
Abstract
Abstract
Background:
Enteric glia cells (EGCs) play an important role in maintaining proper intestinal barrier function. We have shown that vagal nerve stimulation (VNS) increases EGC activation, which is associated with better gut barrier integrity. Enteric neurons communicate with EGCs through nicotinic cholinergic signaling, which may represent a pathway by which VNS activates EGCs. This study sought to define further the mechanism by which VNS prevents intestinal barrier failure using an in vitro model. We hypothesized that a nicotinic cholinergic agonist would increase EGC activation, prevent intestinal nuclear factor kappa-B (NF-κB) activation, and result in better intestinal barrier function.
Methods:
Cultured EGCs were exposed to the nicotinic cholinergic agonist nicotine. Expression of glial fibrillary acidic protein (GFAP) was measured by immunoblot to determine changes in EGC activation. Caco-2 cells were grown to confluence and incubated alone or in co-culture with EGCs. Cells were then stimulated with Cytomix for 24 h in the presence or absence of nicotine, and barrier integrity was assessed by permeability to 4-kDa FITC-dextran. Changes in phosphorylated inhibitor of NF-κb (P-IκBα) and phosphorylated NF-κB (P-NF-κB) were assessed by immunoblot.
Results:
Stimulation with nicotine resulted in EGC activation, as demonstrated by an increase in GFAP expression. Cytomix stimulation increased permeability in Caco-2 cells cultured alone or with EGCs. Treatment of stimulated Caco-2/EGC co-cultures with nicotine reduced permeability similar to control. Nicotine failed to prevent barrier permeability in Caco-2 cells alone. Co-culture of stimulated Caco-2 cells with nicotine-activated EGCs prevented Cytomix-induced increases in P-IκBα and P-NF-κB expression.
Conclusion:
A pharmacologic nicotinic cholinergic agonist increased EGC activation and improved intestinal epithelial barrier function in an in vitro model of intestinal injury. Nicotine-activated EGCs appear to modulate barrier function by preventing the activation of the NF-κB pathway. Therapies aimed at activating EGCs may have important clinical applications for improving intestinal barrier function after injury.
G
On a cellular level, disruption or loss of the tight junctions that link adjacent intestinal epithelial cells results in increased intestinal permeability and gut inflammation. Tight-junction proteins form a barrier protecting the paracellular space between adjacent epithelial cells. These proteins are connected to the actin cytoskeleton of individual intestinal epithelial cells through their interactions with myosin light chain (MLC) [5]. An increase in phosphorylation of MLC causes actin cytoskeletal contraction, alters expression of tight-junction proteins, and results in a loss of normal barrier function [6,7]. We demonstrated that vagal nerve stimulation (VNS) may improve intestinal barrier integrity in severe injury through greater expression of tight-junction proteins [8–11]. However, the signalling mechanism that links stimulation of the vagus nerve and the intestinal epithelium has yet to be elucidated fully.
Enteric glia cells (EGCs), the largest cell subset of the enteric nervous system (ENS), are identified by the unique marker glial fibrillary acidic protein (GFAP) [12]. These EGCs receive and propagate signals both to and from nearby enteric neurons and the intestinal epithelium [12–14]. Evidence suggesting a connection between the vagus nerve and EGCs was demonstrated in vivo recently, as VNS increased intestinal GFAP expression, a common marker of EGC activation [11]. Further, other studies have shown that the anti-inflammatory effects of VNS require the presence of nicotinic cholinergic receptors [15] and that EGCs increase release of barrier-inducing factors in response to several cholinergic agonists [16]. Our laboratory has demonstrated that intraperitoneal injection of nicotine, a nicotinic receptor agonist, protects the intestinal barrier in a severe burn model, replicating the gut-protective effects of VNS [17]. Collectively, these findings suggest that the vagus nerve alters EGC expression through a nicotinic cholinergic signaling mechanism.
This study examined further the potential connections among nicotinic cholinergic signaling, EGCs, and the intestinal barrier using an in vitro co-culture model with EGCs and a well-established intestinal epithelial cell line. We postulated that a nicotinic cholinergic agonist would activate EGCs, and that activated EGCs would improve intestinal barrier function through inhibition of the NF-κB inflammatory pathway.
Materials and Methods
Cell lines
Caco-2 human intestinal epithelial cells and rat EGCs were obtained from the American Type Culture Collection (Manasas, VA). All three cell lines were grown at 37°C in a 5% CO2 humidified atmosphere. Cells were grown in Dulbecco's Modified Eagle Medium (DMEM) with high glucose (GIBCO, Carlsbad, CA) supplemented with 10% fetal bovine serum (FBS)(GIBCO), penicillin G (10,000 U/mL; GIBCO), and streptomycin (10,000 mcg/mL; GIBCO).
Nicotine stimulation
To examine further the link between VNS and EGCs, we sought to perform several cholinergic signaling studies using the agonist nicotine (Sigma, St. Louis, MO). To determine if nicotine increases GFAP expression in EGCs, we created a dosage–response curve using increasing doses of nicotine. Nicotine was diluted in phosphate-buffered saline (PBS) to the following concentrations: 1e-10 M, 1e-8 M, 1e-6 M, 1e-4 M, and 1e-2 M. The EGCs were seeded on six-well plates (100,000 cells/well) and allowed to grow for three days in DMEM. Cells were then placed in serum-free medium and incubated with either PBS or the different concentrations of nicotine for 24 h at 37°C in a 5% CO2 humidified atmosphere. Cells were then subject to lysis and immunoblotting.
Co-Culture Model and Immunostimulation
Caco-2 cells (80,000/well) were seeded onto permeable filters with 4-mcm pore size in 12-well Transwell bicameral chambers (Corning Inc, Corning, New York). A population of EGCs (30,000 cells/well) were seeded in the basal well of the 12-well Transwell dish. Medium was changed daily throughout the co-culture process. After the incubation period, cells were subject to permeability assays or lysis for immublotting.
For the co-culture model, cells were seeded on day 0 and allowed to grow for two days in DMEM. The EGCs were plated on day 1 and allowed to grow for one day alone in DMEM. Cells were then co-cultured on day 2 and grown together for another three days. On day 3, Caco-2 cells were incubated in Enterocyte Differentiation Medium (BD Bioscience, Bedford, MO) to help induce Caco-2 differentiation. On day 4, cells were placed in serum-free medium, and selected wells were treated with PBS or nicotine (1e-3M) for 10 min, followed by incubation at 37°C in a 5% CO2 humidified atmosphere with either PBS or Cytomix (TNF-α [100 ng/mL; Sigma], IFN-γ [100 ng/mL; Pierce, Rockford, IL], and IL-1β [100 ng/mL; Sigma]) for 24 h for permeability assays or 10 min or 2 h for lysis and immublots.
Permeability assay
An in vitro permeability assay was performed to assess epithelial monolayer barrier function. After the 24-h Cytomix incubation period, 200 mcL of 4-kDa FITC-Dextran (10 mg/mL; Sigma) was added to the Transwell insert on the apical side of the monolayer. After 4 h of incubation, 100-mcL aliquots of DMEM were obtained from the basal chamber. Fluorescence was measured in a spectrometer (SpectraMax, Molecular Devices, Sunnyvale, CA) and compared with a standard curve of known FITC-Dextran concentrations diluted in PBS and DMEM.
Immunoblotting
Cells were washed with PBS and placed in 0.25% trypsin–ethylenedaminetetraacetic acid (GIBCO). Once cells were in suspension, they were placed in a centrifuge at 14,000 rpm for 5 min. The supernatant liquid was removed and the tubes placed on ice. Then 250 mcL of 4% sodium dodecyl sulfate (SDS) in PBS lysis buffer and 5 mcL of protease inhibitor (Pierce) were added to the tubes, followed by sonication to lyse the cellular pellet. Total protein samples were stored at −80°C for later analysis.
The total protein concentration of the samples was determined by the bicinchoninic acid (BCA) protein assay using the microplate procedure (Pierce). Samples containing 10 mcg of protein were placed in SDS sample buffer and boiled for 5 min. Proteins were separated by sodium dodecyl sulfate (SDS)-polyacrylamide gel electrophoresis using 8–16% Tris-glycine polyacrylamide gradient gel and transferred to nitrocellulose membranes (Invitrogen). Membranes were washed with Tris-buffered saline/Tween 20 (TBST) and blocked with 5% bovine serum albumin (BSA) in TBST for 1 h. Membranes were then incubated in primary antibodies specific for phosphorylated NF-κBp65 (P-NF-κBp65; 1:250; Santa Cruz Biotechnology, Santa Cruz, CA), phosphorylated inhibitor of κB-α (P-IκBα; 1:250; Cell Signaling, Danvers, MA), inhibitor of κB-α (IκBα; 1:250; Cell Signaling), GFAP (1:500; Sigma), or β-actin (1:500; Cell Signaling) overnight at 4°C in 5% BSA in TBST. Membranes were washed and incubated with horseradish peroxidase-linked anti-rabbit IgG (1:2,000; Cell Signaling) or anti-mouse IgG (1:2,000, Cell Signaling) in 5% BSA in TBST for 1 h at room temperature. The Pierce Supersignal West Pico Chemiluminescent Kit was applied to the membranes for antibody detection through the Xenogen IVIS Lumina (Caliper Life Science, Hopkinton, MA) imaging system. Mean pixel density was determined using the UN-SCAN-IT Gel Digitizing software (Silk Scientific, Orem, UT). Band densities were compared with the β-actin band densities in each lane as a loading control. Data are expressed as the relative band density compared with control for each experiment.
Statistical analysis
All values are expressed as mean±standard error of the mean (SEM). Statistical significance was determined using analysis of variance (ANOVA) with Student-Newman-Keuls correction. A p value<0.05 was considered statistically significant.
Results
EGCs are activated when stimulated with nicotine
To define further the connection between VNS and changes in EGC phenotype, we determined if the nicotinic cholinergic agonist, nicotine, could activate EGCs. We incubated EGCs with increasing concentrations of nicotine and examined GFAP expression by immunoblot (Fig. 1). The dosage curve revealed an increase in GFAP expression at nicotine concentrations greater than 1e-4 M, suggesting that a nicotinic cholinergic agonist can activate EGCs in vitro.
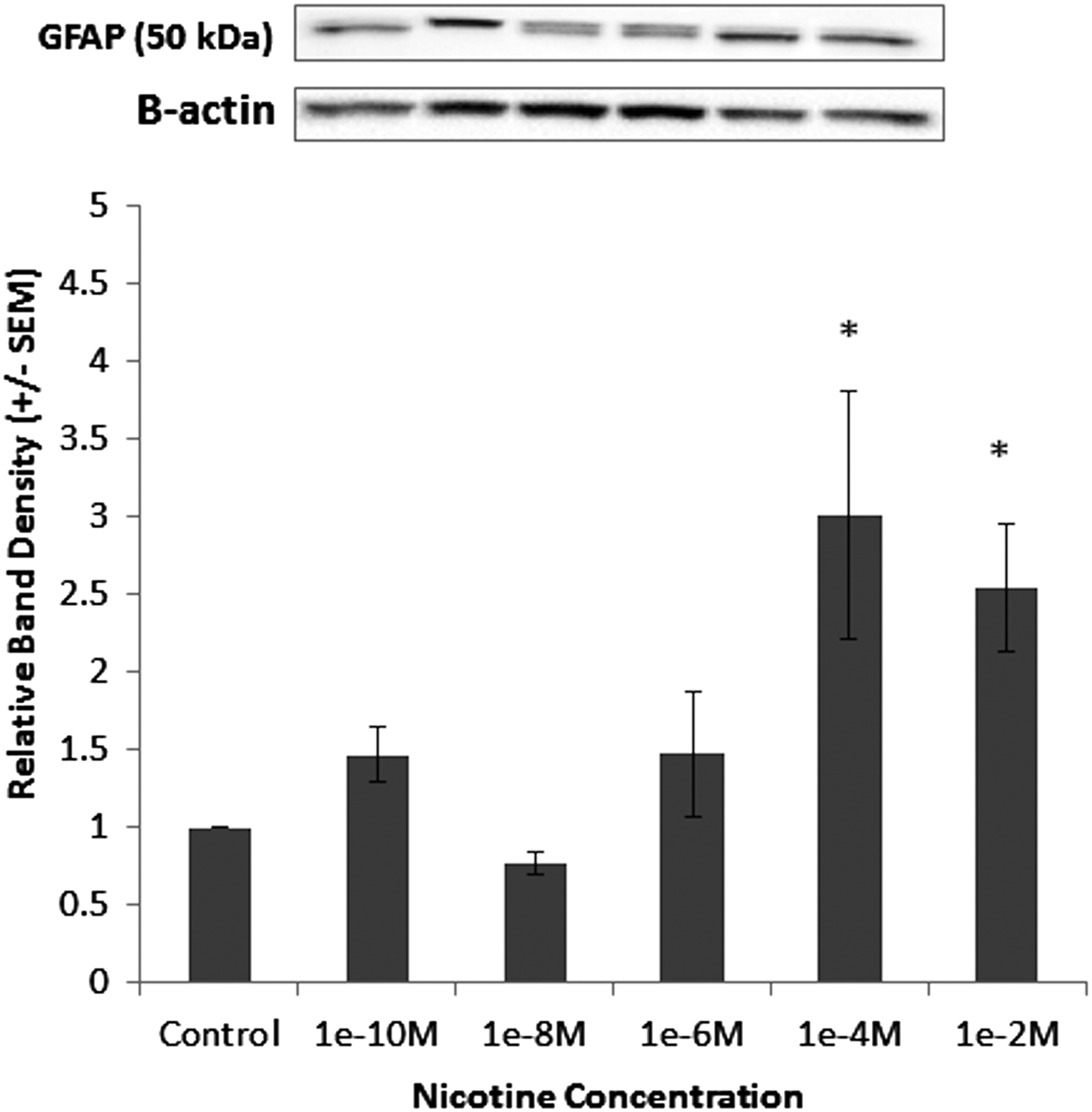
Nicotine increases expression of glial fibrillary acidic protein (GFAP) in enteric glial cells (EGCs). Cells were grown to confluence and incubated with either increasing concentrations of nicotine or phosphate-buffered saline. Concentrations of GFAP were determined by immunoblot. Representative band densities of immunoblots of GFAP in EGCs. Graph representing relative changes in GFAP normalized to control in EGCs after exposure to increasing concentrations of nicotine. Data are expressed as relative band densities/control±standard error of the mean. *p<0.05 vs. control using analysis of variance.
Activated EGCs improve barrier function in caco-2 cells
To determine if nicotine-activated EGCs improved barrier function, an in vitro permeability assay was performed using FITC-Dextran. Caco-2/EGC co-cultures stimulated with Cytomix for 24 h demonstrated an increase in barrier permeability. The addition of nicotine to stimulated monolayers prevented the Cytomix-induced increase in permeability (Fig. 2A). Thus, it appears that nicotine-activated EGCs improve barrier function in Caco-2 cells. To rule out the possibility that nicotine may be having a direct effect on the epithelial cells, we added nicotine to Caco-2 monolayers in the absence of EGCs. Stimulation of the epithelial monolayers with Cytomix for 24 h again caused an increase in permeability. However, incubation of stimulated monolayers with nicotine failed to prevent a Cytomix-induced increase in permeability (Fig. 2B). Thus, nicotine appears to have no barrier-inducing effects on Caco-2 cells directly, but instead acts through EGCs.

Nicotine stimulation prevents Cytomix-induced barrier permeability only in presence of enteric glial cells (EGCs). Caco-2 cells were grown to confluence in the presence or absence of EGCs. Selected cells were incubated with 1 mM nicotine or phosphate-buffered saline (PBS). Cells were then stimulated with Cytomix or PBS for 24 h prior to measurement of permeability to 4-kDa fluorescein isothiocyanate (FITC)-dextran. (
Activated EGCs inhibit NF-κB activation
After determining the effects of nicotine-activated EGCs on Caco-2 monolayers, we sought to determine if these effects were attributable to an inhibition of the NF-κB pathway. Thus, we first measured changes in P-IκBα expression after 10 min of Cytomix-stimulation by immunoblot, as increases in P-IκBα marks it for degradation, increasing NF-κB activation [18–20]. Cytomix-stimulation of Caco-2 cells co-cultured with EGCs in the absence of nicotine resulted in a two-fold increase in P-IκBα expression (Fig. 3B). However, co-culture with nicotine-activated EGCs prevented the Cytomix-induced increase in P-IκBα expression.
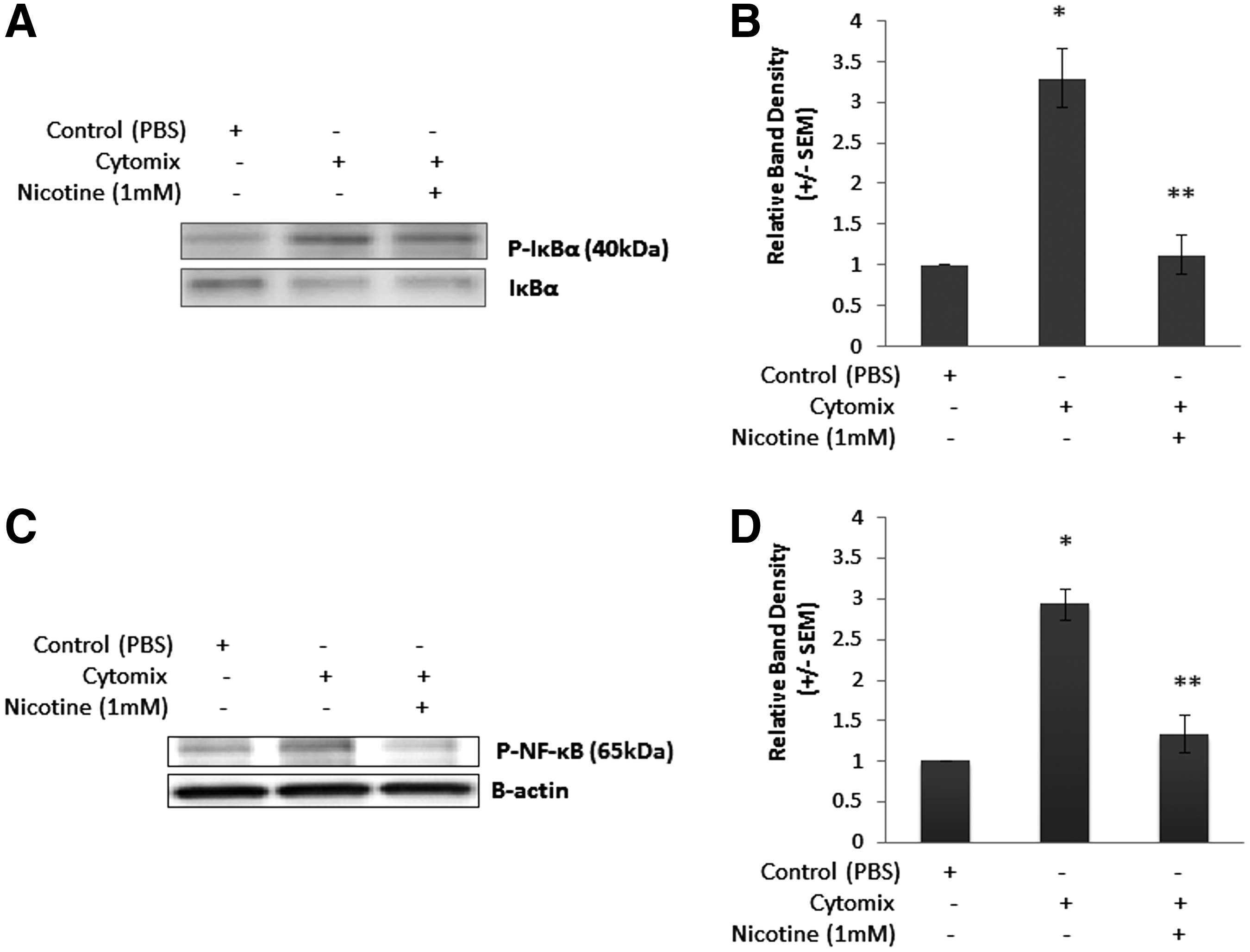
Activated enteric glial cells (EGCs) reduced NF-κB inflammatory signaling in Cytomix-stimulated Caco-2 cells. Caco-2 cells were grown to confluence in co-culture with EGCs. Selected cells were incubated with 1 mM nicotine or phosphate-buffered saline (PBS). Cells were then stimulated with Cytomix or PBS for 24 h. Expression of P-IκBα and P-NF-κBp65 was determined by immunoblot. (
Next, we examined changes in P-NF-κBp65 in Caco-2 whole cell lysates after 10 min of Cytomix-stimulation by immunoblot (Fig. 3C), which is indicative of NF-κBp65 activation [21]. Cytomix-stimulation of Caco-2 cells co-cultured with EGCs in the absence of nicotine resulted in a two-fold increase in P-NF-κBp65 expression (Fig. 3D). However, co-culture with nicotine-activated EGCs prevented the Cytomix-induced increase in P-NF-κBp65 expression. Thus, it appears that nicotine-activated EGCs reduce Caco-2 monolayer permeability through an inhibition of the NF-κB pathway.
Activated EGCs reduce phosphorylated myosin light chain (P-MLC) expression in caco-2 cells
After determining the effects of activated EGCs on the NF-κB pathway, we sought to determine if these effects lead to an improvement in tight junction protein expression. Thus, we examined P-MLC expression in Caco-2 cells after 2 h of Cytomix-stimulation, as increases in NF-κB activation are associated with increases in MLCK transcription [18,22,23]. Furthermore, increases in P-MLC are associated with increased actin cytoskeletal contraction and tight junction breakdown [6,7,9,18,24]. Cytomix-stimulation of Caco-2 cells in co-culture with EGCs in the absence of nicotine resulted in an increase in P-MLC expression, which is associated with tight junction breakdown (Fig. 4). However, co-culture of Cytomix-stimulated Caco-2 cells with nicotine-activated EGCs significantly reduced P-MLC expression levels: to control values (Fig. 4). Therefore, it appears that nicotine-activated EGCs improve Caco-2 barrier function through a reduction in P-MLC expression.
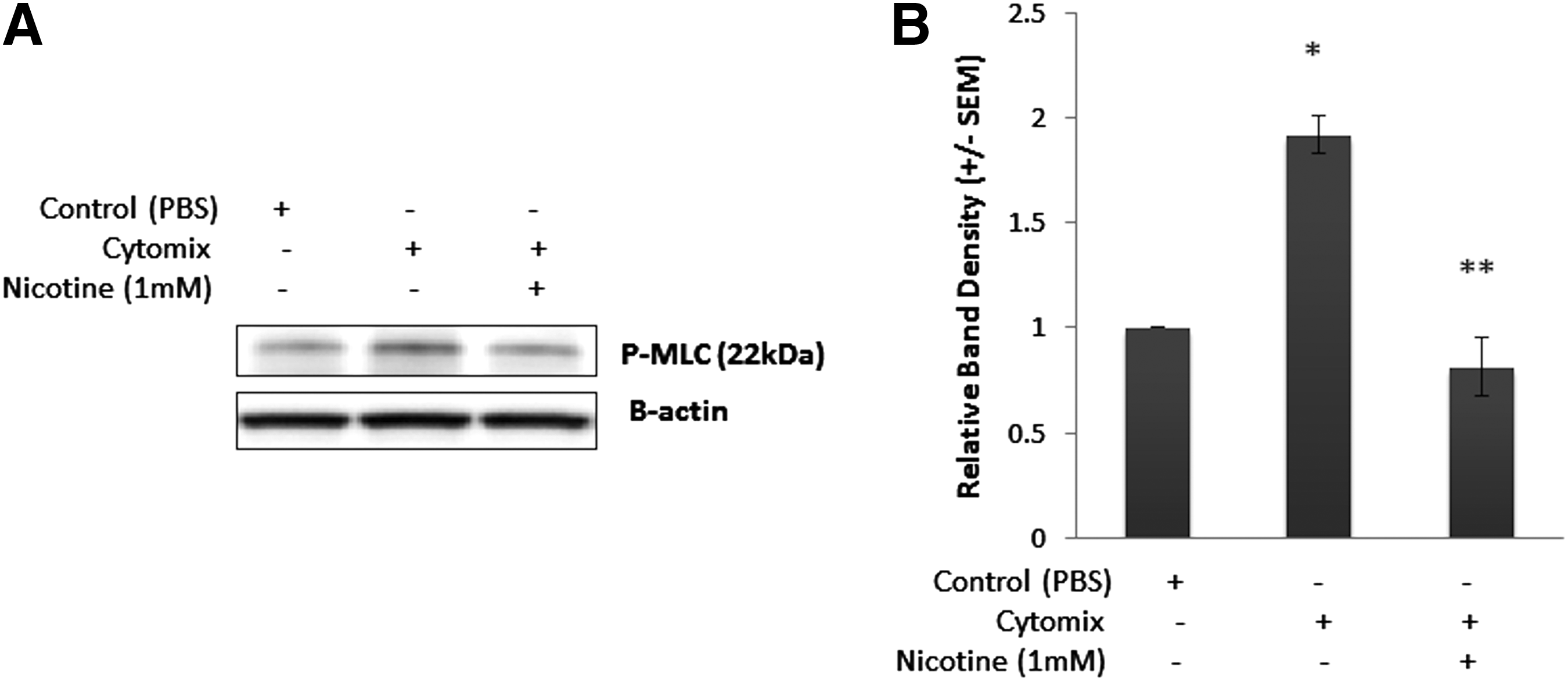
Nicotine-activated enteric glial cells (EGCs) improve Caco-2 barrier function through a reduction in phosphorylation of myosin light chain (P-MLC). Confluent Caco-2 cells were co-cultured with EGCs and incubated with phosphate-buffered saline or nicotine (1 mM) prior to stimulation with Cytomix for 24 h. Changes in P-MLC expression were measured using immunoblotting. (
Discussion
Because gut barrier breakdown is associated with systemic inflammation, we postulate that therapies that prevent barrier breakdown may have important clinical applications in improving patient outcome after severe injury. Recent evidence suggested that VNS may be a useful strategy for attenuating gut barrier failure in patients who have been injured severely [2,8–11]. Our laboratory has focused on studying the vagus nerve as a potential modulator of the intestinal barrier and has found that VNS improves gut barrier function through activation of EGCs and modulation of intestinal tight-junction proteins [11]. In this study, we examined more closely the signaling events that mediate VNS-induced gut protection, specifically focusing on the potential of EGCs to propagate the barrier-protective effects of VNS. We studied the nicotinic cholinergic agonist nicotine, as studies suggest that the vagus nerve propagates several of its anti-inflammatory signals through nicotinic cholinergic mechanisms. Borovikova et al. demonstrated that cholinergic agonists such as acetylcholine, nicotine, and muscarine all replicate the anti-inflammatory effects of VNS [25].
In this series of experiments, we have established a potential connection between the vagus nerve and EGCs by showing that the nicotinic cholinergic agonist, nicotine, increases EGC activation, as measured by changes in GFAP expression. MacEachern et al. also demonstrated that nicotinic cholinergic agonists activate EGCs, which resulted in improved intestinal epithelial ion transport [16]. Furthermore, we were able to show that nicotine-activated EGCs improve barrier function in epithelial monolayers stimulated with pro-inflammatory cytokines through inhibition of the NF-κB pathway and a reduction in P-MLC expression. The barrier-protective effects of nicotine were lost when Caco-2 cells were exposed to nicotine in the absence of EGCs, suggesting that nicotine exerts its effects through EGC activation rather than by acting directly on the intestinal epithelial cell. This supports our data published previously, here showing similar effects at a higher dose of nicotine [17]. Combined with data presented in this study, it is evident increasingly that VNS can signal to EGCs, and that this may be mediated through a nicotinic cholinergic signaling mechanism.
Gut barrier breakdown is associated with intestinal epithelial-cell nuclear factor-κB (NF-κB) activation [18,22,23]. In normal conditions, NF-κB is sequestered in the cytoplasm by its bound protein inhibitor of κB-α (IκBα). However, pro-inflammatory stimuli of intestinal epithelial cells results in phosphorylation of IκBα, marking it for ubiquination and degradation, freeing NF-κB to translocate to the nucleus and initiate transcription of myosin light chain kinase (MLCK) [18,22,23]. Increases in MLCK expression result in increased phosphorylation of MLC, inducing actin cytoskeletal contraction and breakdown of the tight-junction proteins ZO-1 [6,7,18,24] and occludin [9]. Thus, activation of the NF-κB pathway increases intestinal permeability through tight-junction breakdown.
We demonstrated that inflammation-induced increases in NF-κB activation are associated with increased epithelial permeability and P-MLC expression in vitro. To simulate severe injury in the model, we utilized the pro-inflammatory cytokines TNF-α, IFN-γ, and IL-1β, collectively referred to as Cytomix, as they have been validated in multiple studies to simulate the effects of inflammation-induced intestinal barrier failure [26–29]. Other studies have shown similar correlations between intestinal permeability and increases in NF-κB activation and P-MLC expression in vivo. In a murine severe burn model, increases in intestinal P-IκBα and P-NF-κB resulted in increases in MLCK activity [18], which led to increases in P-MLC expression. Findings here suggest that increased intestinal NF-κB activation and subsequent increase in P-MLC shown in this study results in the loss of barrier function in intestinal epithelial cells.
To examine further the importance of activated EGCs themselves, we determined their effects on injured intestinal epithelial cells. Our results indicate that EGCs reduce Cytomix-induced epithelial permeability by inhibiting NF-κB activation and decreasing P-MLC expression. The barrier-inducing effects of EGCs have been well validated in many other studies [30–32]. The EGCs have a dramatic effect on the global gene expression of intestinal epithelial cells, increasing expression of cell adhesion, differentiation, and motility genes, while decreasing subsequently expression of proliferation genes [32]. Additionally, in vitro studies by Neunlist et al. revealed a similar correlation in that EGCs reduce epithelial cell proliferation and increase cell surface area [30]. Evidence in this and numerous other studies suggest that the primary method of communication between EGCs and the intestinal epithelium is through soluble secreted mediators [12–14,16,33].
Although this finding certainly provides an important link between EGCs and the intestinal epithelium, the intracellular signaling mechanism in these models has yet to be identified. However, results from this series of experiments suggest that activated EGCs inhibit intestinal epithelial cell NF-κB activation, resulting in improved epithelial barrier function through a reduction in P-MLC expression. Inhibiting the NF-κB inflammatory pathway may improve gut barrier function, as it has been shown in both our laboratory and others that binding of the NF-κB transcriptional factor in intestinal epithelial cells is associated with an increase in P-MLC expression, resulting in tight-junction disruption [18,22]. The mechanism by which EGC activation causes inhibition of NF-κB nuclear translocation has yet to be fully defined and should be a point of further study.
Taken together, our findings suggest that the barrier protective effects of VNS are a result of nicotinic cholinergic signaling, which results in activation of EGCs (Fig. 5). Our data suggest that EGC activation improves intestinal barrier integrity by inhibiting the pro-inflammatory NF-κB pathway. Defining further the mechanism by which EGCs limit intestinal barrier breakdown after injury may lead to novel treatment strategies aimed at limiting the systemic inflammatory response to severe injury.
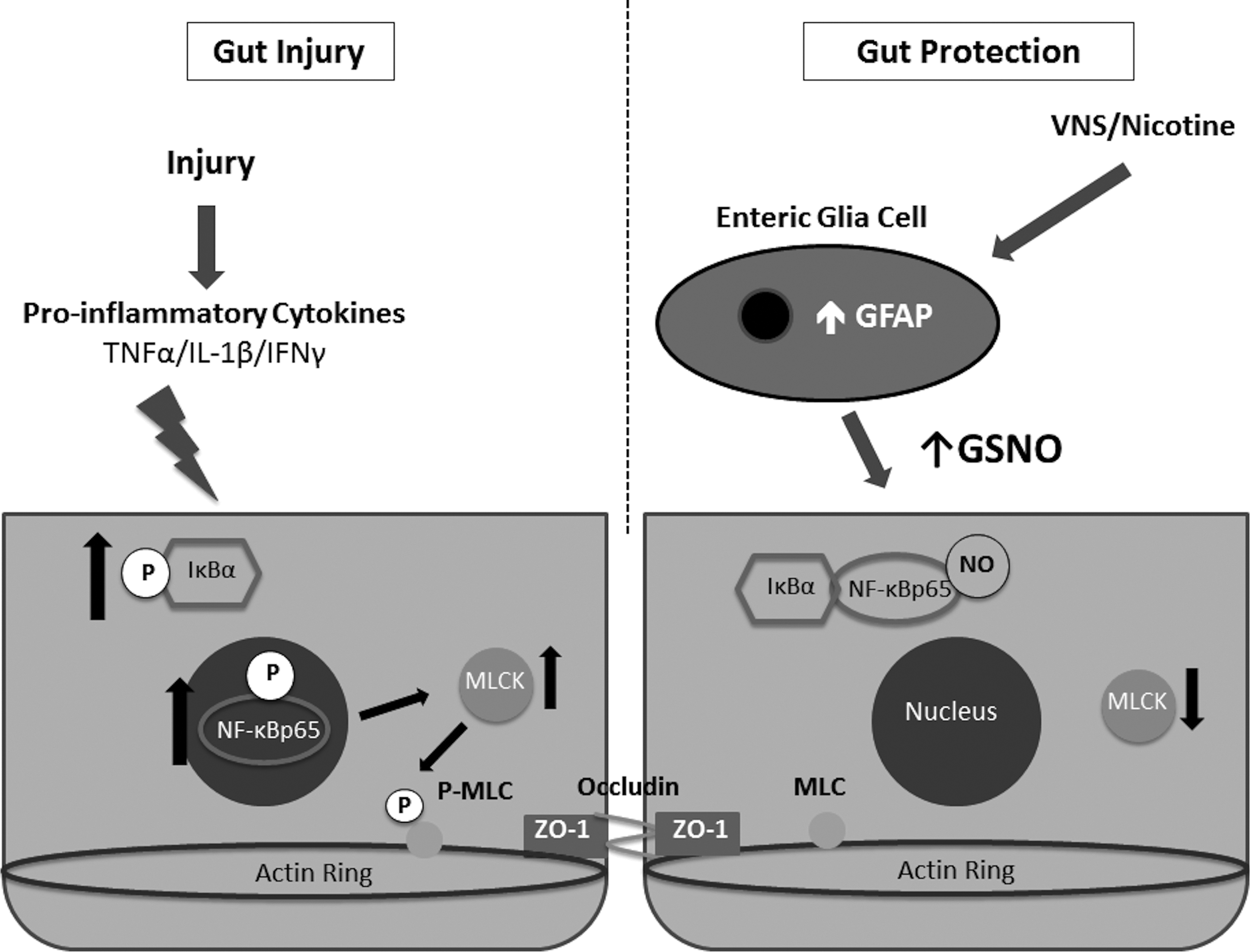
Diagram demonstrating mechanism by which vagal nerve stimulation provides gut barrier protection after injury.
Footnotes
Acknowledgments
The authors thank Ann-Marie Hageny and James Putnam for their technical assistance.
Author Disclosure Statement
The authors have no disclosures to report.