental cues (i.e., RA, CXCL12), and removal of fibroblasts via phagocytosis may be detrimental to the growth and health of the organoid component. Experiments to selectively ablate BAMs (such as clodronate depletion) in the LMNO system would increase our understanding of how BAMs may function in this fusion platform.
A major limitation of the LMNO system is the inability to sustain cultures long-term. While we were able to culture LMNOs for 1–2 months, we observed caspase-3 staining indicating apoptotic cells. Though apoptosis is observed in many cases of development and prior neural organoid systems, there is a major possibility that cells in the LMNO system undergo apoptosis due to a lack of oxygen and nutrient availability. Neural organoids are traditionally grown using a shaking or rolling culture system and can be sustained beyond 1 year. 30,31 Here, we cultured LPM to organoids in a static culture system to minimize disruption to the fusion process. The lack of circulating/diffusing nutrients may have led to increased levels of apoptosis and overall poor tissue health. Future systems should employ ways to improve nutrient and oxygen circulation—for example, seeding LMNO fusions in a microfluidic chamber with a constant flow of media. Further experiments should be conducted to assess how these improved culture systems affect overall organoid health.
Footnotes
Acknowledgment
The authors would like to thank Stellan Riffle for providing technical support during organoid generation.
Author Disclosure Statement
The authors have nothing to disclose and declare no competing financial interests.
Funding Information
This work was supported by National Institutes of Health grants 1R35 GM128915-01NIGMS (V.G.), 1RF1MH123971-01 (V.G. and J.A.S.), 2R35GM128915-06 (V.G.), 1R01NS131823 (J.A.S.), 1F31HD114431-01 (C.B.), F99NS125829 (G.L.R.), F31NS125875 (H.E.J.), and T32HD007186 (H.E.J.). All SIM and spinning disk confocal microscopy imaging and image analysis were performed in part using the Vanderbilt Cell Imaging Shared Resource, which is supported by NIH grants 1S10OD012324-01 and 1S10OD021630-01.
Supplementary Material
Supplementary Figure S1
Supplementary Figure S2
Supplementary Figure S3