
Abstract
Cleft lip and palate (CLP), one of the most frequent congenital malformations, affects the alveolar bone in the great majority of the cases, and the reconstruction of this defect still represents a challenge in the rehabilitation of these patients. One of the current most promising strategy to achieve this goal is the use of bone marrow stem cells (BMSC); however, isolation of BMSC or iliac bone, which is still the mostly used graft in the surgical repair of these patients, confers site morbidity to the donor. Therefore, in order to identify a new alternative source of stem cells with osteogenic potential without conferring morbidity to the donor, we have used orbicular oris muscle (OOM) fragments, which are regularly discarded during surgery repair (cheiloplasty) of CLP patients. We obtained cells from OOM fragments of four unrelated CLP patients (CLPMDSC) using previously described preplating technique. These cells, through flow cytometry analysis, were mainly positively marked for five mesenchymal stem cell antigens (CD29, CD90, CD105, SH3, and SH4), while negative for hematopoietic cell markers, CD14, CD34, CD45, and CD117, and for endothelial cell marker, CD31. After induction under appropriate cell culture conditions, these cells were capable to undergo chondrogenic, adipogenic, osteogenic, and skeletal muscle cell differentiation, as evidenced by immunohistochemistry. We also demonstrated that these cells together with a collagen membrane lead to bone tissue reconstruction in a critical-size cranial defects previously induced in nonimmunocompromised rats. The presence of human DNA in the new bone was confirmed by PCR with human-specific primers and immunohistochemistry with human nuclei antibodies. In conclusion, we showed that cells from OOM have phenotypic and behavior characteristics similar to other adult stem cells, both in vitro and in vivo. Our findings suggest that these cells represent a promising source of stem cells for alveolar bone grafting treatment, particularly in young CLP patients.
Introduction
An alternative strategy would be to utilize bone marrow stem cells (BMSC) supplied with biomaterials, such as collagen sponge and platelet-rich plasma, which have already shown promising results in preclinical and clinical trials.5,9–14 However, isolation of BMSC for tissue bioengineering is as invasive as obtaining medullar bone. Therefore, it is important to identify alternative stem cell sources for alveolar cleft reconstruction in newborn patients that rely on less-invasive approaches.
One potential approach is to harvest stem cells from tissues obtained through standard treatment of CLP patients. The first surgical procedure performed in the rehabilitation of CLP patients is cheiloplasty, where small fragments of orbicular oris muscle (OOM) are regularly discarded. Muscle tissue contains a heterogeneous cell population, but is considered to be a promising source of stem cells. Muscle precursor cells (MPC) and muscle-derived stem cells (MDSC) are of particular interest, both with the capacity of differentiating into other cell types, such as adipocytes, osteocytes, and chondrocytes.15–22
The multipotency of MDSC prompted further investigation of the possible utilization of these muscle fragments as sources of stem cells for primary or secondary bone grafting protocols in the near future. This study describes the isolation and characterization of cells from OOM tissue of CLP patients, their antigen expression profile, and their ability to differentiate in vitro. We also show that these cells, here denominated “cleft lip and palate muscle-derived stem cells (CLPMDSC),” along with a collagen membrane (CM), successfully healed a critical-sized cranial defect in Wistar nonimmunosuppressed (NIS) rat model, suggesting their potential use in the reconstruction of the alveolar defect in CLP patients.
Materials and Methods
Isolation of stem cells from OOM of CLP patients
Stem cells were derived from OOM fragments obtained during surgical cheiloplasty performed at SOBRAPAR Hospital, Campinas, Brazil, in CLP patients. This study included tissue samples from four 3-month-old CLP patients, who were enrolled upon signed informed consent by their parents in accordance to the guidelines of the Ethical Committee of the Institute of Biosciences, at the University of São Paulo. We included only donors who did not present systemic diseases and/or oral infections. OOM cells were obtained by the previously described preplating technique15,16,19,23–25 and identified as CLPMDSC. Surrounding connective tissue was carefully removed, the samples then thoroughly washed with sterile phosphate-buffered saline (PBS) supplemented with 4% antibiotics (100 units/mL penicillin and 100 μg/mL streptomycin; Invitrogen, Carlsbad, CA), and digested with trypsin solution (TrypLe; Invitrogen) for 1 h at 37°C. Once digested, the tissue was transferred with minimal dissection into 35-mm Petri dishes (Corning, NY) containing D-MEM/F12 culture medium (Invitrogen) with 15% fetal bovine serum (FBS; Hyclone, Hyclone Laboratories, Logan, UT), 2 mM L-glutamine, 2 mM nonessential amino acids (Invitrogen), 100 units/mL penicillin, and 100 μg/mL streptomycin (Invitrogen). After 2 weeks, cells were washed with PBS, then dissociated in trypsin solution and seeded at 104 cells per 25 cm2 for the first passage. In order to prevent cell differentiation, cultures were maintained semiconfluent and subcultured every 4–5 days with daily medium changes.
Flow cytometry
Analysis of surface antigens was performed in all cell strains. Cells were harvested with TrypLe (Invitrogen), washed with PBS, and incubated at 4°C for 30 min with the following anti-human antibodies: CD29-PE CY5, CD90 (Thy-1), CD14, CD34, CD45-FITC, CD105, CD117, CD 31-PE (Becton Dickinson, Franklin Lakes, NJ), SH3, and SH4 (Case Western Reserve University, Cleveland, OH). After the wash, unconjugated primary antibodies were incubated with anti-mouse-PE secondary antibody (Guava Technologies, Hayward, CA) for additional 15 min at 4°C. Finally, the cell suspension was washed with PBS, and 104 labeled cells were acquired with an EasyCyte flow cytometer (Guava Technologies). Control samples were incubated with PBS instead of primary antibody, followed by incubation with anti-mouse-PE secondary antibody. All the generated plots were analyzed in Guava ExpressPlus software (Guava Technologies).
Cell differentiation procedures
To evaluate the properties of mesenchymal stem cells (MSC), CLPMDSC (fourth passage) were subjected to in vitro myogenic, chondrogenic, adipogenic, and osteogenic differentiations.
Myogenic differentiation
Cells were cultured in DMEM-HG supplemented with 10% FBS (Hyclone), 5% horse serum (Sigma-Aldrich, St. Louis, MO), 0.1 μM dexamethasone, 50 μM hydrocortisone, and 1% antibiotic (100 units/mL penicillin and 100 μg/mL streptomycin; Invitrogen) for 60 days.25–29 Differentiated CLPMDSC were stained with the following antibodies: monoclonal anti-α-sarcomeric actin, clone 5C5 (Sigma); monoclonal anti-myosin skeletal (Sigma); monoclonal anti-titin clone T11 (Sigma). Secondary antibodies used were CyTM3 goat anti-mouse (CyDye Fluorolink) and FITC goat anti-rabbit (Santa Cruz Biotechnology, Santa Cruz, CA), through a previously described protocol. 30 Samples were examined via confocal microscopy (Advanced Imaging Microscopy Release 3.5; Carl Zeiss, Jena, Germany).
Adipogenic differentiation
Cells were seeded into 6-well plates (Corning), at a density of 2 × 105 cells/well, in DMEM-HG supplemented with 10% FBS (Hyclone), 1 μM dexamethasone (Sigma), 100 μM indomethacin (Sigma), 500 μM 3-isobutyl-1-methylxanthine (IBMX), and 10 μg/mL insulin. Fifteen days after induction, Oil Red-O (Sigma) staining was used to identify intracellular accumulation of lipid-rich vacuoles. 12 Briefly, cells were fixed with 4% paraformaldehyde in PBS for 30 min, washed in PBS, and stained with a working solution of 0.16% Oil Red-O in PBS for 20 min.25–29
Chondrogenic differentiation
A pellet culture system was used for chondrogenesis induction. About 2.5 × 105 cells were transferred to a 15 mL centrifuge tube containing chondrogenic medium consisting of DMEM-HG supplemented with 1% linoleic acid (ITS-Premix; BD, Franklin Lakes, NJ), 1% 10 μM dexamethasone, 1% 100 mM sodium pyruvate, 1% 5 mM ascorbic acid-2 phosphate, and 10 ng/mL TGFB1 (R&D Systems, Minneapolis, MN), then centrifuged at 500 g for 5 min in order to pellet cells. On day 1, roughly spherical pellets were formed in each tube. On day 21, the pellets were fixed as described elsewhere. 29 Tissue samples were fixed in 10% formalin for 24 h (4°C) and paraffin embedded. For morphological analysis, 5-μm sections stained with toluidine blue were examined under AxioVert II light microscope (Carl Zeiss). 29 Primary antibodies used were mouse anti-human Collagen II (Abcam) and Aggrecan I (Abcam, Cambridge, MA), detected with CyTM3 goat anti-mouse (CyDye Fluorolink), and examined through confocal microscopy (Advanced Imaging Microscopy Release 3.5; Carl Zeiss).
Osteogenic differentiation
Cells were cultured in osteogenic medium containing DMEM-LG with 0.1 μM dexamethasone and 50 μM ascorbic acid-2 phosphate. On day 9, 10 mM β-glycerolphosphate was added to induce mineralization, and on day 21, von Kossa staining was performed in order to identify accumulation of mineralized calcium phosphate. Briefly, cells were stained with 1% silver nitrate (Sigma) for 45 min under ultraviolet light, followed by 3% sodium thiosulfate (Sigma) for 5 min, and then counterstained with Van Gieson.25–29
Karyotype analysis
Chromosomal G-banding staining was done according to standard protocol and nomenclature following the “International System for Human Cytogenetic Nomenclature” (ISCN). At least 100 metaphase spreads were counted.
Animal model to evaluate the use of undifferentiated CLPMDSC for bone healing
The Animal Research Ethics Committee at the Institute of Biosciences, University of São Paulo, approved the protocol for animal use in the present study. A total of six NIS Wistar rats (9-month-old males, body weight 320–420 g each) were selected. The animals were anesthetized with intraperitoneal injections (0.3 mL/100 g body weight) containing ketamine hydrochloride (5%) and xylazine (2%). The rats were then positioned in a cephalostat during the procedure. A midline skin incision from the nasofrontal area to the external occipital protuberance was performed. The skin and underlying tissues were reflected laterally to expose the full extent of the calvaria. Two symmetrical full-thickness 5 × 8 mm cranial defects 31 were made on each parietal region using a micromotor drill under constant irrigation with sterile physiologic solution to prevent bone overheating. The dura mater was maintained intact. CM (8 × 5 mm) (Oral Medica, São Paulo, Brazil) alone was positioned on the left side and CM with undifferentiated CLPMDSC on the right side. Animals were sutured with nylon-4 sutures (Ethicon, São Paulo, Brazil), and euthanized 20 (n = 4) or 30 days (n = 2) after cell transplantation.
Cell preparation for transplantation procedure
We used a CM carrier as a framework to seed undifferentiated human CLPMDSC, as previously described. 28 A total of 106 undifferentiated human MDSC from the previously characterized strain (CLPMDSC) were seeded onto CM placed on a 35-mm plate (6-well plate; Corning). The cells were supplemented with 2.5 mL of medium used for undifferentiated CLPMDSC and incubated at 37°C and 5% CO2 for 24 h prior to transplantation in order to adhere to the CM. Membranes with adherent cells were washed twice with PBS, transferred to the right cranial bone defect, and the cell-bearing CM surface was positioned in direct contact with the dura mater.
Histological preparation
Sections from the 20th and 30th postsurgery days were obtained for histological assessment. Tissue samples were fixed in 10% formalin solution for 24 h, decalcified in 5% formic acid for 48 h, and paraffin embedded. For morphological study, 5-μm sections stained with hematoxylin and eosin (HE) were examined under an AxioVert II light microscope (Carl Zeiss).
Analysis of the presence of human cells in the new bone
DNA analysis
Tissue from the left (membrane only) and right (membrane + cells) critical calvarial defects extracted from a rat euthanized 20 days after transplantation for histological assessment was scraped from the histological slides for DNA extraction, done through QIAamp DNA Mini KIT (Qiagen, Venlo, The Netherlands). For the human DNA amplification, PCR reactions were performed with EH10 primers (EH10F, 5′CGGGGTACCGGAGTGTAGGGCCAGAAACA3′/EH10R, 5′GGAAGATCTACTGCTTCCTTTGGTCGTGA3′), annealing temperature of 60°C and repeated for 35 cycles.
Immunohistochemistry
The sections were deparaffinized with two 5-min washes in xylene, hydrated in graded ethanol series and then rinsed in distilled water. The samples were fixed for 20 min at room temperature with 4% paraformaldehyde in PBS and washed three times with 100 mM glycine in PBS.
For antigen retrieval, slides were incubated for 40 min in citrate buffer (95–100°C) and then cooled for 20 min at room temperature, rinsed in phosphate buffered saline (PBS), and blocked for 1 h in immunofluorescent blocking buffer (IBB-5% BSA, 10% FBS, 1 × PBS, and 0.1% Triton X-100). Samples were then incubated for 1 h at room temperature with 1:100 mouse anti-human nuclei monoclonal antibody (HuNu; Chemicon, Temecula, CA), washed with PBST, and incubated with secondary antibody (1:600 Alexa Fluor 488 anti-mouse IgG; Invitrogen) for 1 h at room temperature. Tissue was counterstained with Alexa Fluor 568 Phalloidin (Invitrogen) and mounted using ProLong Anti-fade (Invitrogen).
Results
Isolation of CLPMDSC
Cells from OOM fragments started growing 10 days after plating. These cells generated primary adherent cultures with fibroblast-like morphology (Fig. 1A). The number of cells from OOM decreased slightly after freezing and thawing, and the remaining viable cells were successfully expanded during the consecutive days (data not shown). They were successfully grown up to the 18th passage, with semiconfluence appearance and without any signs of spontaneous differentiation. We did not observe any chromosomal abnormalities at the 1st or at the 18th passage (46, XX; Fig. 1B).
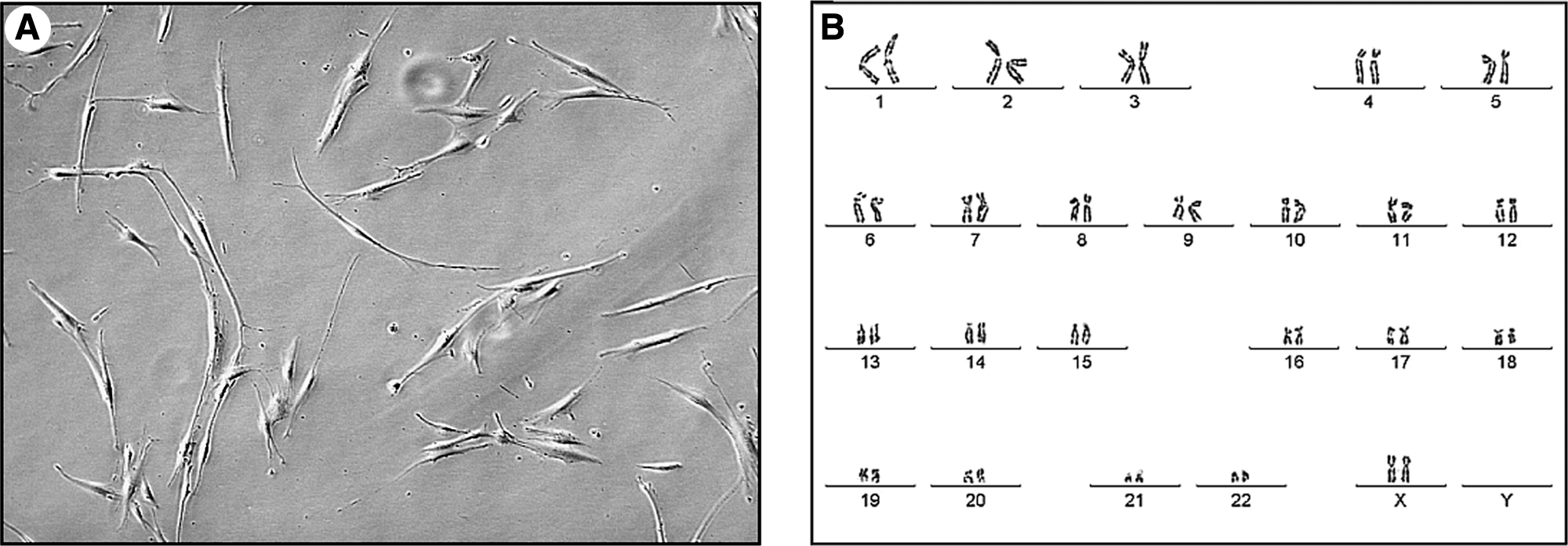
CLPMDSC strain displaying fibroblast-like morphology (
Immunophenotypic analysis
Ex vivo–expanded CLPMDSC expressed cell adhesion markers (CD29 and CD90) and MSC markers (SH3, SH4, and CD105) in the majority of the cells (>92%), while they did not express the hematopoietic makers (CD14, CD45, CD34, and CD117) and endothelial marker (CD31) (Fig. 2).

Flow cytometry analysis of CLPMDSC. Immunophenotype of adherent cells isolated from OOM (CLPMDSC). Values represent the mean percentage of all assessed cells positively stained by the indicated antigens (top of each graph) and analyzed by flow cytometry. Graphs show relative number of cells (counts) versus fluorescence intensity. Unmarked cells (control) were used as negative controls in both nonconjugated and conjugated antibodies, and nonspecific binding (isotype standard) was used only to nonconjugated antibodies. Abbreviations: CD, cluster of differentiation.
CLPMDSC exhibit multilineage potential in vitro
In order to evaluate their plasticity in vitro, the cultured cells were induced to differentiate into the following mesodermal cell lines: adipogenic, chondrogenic, myogenic, and osteogenic.
The cells induced to differentiate into myogenic strains became elongated with multiple nuclei. They expressed three skeletal muscle markers (myosin, Fig. 3A; actin, Fig. 3B; myosin and actin, Fig. 3C; titin, Fig. 3D), while cells that were not induced were negative for these markers (Fig. 3A′, B′, and D′).
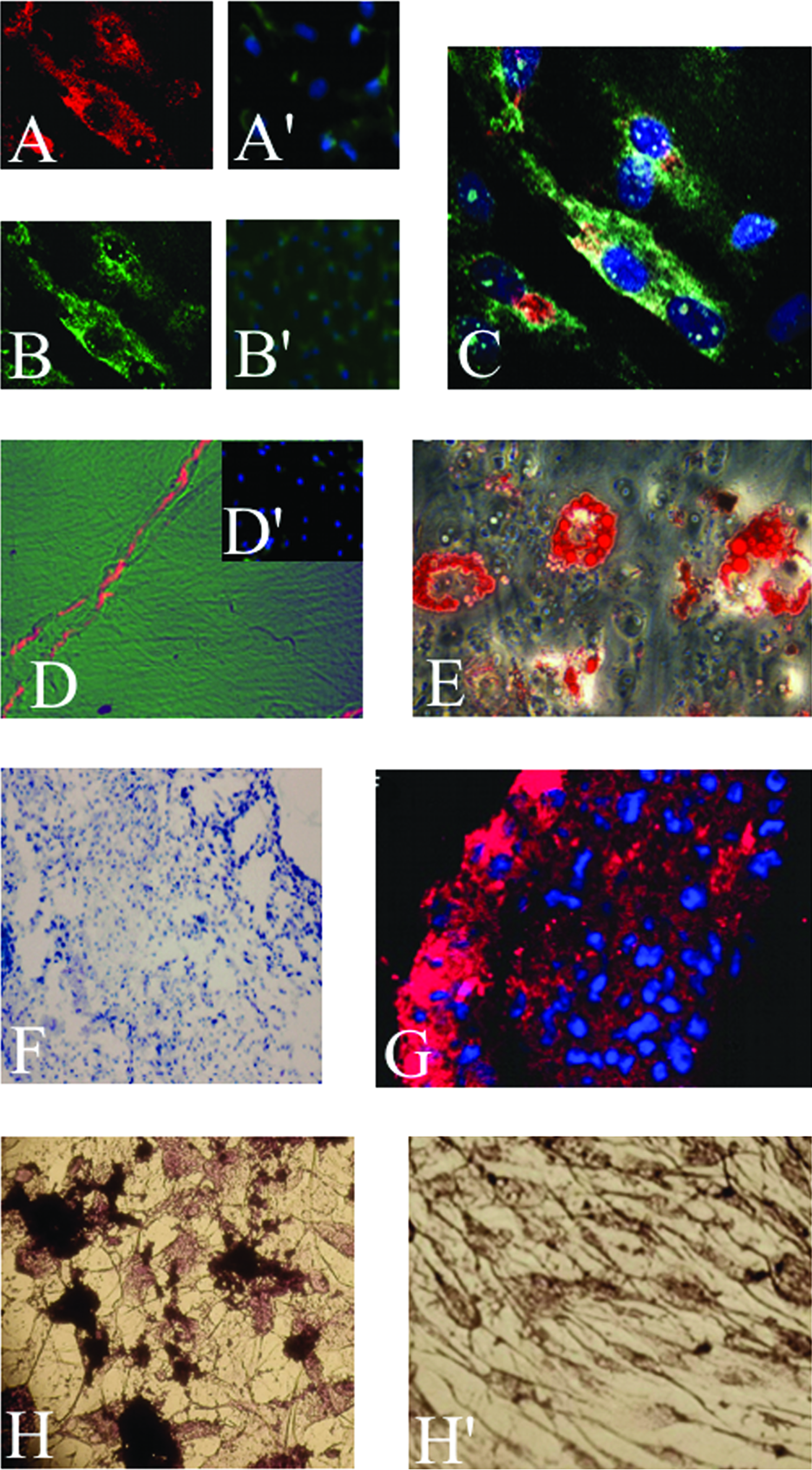
In vitro staining of CLPMDSC. Myogenic differentiation evidenced by actin (
The cells cultured in adipogenic medium exhibited multiple intracellular lipid-filled droplets visualized through Oil Red-O staining (Fig. 3E) and an increase of PPARγ2 through quantitative real-time PCR (Supplemental Fig. 1, available online at www.liebertonline.com/ten). No lipid droplets were observed in undifferentiated control CLPMDSC (data not shown).
In the chondrogenic assay, the cells in suspension formed round pellets with 1–2 mm in diameter. After 21 days in culture, toluidine blue–stained frozen sections were obtained from the pellets. These displayed irregular groups of cells embedded in a heterogeneous metachromatic-staining matrix surrounded by a thin capsule (Fig. 3F). Further, they expressed the chondrogenic markers Aggrecan I (data not shown) and collagen II (Fig. 3G) through immunohistochemical analysis. An increase in Aggrecan I was also observed through qRT-PCR (Supplemental Fig. 1).
A distinct morphological change was observed in the cells on the 9th and 21st day after bone-inducted differentiation. In addition, osteogenic differentiation was confirmed by observation of calcium mineralization through von Kossa staining (Fig. 3H) and, on the 9th day of growth, by an increase of c-fos detected through qRT-PCR (Supplemental Fig. 1). Noninduced cell cultures did not exhibit any calcium deposits (Fig. 3H′).
In vivo osteogenic potential of CLPMDSC
We have evaluated the in vivo osteogenic potential of these cells through the analysis of CLPMDSC in six animals using a previously established model in our laboratory. 28 None of the six experimental animals died of infection or any other complication as a result of surgery or cell transplantation process. Histological examination of the cranial defect 20 days postsurgery in four animals revealed intense deposition of new bone on the right side (Fig. 4A–C). We observed that the gap had been filled with woven bone intermixed with granulation tissue. On the other hand, the side containing CM alone had the defect filled with loose connective tissue exhibiting chronic inflammatory infiltrate, intermingled with remnants of the membrane (Fig. 4A′–C′). At 30 days postsurgery, a histological evaluation of the right side, which contained both the CLPMDSC and the CM, revealed reduction of granulation tissue, while bone was in an advanced maturation stage, with some lamellae formation in the two studied animals (Fig. 4D). On the left side, with CM only, it was possible to observe early bone neoformation just on the edge of the cranial defect (Fig. 4D′).

DNA amplification was successfully obtained only when we used DNA extracted from the right side, where the CLPMDSC had been transplanted (Fig. 5A). We also observed positive staining for human nuclei through immunohistochemical analysis only on the right side (Fig. 5B, B′). Thus, these results confirm that the human cells are part of the new bone formed in the critical cranial defect.

(
Discussion
We here report for the first time the establishment of OOM stem cell primary lineages obtained from four CLP subjects (aged 3 months) using the direct plating technique. These cells presented a fibroblast-like morphology and behavior consistent with other MSC obtained from bone marrow, 12 dental pulp,27,28 umbilical cord vein, 29 and fat. 26 Their chromosomes were also stable, as a normal karyotype was observed on both the 1st and 18th passages.
The high expression of mesenchymal (CD105, SH3, and SH4) and adhesion markers (CD29 and CD90) and the lack of expression of hematopoietic or endothelial markers observed in CLPMDSC are similar to the ones seen in primary MSC obtained from other sources, such as fat, dental pulp, bone marrow, and umbilical cord vein.25–29
Immunophenotype comparison between the CLPMDSC and other cell lineages obtained from muscle is difficult since the markers used for cell characterization vary in different studies.20–22 MPC express genes related to myogenic commitment, such as α-sarcomeric, actin, myosin heavy chains, and desmin, while MDSC are positive for CD105 and negative for CD45, CD34, and CD31 and only some lineages are actin positive.20–22 Although the number of cell markers tested on CLPMDSC, and on other MD cells, is still limited, the immunophenotype of OOM stem cells was more comparable to the MDSC lineage than to MPC. The OOM stem cells were also negative for titin, actin, and myosin.
The in vitro plasticity of CLPMDSC into four mesodermal tissues (adipocytes, muscle, chondrocytes, and osteocytes) was consistent with the observed lineage-specific differentiation of bone marrow, dental pulp, fat, muscle, and other adult stem cells.12,25–29 These results thus reinforce the mesenchymal properties of CLPMDSC.
CLPMDSC differentiate into bone tissue in vitro without the addition of any BMP. This observation further suggests that CLPMDSC are more comparable to MDSC than to MPC, as these last ones depend on BMP2 to successfully differentiate into in vitro bone tissue.19–22
The high osteogenic potential of the CLPMDSC was further observed through their ability, along with the CM, to reconstitute a critical cranial defect in NIS male Wistar rats. The newly formed bone tissue is, in part, of human origin since we were able to amplify its DNA using primers specific for human genes. In addition to that, positive staining for human nucleus was exclusively obtained on the right-side critical defects, where the CM containing the CLPMDSC was transplanted. The in vivo osteogenic potential of MDSC from the gluteus medius muscle of adult humans and hydroxyapatite scaffold in nude mice has also been recently reported. 19 However, as a particular note, we were able to correct a critical cranial bone defect, which does not spontaneously heal itself, in rats. Therefore, we suggest that the use of CLPMDSC combined with biomaterials could be a promising alternative in rehabilitating the alveolar bone of CLP patients, as it would eliminate the need for surgical intervention to obtain the rib or iliac bone, or BMSC for primary or secondary grafts.
Our present, as well as previously published, 28 work further suggests that human SC transplanted into rats do not suffer rejection or provoke any immunological response. This is not unexpected, as there are several reports showing that MSC have immunoprivileged properties. 32 These observations imply the potential application of heterologous CLPMDSC, or MSC derived from other sources, in patients for alveolar bone grafting procedures. The mechanism through which CLPMDSC induces in vivo bone differentiation needs to be investigated. It is possible that these cells play an important role in triggering the healing process in cells of the dura mater or surrounding bone tissue. This last assumption is supported by the finding that transplanted BMSC require the presence of an organized framework to which they can adhere and proliferate for periods long enough to ensure differentiation and osteogenesis.33–36 Differentiation of human CLPMDSC can also depend on interactions between them and CM, which occur on the surface and are the direct result of the unique chemical environment that is therefore created. 35 Finally, both human and rat cells can form bone, although the contribution of human stem cells was essential because CM alone was not capable of closing the critical-size defect.
In conclusion, we demonstrated that MDSC can be easily obtained from OOM fragments, which are currently discarded during cheiloplasty, allowing the isolation of these cells without the need of additional surgeries, thus, representing a noninvasive source of stem cells to be used in bone reconstruction.
Footnotes
Acknowledgments
The authors would like to express their gratitude to Professor Cassio Raposo do Amaral (1943–2005) for his incentive and fruitful ideas on new insights in rehabilitating craniofacial malformation; Professor Mayana Zatz for her incentive on stem cells research, Dr. Bechara Kachar, M.D., who allowed the use of his facility at NIH; Dr. Mariz Vainzof for the muscle antibodies; Mrs. Constancia Urbani for secretarial assistance, and the following colleagues for assistance: Carlos Maranduba, Ph.D.; Alexandre Kerkis, Ph.D.; Fernanda S. Jehee, Ph.D.; Rogerio Castilho, Ph.D.; Marta Canovas, Erika Yeh, and Natassia Vieira. FAPESP/CEPID and CNPq sponsored this study.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.