
Abstract
Abstract
Background:
The immune system protects the host against dangerous pathogens and toxins. The central nervous system is charged with monitoring and coordinating appropriate responses to internal and external stimuli. The inflammatory reflex sits at the crossroads of these crucial homeostatic systems. This review highlights how the vagus nerve-mediated inflammatory reflex facilitates rapid and specific exchange of information between the nervous and immune systems to prevent tissue injury and infection.
Methods:
Review of the pertinent English-language literature. Nearly two decades of research has elucidated some of the essential anatomic, physiologic, and molecular connections of the inflammatory reflex. The original descriptions of how these key components contribute to afferent and efferent anti-inflammatory vagus nerve signaling are summarized.
Results:
The central nervous system recognizes peripheral inflammation via afferent vagus nerve signaling. The brain can attenuate peripheral innate immune responses, including pro-inflammatory cytokine production, leukocyte recruitment, and nuclear factor kappa β activation via α7-nicotinic acetylcholine receptor subunit-dependent, T-lymphocyte-dependent, vagus nerve signaling to spleen. This efferent arm of the inflammatory reflex is referred to as the “cholinergic anti-inflammatory pathway.” Activation of this pathway via vagus nerve stimulation or pharmacologic α7 agonists prevents tissue injury in multiple models of systemic inflammation, shock, and sepsis.
Conclusions:
The vagus nerve-mediated inflammatory reflex is a powerful ally in the fight against lethal tissue damage after injury and infection. Further studies will help translate the beneficial effects of this pathway into clinical use for our surgical patients.
Whereas “appropriate” or healthy immune function is essential, over-activation of the immune system is potentially lethal to the host. Inadequate restraint of local cytokine responses can release toxic inflammatory mediators into the general circulation, causing systemic inflammation and vital organ damage [2]. As is often the case with sepsis, the inciting pathogenic insult may be eradicated while the immune system continues to assault host tissues [3]. Indeed, the “cytokine theory of disease” states that cytokines are both necessary and sufficient to cause the clinical signs and symptoms of the systemic inflammatory response to infection [4].
Considering the importance of immune homeostasis, it is not surprising that the immune system has developed strategies for keeping pro-inflammatory responses in check [5,6]. The best characterized of these counter-regulatory pathways is the hypothalamic–pituitary–adrenal axis (HPA), first described more than 60 years ago by the renowned endocrinologist Hans Selye [7]. In this classical neuroendocrine pathway, the paraventricular nucleus of the hypothalamus secretes corticotropin-releasing hormone (CRH) in response to physical and emotional stresses or trauma. The CRH then enters the hypophyseal portal blood supply to stimulate secretion of adrenocorticotropic hormone (ACTH) from the anterior pituitary gland. The ACTH enters the circulation and stimulates the adrenal cortex to secrete glucocorticoids, which inhibit cytokine production and suppress systemic inflammation.
Although the HPA axis is highly effective, humoral pathways are inherently limited by the relatively slow, non-specific nature of circulating mediators. From a teleologic perspective, it would appear advantageous for the immune system to emulate the nervous system, which responds to environmental changes with unparalleled speed and accuracy [8]. More than a decade of investigation has demonstrated that the immune system may do just that [9]. Ironically, this previously unrecognized mechanism involves the most intensely studied of all neural pathways, the vagus nerve. Currently referred to as the “inflammatory reflex,” this pathway can provide an unrivaled degree of rapid, coordinated, and specific communication between the nervous and immune systems (Fig. 1). Herein is reviewed the anatomic, physiologic, and molecular connections that comprise the inflammatory reflex, with particular emphasis on how the efferent arm of this pathway regulates immune responses after tissue injury and infection.
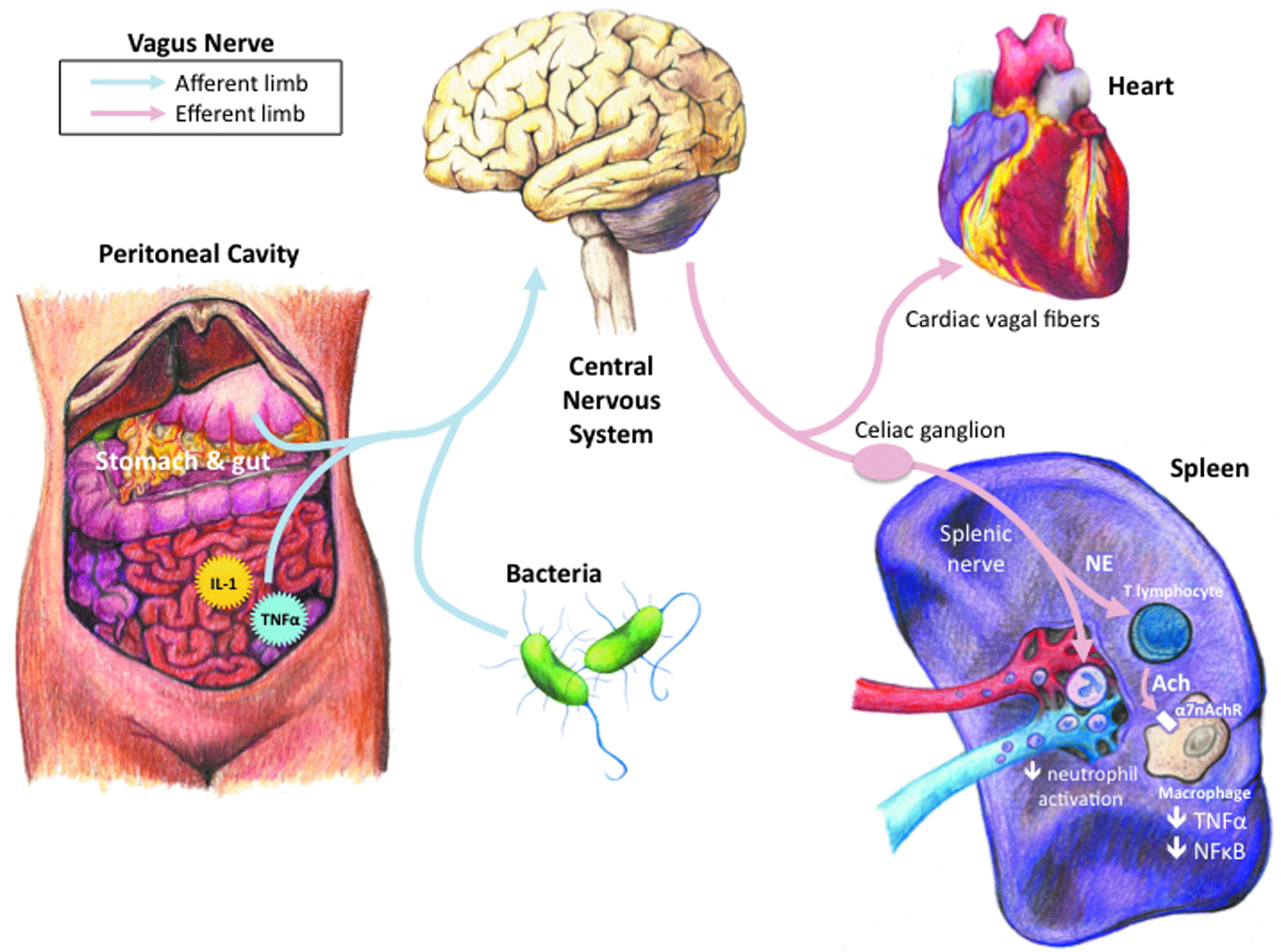
Communication between the nervous and immune systems.
The Inflammatory Reflex
Afferent (sensory) arm
Derived from the Latin word meaning “to wander,” the vagus nerve is the longest of the twelve cranial nerves. It innervates visceral organs extending from the lungs proximally to the large intestine distally. Vagus nerve fibers are predominantly afferent or sensory, which supports an essential role in helping the brain monitor the peripheral environment [10]. The vagus nerve can respond to environmental stimuli through various receptors, including those for mechanical pressure, stretch, increases and decreases in temperature and osmotic pressure, and pain [10]. The vagus nerve also has receptors for chemical stimuli, including acids, glucose, amino acids, fatty acids, and the peptides cholecystokinin (CCK) and somatostatin [10].
The vagus nerve can monitor peripheral immune responses directly via cytokines. Intraperitoneal injection of the pro-inflammatory cytokine interleukin-1 (IL-1) elicits a central nervous system-mediated acute-phase response consisting of fever, hyperalgesia, and elevated circulating glucocorticoid concentrations [11]. Watkins et al. first demonstrated that intra-peritoneal injection of IL-1 fails to elicit the expected systemic acute-phase response after bilateral subdiaphragmatic vagotomy [12]. Whereas this neural reflex is important for detecting low degrees of cytokine production, higher IL-1 concentrations can initiate an acute-phase response following bilateral subdiaphragmatic vagotomy [13]. These findings suggest that the vagus nerve can sense small changes in local cytokine production and allow the inflammatory reflex to respond rapidly and precisely to peripheral inflammation.
Afferent vagus nerve recognition of peripheral cytokines initiates a humoral acute-phase response originally thought to occur within 60–90 min [14]. However, recent studies indicate that this vagal reflex may respond far more rapidly. Fairchild et al. observed that mice injected intraperitoneally with large inocula of bacteria or fungi develop bradydysrhythmias within 1–3 min [15]. These pathogen-induced dysrhythmias are characteristic of cardiac vagus nerve firing and are terminated by the muscarinic receptor antagonist atropine [15]. Intense activity of the c-Fos proto-oncogene protein in neurons of vagal sensory ganglia and the brain stem indicates that the pathogens signal the brain via the afferent vagus nerve [15]. If efferent vagal firing of the inflammatory reflex occurs along with cardiac vagus nerve firing, this mechanism can be considered a completely neural-based immunomodulatory pathway.
Efferent (effector) arm
The cholinergic anti-inflammatory pathway is the motor arc of the inflammatory reflex, as acetylcholine (Ach) is considered the principal vagus nerve neurotransmitter. Borovikova et al. first elucidated the molecular basis for cholinergic anti-inflammatory signaling by showing that acetylcholine significantly reduces pro-inflammatory cytokine production in endotoxin-stimulated macrophages [16]. Subsequent experiments found that electrical stimulation of the vagus nerve significantly reduces tumor necrosis factor (TNF) synthesis in the liver and heart during lethal systemic inflammation [16–18]. The net result of vagus nerve stimulation is a decrease in circulating pro-inflammatory cytokine concentrations and vital organ damage [19]. In contrast, interruption of cholinergic signaling via surgical cervical vagotomy significantly exacerbates pro-inflammatory cytokine production [16,19].
Further studies identified the α7 subunit of the nicotinic ACh receptor (α7nAChR) as essential to cholinergic anti-inflammatory activity [20]. Acetylcholine can reduce cytokine synthesis and release by preventing nuclear factor (NF)-κβ from translocating into the nucleus, either through inhibiting phosphorylation of the inhibitory protein I-κβ or by activating the Jak2-STAT3 anti-inflammatory signaling pathway [21,22]. However, macrophages from α7nAChR-knockout animals do not respond to ACh stimulation [20]. Vagus nerve stimulation significantly reduces organ TNF synthesis in wild-type mice [20]. In contrast, vagus nerve stimulation fails to inhibit organ TNF production in α7nAChR-knockout animals and thus cannot regulate systemic pro-inflammatory cytokine concentrations [20]. In fact, α7nAChR-knockout mice generate significantly more organ and systemic pro-inflammatory cytokines than control animals, which is similar to the response observed after surgical vagotomy [20]. This suggests that the inflammatory reflex maintains tonic control over pro-inflammatory cytokine production.
Cholinergic stimulation has been shown to reduce pro-inflammatory cytokine production and prevent lethal tissue injury in multiple models of local and systemic inflammation and sepsis, including acute lung injury; arthritis; burns; cerebral ischemia; colitis; encephalomyelitis; hemorrhagic shock; intracerebral hemorrhage; myocardial, renal, and splanchnic ischemia-reperfusion injury; pancreatitis; polymicrobial abdominal sepsis; and post-operative ileus [23–42]. Cholinergic pathway activation is possible via vagus nerve stimulation or pharmacologically via administration of selective α7nAChR agonists or inhibitors of acetylcholinesterase [43,44]. Cholinergic agents can recapitulate the anti-inflammatory properties of vagus nerve stimulation and rescue animals from lethal polymicrobial sepsis in a clinically relevant time [45]. Vagus nerve stimulation is effective both with and without electricity, and transcutaneous mechanical vagus nerve stimulation can inhibit pro-inflammatory cytokine production and rescue animals from lethal polymicrobial sepsis, even when treatment is delayed for 24 h after the onset of disease [46].
The macrophages responsible for the initial response to circulating pathogens and toxins reside in organs of the reticuloendothelial system, including the lung, liver, and spleen [47]. Toxic bacterial mediators, such as lipopolysaccharide (LPS), cause tissue macrophages to secrete pro-inflammatory cytokines. We found that TNF production in the spleen is a primary source of elevated systemic TNF concentrations during lethal endotoxemia [47]. Splenectomy significantly reduces circulating TNF concentrations in murine endotoxemia [47]. Vagus nerve stimulation fails to regulate systemic TNF production in splenectomized animals [47]. Interruption of the common celiac branch of the abdominal vagus nerve abolishes vagal anti-inflammatory effects, suggesting that cholinergic signaling targets the spleen via this specific branch of the vagus nerve [47]. Not only does splenectomy interfere with vagus nerve stimulation, but pharmacologic cholinergic agonists fail to inhibit pro-inflammatory cytokine production or reduce the mortality rate in splenectomized animals [47]. In fact, cholinergic stimulation in splenectomized animals exacerbates pro-inflammatory cytokine responses and accelerates sepsis lethality [47]. These findings indicate that the spleen is a crucial anatomic and physiological interface between brain-mediated anti-inflammatory vagus nerve signaling and peripheral inflammatory responses.
At first glance, targeting cytokine production in the spleen via the vagus nerve is problematic because neuro-anatomists have had difficulty identifying markers of cholinergic signaling in the spleen [48–50]. It is generally accepted, however, that the spleen receives solely sympathetic innervation via catecholaminergic neural fibers emanating from the celiac and superior mesenteric ganglia that traverse the splenic nerve [51,52]. This apparent discrepancy between the anatomic and physiologic roles of vagal innervation of the spleen can be explained by studies suggesting that cholinergic signals travel via the common celiac vagal branches to synapse with catecholaminergic neurons in the celiac and superior mesenteric ganglia before reaching the spleen [53]. Thus, transection of the splenic nerve abolishes the anti-inflammatory effects of cervical vagus nerve stimulation [53]. Moreover, depletion of norepinephrine in the spleen abrogates the cytokine-suppressing effects of vagus nerve stimulation [53]. It now appears that the cholinergic anti-inflammatory pathway requires some degree of catecholaminergic signaling to modulate cytokine production in the spleen.
Although crosstalk between the vagus and splenic nerves can help explain cholinergic modulation of cytokine synthesis in the spleen, it does not account for the crucial role of α7nAChR on the surface of cytokine-producing tissue macrophages. How can norepinephrine release a signal via α7nAChR to inhibit cytokine production by resident macrophages in the spleen? First, lymphocytes can synthesize and release ACh [54, 55]. Second, splenic lymphocytes harvested from mice after subdiaphragmatic vagotomy produce significantly more pro-inflammatory cytokines after stimulation in vitro [56]. Third, adoptive transfer of splenic CD4+ T cells from vagotomized mice into naïve animals exacerbates pro-inflammatory cytokine production during experimental colitis [57].
The precise role of T lymphocytes in vagus modulation of splenic cytokine production is best explained by the recent findings of Rosas-Ballina et al., who identified a pool of memory T lymphocytes in the spleen that release ACh in response to stimulation with norepinephrine [58]. In nude mice, which lack T lymphocytes, vagus nerve stimulation fails to reduce serum TNF concentrations [58]. Adoptive transfer of T lymphocytes into nude mice restores anti-inflammatory vagus nerve signaling, and transferred T cells are seen in the vicinity of white pulp nerve fibers [58]. In contrast, adoptive transfer of T lymphocytes unable to synthesize ACh fails to restore cytokine inhibition [58]. These findings suggest that T lymphocytes “translate” catecholaminergic splenic nerve activity into cholinergic signals to modulate cytokine production via the α7nAChR on splenic macrophages.
It is worth noting that the immunomodulatory effects of cholinergic stimulation extend beyond regulation of pro-inflammatory cytokine synthesis. Cholinergic stimulation also inhibits chemokine production and adhesion molecule expression on the surface of activated endothelial cells [59]. Cholinergic inhibition of endothelial cell activation is associated with blockade of TNF-induced NF-κβ entry and activation of the Jak2/STAT3 pathway [59,60]. This inhibitory effect of cholinergic stimulation is reversed by mecamylamine, a selective nAChR antagonist [59]. In a murine air pouch model of systemic leukocyte recruitment, both electrical vagus nerve stimulation and cholinergic agonists significantly inhibit neutrophil trafficking to sites of local inflammation [59].
Whereas endothelial cells express nicotinic ACh receptors, it is unclear how cholinergic stimulation in vivo regulates leukocyte trafficking to sites of local inflammation because the vagus nerve does not innervate endothelial cells. Consistent with the importance of the spleen to cholinergic regulation of cytokine production, cholinergic regulation of leukocyte trafficking also requires the spleen [61]. Cholinergic stimulation significantly reduces the concentration of CD11b, a beta (2) integrin involved in cell adhesion and leukocyte chemotaxis, on the surface of neutrophils, but only in the presence of the spleen [61]. Vagus nerve stimulation and systemic administration of nicotine fail to regulate neutrophil activation in the absence of the spleen [61]. These studies suggest that the cholinergic anti-inflammatory pathway deactivates circulating neutrophils passing through the spleen on their way to sites of tissue inflammation. Conceptually, this centralized mechanism could allow the brain to influence nearly every tissue and cell of the body through the “education” of circulating immunocompetent cells in the spleen.
The protective effects of the inflammatory reflex against external tissue injury may extend beyond classical immunomodulation. If one considers the immediate dangers to the host of traumatic injury, prevention of excessive bleeding from a wound is paramount to survival. To this end, Czura et al. found that vagus nerve stimulation significantly reduces blood loss and time to cessation of bleeding after minor peripheral tissue trauma in swine [62]. Cholinergic stimulation is associated with increases in local but not systemic thrombin activation at the site of tissue injury [62]. More recently, we observed that cholinergic stimulation significantly reduces blood loss after potentially lethal, large-volume liver hemorrhage in rats (manuscript in preparation). Cholinergic stimulation also significantly increases local thrombin activation at the site of hepatic injury. These findings suggest that the inflammatory reflex protects the host against tissue injury through specific regulation of both inflammation and hemostasis. In fact, cholinergic regulation of hemostasis is not altogether surprising, considering the significant crosstalk (albeit transiently) between the inflammatory and coagulation cascades. Indeed, the only therapy ever approved clinically for severe sepsis, recombinant-activated protein C, is also a potent anti-coagulant [63].
Clinical Correlates of the Inflammatory Reflex
Before the advent of modern anti-ulcer therapy, treatment of intractable peptic ulcer disease often required surgical ligation of the abdominal vagus nerve. Given our current understanding of the inflammatory reflex, one could expect these patients to have deficits in afferent or efferent vagus nerve signaling. Whereas thousands of these types of operations have been performed, outcome data for this patient population after systemic inflammatory diseases and sepsis are limited [64]. To confound any analysis further, it is unclear if compensatory mechanisms are upregulated chronically after vagotomy. This phenomenon is observed in vagotomized mice that can exhibit enhanced sensitivity to inflammation that dissipates over time [65,66].
Although surgical vagotomy is utilized less commonly, advances in the fields of epilepsy and depression demonstrate that vagus nerve stimulation using an implantable stimulator is an effective and safe therapy for medically refractory symptoms [67,68]. It is possible these patients also experience increases in inflammatory reflex signaling via electrical activation of the cervical vagus nerve. In fact, the stimulation parameters used clinically to control epilepsy and experimentally to inhibit systemic inflammation are similar [69,70]. To date, clinical studies of inflammation in these patients are limited. De Herdt et al. measured concentrations of pro-inflammatory cytokines produced by LPS-stimulated peripheral blood mononuclear cells isolated from 10 patients both at baseline and after six months of vagus nerve stimulation [71]. They observed a significant decrease in IL-8 induction, but no significant changes in TNF, IL-1, or IL-6 concentrations, following vagus nerve stimulation [71]. Another study of basal cytokine expression in eight epilepsy patients before and after three months of vagus nerve stimulation found no significant differences in the serum concentrations of TNF, IL-6, or C-reactive protein [71]. Of note, these individuals were not studied under conditions of inflammation, and there is no experimental evidence that vagus nerve stimulation can regulate basal cytokine production.
Another active area of research relates to the role of nutrition in critically ill patients. Enteral feedings are associated with better outcomes and fewer complications than total parenteral nutrition [72]. In contrast to parenteral nutrition, enteral nutrition may help maintain gut luminal integrity and prevent translocation of intra-luminal bacteria into the general circulation [73,74]. However, recent experimental studies suggest that the benefits of enteral nutrition may derive from activation of the cholinergic anti-inflammatory pathway. Luyer et al. reported that enteral nutrition in rats activates gut intra-luminal CCK receptors that transmit signals via the afferent vagus nerve to the brain [75]. This gut–vagus–brain feedback loop results in decreased systemic pro-inflammatory cytokine production after lethal traumatic hemorrhage [75]. Further studies found that enteral nutrition can activate this vagal reflex pathway and significantly reduce systemic TNF concentrations during gram-negative sepsis [76]. In theory, patients receiving enteral nutrition may derive benefit from activity within the inflammatory reflex, whereas total parenteral nutrition may not stimulate CCK receptors or activate anti-inflammatory vagus nerve activity [77].
Heart rate variability is an evolving area of research that may provide additional insights into the neurologic basis of inflammation. “Heart rate variability” refers to changes or differences in the beat-to-beat interval (time), as measured during a normal heart rhythm [78]. Classical physiology dictates that increased efferent vagus nerve activity leads to a slowing of the heart rate, and thus greater heart rate variability, via inhibition of the sinoatrial node. More than 20 years of clinical studies of adult and pediatric patients with disorders of systemic inflammation and sepsis indicate that depressed heart rate variability is a significant negative prognostic factor that correlates with elevated cytokine concentrations [79–90]. Experimentally, pharmacologic activation of the cholinergic anti-inflammatory pathway is associated with increases in heart rate variability [91]. Taken together, these data suggest it is possible that reduced heart rate variability, reflecting diminished cardiac vagal activity, is also a marker for diminished activity of the cholinergic anti-inflammatory pathway [92]. Insufficient cholinergic signaling could result in lethal systemic inflammation from excessive cytokine synthesis.
Finally, the role of the spleen in the pathogenesis of systemic inflammation and sepsis may need re-evaluation as well. It is generally accepted that splenectomized patients are at higher risk of rapidly progressive bacterial infections, a syndrome known as “overwhelming post-splenectomy sepsis” (OPSS). The pathogenesis of OPSS is unclear but is believed to be an inadequate immune response to encapsulated bacteria [93]. Experimental studies indicate that the spleen is an essential target of the cholinergic anti-inflammatory pathway and the primary source of systemic pro-inflammatory cytokine production during lethal systemic inflammation [47]. In view of these findings, one may consider that perhaps instead of failing to respond to bacterial invasion, splenectomized patients actually fail to inhibit pathogen-generated excessive cytokine production because of a non-functioning inflammatory reflex.
Conclusion
The care of surgical patients is inextricably linked to the mammalian immune system's long-standing battle against infection and tissue injury. Recent studies suggest that patient outcomes are improving, albeit slowly, in sepsis and sepsis-related organ dysfunction [94–96]. Notwithstanding, the advancing age of our patients, increasing complexity of medical co-morbidities, and rapid spread of multi-drug-resistant organisms are but several reminders that these encouraging trends may not continue. The elucidation of the inflammatory reflex helps in understanding how the immune system has evolved to protect the host. Just as bacteria exploit weaknesses in the immune system to ensure their survival, we must learn to harness the efficiency and specificity of endogenous, protective immunologic pathways. It is to be hoped that future clinical studies will show that cholinergic stimulation is a safe and beneficial means of controlling dangerous inflammation in our surgical patients.
Footnotes
Author Disclosure Statement
No conflicting financial interests exist.