
Abstract
Biologic scaffolds composed of extracellular matrix (ECM) are utilized in numerous regenerative medicine applications to facilitate the constructive remodeling of tissues and organs. The mechanisms by which the host remodeling response occurs are not fully understood, but recent studies suggest that both constituent growth factors and biologically active degradation products derived from ECM play important roles. The objective of the present study was to determine if degradation of ECM scaffold materials in vitro by methods that are biochemically and physiologically relevant can yield products that possess chemotactic and/or mitogenic activities for fully differentiated mammalian endothelial cells and undifferentiated multipotential progenitor cells. ECM harvested from porcine urinary bladder was degraded enzymatically with pepsin/hydrochloric acid or papain. The ECM degradation products were tested for chemoattractant properties utilizing either 48-well chemotaxis filter migration microchambers or fluorescence-based filter migration assays, and were tested for mitogenic properties in cell proliferation assays. Results showed that ECM degradation products possessed chemotactic and mitogenic activities for multipotential progenitor cells and that the same degradation products inhibited both chemotaxis and proliferation of differentiated endothelial cells. These findings support the concept that degradation products of ECM bioscaffolds are important modulators of the recruitment and proliferation of appropriate cell types during the process of ECM scaffold remodeling.
Introduction
Previous studies have shown that nonphysiologic methods of ECM degradation such as acid and heat can produce low molecular weight molecules that have angiogenic, 23 chemoattractant,23–25 and antimicrobial26,27 properties. Interestingly, degradation of the ECM scaffold is necessary to realize these biologic effects. It has been clearly shown that intact ECM has no antimicrobial effects, while degradation products of the same ECM are able to strongly inhibit bacterial growth.26–28 Similarly, if ECM scaffolds are chemically crosslinked such that they are resistant to degradation, the host remodeling response is markedly altered toward fibrous encapsulation and chronic inflammation rather than constructive remodeling. 29 It is clear that the products and biologic effects of ECM degradation are different from the components of the intact ECM.
Although the ECMs that are derived from different tissues vary in their three dimensional ultrastructures, all known mammalian ECMs are very similar in biochemical composition. Urinary bladder extracellular matrix (UBM) was chosen as a representative ECM for this study.
Papain and pepsin were selected as well-characterized enzymes to digest the UBM. The physiologic roles and methods of cleavage of each of these enzymes are well known. Papain is a cysteine protease with several physiologic roles, one of which is remodeling of extracellular matrix. 30 Both pepsin and papain are affordable and relatively easy-to-use for the purpose of in vitro ECM degradation.
The present study investigated the chemotactic and mitogenic activities of ECM degradation products that result from the exposure of urinary bladder–derived ECM to either papain or pepsin. Two separate populations of progenitor cells and four separate differentiated endothelial cell populations were used to evaluate biologic activities of the ECM degradation products.
Materials and Methods
Overview of experimental design
ECM was harvested from porcine urinary bladder 31 and then subjected to enzymatic digestion by exposure to either pepsin or papain. The degradation products were evaluated for chemotactic and mitogenic activities toward progenitor cells and endothelial cells (Table 1).
Cell types assayed included MRL blastema cells (MRL-B), multilineage progenitor cells (MLPCs), human aortic endothelial cells (HAEC), human microvascular endothelial cells from bladder and lung (HMVEC-bladder and HMVEC-lung, respectively), and bovine adrenal capillary endothelial cells (BEC). Concentrations of ECM digests used in these assays were as follows: ECM-pepsin digest, 200–500 μg dry weight/mL for Neuro Probe 48-well microchemotaxis chamber migration assay, 1 mg dry weight/mL for fluorescence-based migration assay, and 78.9 μg total protein/mL for proliferation assay; ECM-papain digest, 500 μg total protein/mL for Neuro Probe chamber migration assay.
Urinary bladder matrix preparation
Porcine urinary bladders were harvested from market-weight pigs (110–130 kg) immediately after euthanasia. Urinary bladder matrix (UBM) was prepared as previously described. 31 Briefly, connective tissues were removed from the serosal surface of the bladder. The tunica serosa, tunica muscularis externa, tunica submucosa, and most of the tunica muscularis mucosa were then mechanically delaminated, and the luminal urothelial cells of the tunica mucosa were dissociated by soaking in a 1.0 N saline solution. The remaining tissue consisted of only the basement membrane, the subjacent tunica propria of the tunica mucosa, and the resident cell population of those two layers. The matrix was decellularized by treating with 0.1% peracetic acid/4% ethanol for 2 h and rinsing with phosphate-buffered saline (PBS) and deionized water. Complete decellularization was confirmed using 4′-6-diamidino-2-phenylindole (DAPI) nuclear staining and hematoxylin and eosin staining. The ECM was then lyophilized and frozen. The frozen ECM was comminuted into a particulate form using a Waring commercial blender and a Wiley Mill with a #60 mesh screen. 32
Pepsin digestion
UBM was digested with pepsin by mixing 1 g of lyophilized, powdered UBM with 100 mg pepsin (Sigma-Aldrich, St. Louis, MO) in 100 mL 0.01 N hydrochloric acid. This solution was stirred constantly at room temperature for 48 h, neutralized to a pH of 7.4 (pepsin is inactivated at neutral pH), and then assayed for chemotactic and mitogenic activities at doses ranging from 10 to 1000 μg dry weight/mL in the chemotactic assays and from 15.8 to 78.9 μg total protein/mL in the proliferation assays. One hundred milligrams of pepsin in 100 mL 0.01 N hydrochloric acid, without UBM, was prepared in parallel, incubated for 48 h, neutralized, and used as a control in the chemotactic and proliferation assays.
Papain digestion
One gram of lyophilized, powdered UBM was placed in a flask containing 100 mL of PBS with 1 U/mL of papain (Sigma-Aldrich). The flask was covered to minimize evaporation, placed inside an incubator at 37°C, and kept under constant stir for a total of 24 h. After the 24 h digestion period, the solution was brought to room temperature and spun at 1000 rpm for 6 min, and the supernatant was collected. For each milliliter of solution, 1 mL of 1 mM E-64 protease inhibitor (Sigma-Aldrich) was added to inactivate the papain. 1 U/mL of papain in PBS, with no UBM, was incubated at 37°C, centrifuged, and inactivated with 1 mM E-64 in parallel, and was used as a control in the chemotactic assay. Intact ECM was not used as a control with either the pepsin or papain digestion products because elution of variable amounts of intact growth factors such as vascular endothelial growth factor (VEGF) and basic fibroblast growth factor (bFGF) would add unknown variables to the experimental design. 33
Cell culture
MRL blastema cells (MRL-B cells) were isolated from the ears of MRL/MpJ mice (Jackson Laboratories, Bar Harbor, Maine). 34 A 2.0-mm hole was punched through the ear of each mouse. The MRL/MpJ mice have been described as having exceptional healing/regenerative properties. 35 At the site of the ear hole punch in the MRL/MpJ mice, a blastema-like structure forms, and if the cells are allowed to accumulate, differentiate, and organize, then the result is ear hole closure and tissue regeneration. 36 At 11 days after creating the ear hole, there was evidence of partial closure, and cells were isolated from the healing edge of the defect. Cells were grown in Dulbecco's modified Eagle's medium (DMEM, Cat. no. D6429; Sigma-Aldrich) supplemented with 10% fetal calf serum (FCS), 2 mM glutamine, 100 U/mL penicillin, and 100 μg/mL streptomycin in 95% air/5% CO2 at 37°C.
Multilineage progenitor cells (MLPCs), a primary cell line of human umbilical cord blood origin that is capable of differentiation into cell types representing the three germinal layers, were purchased from BioE (St. Paul, MN) and cultured in mesenchymal stem cell growth medium (MSCGM) (Cambrex, East Rutherford, NJ) under a humidified atmosphere in 95% air/5% CO2 at 37°C.
Both MLPC and MRL-B cells are positive for molecules associated with progenitor cell status and with tissue regeneration.34,37 Specifically, both cell populations showed expression of Tenascin-C, Thy1, Dlk/Pref-1, Msx1, Thrombospondin, and Tbx5 (Fig. 1 and Table 2).
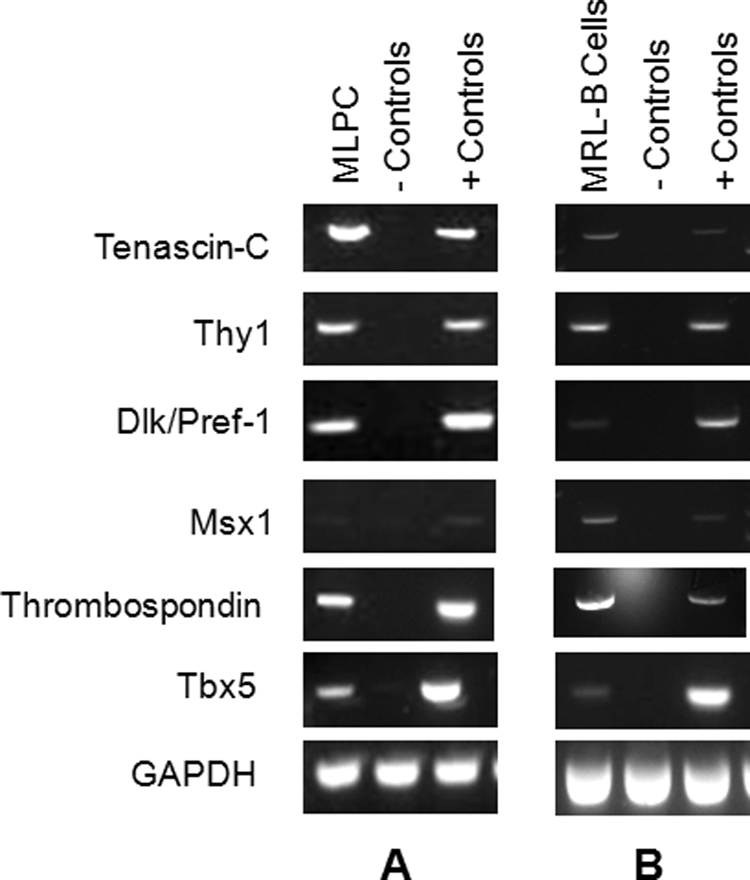
Both multilineage progenitor cells (MLPCs) and MRL day 11 blastema cells (MRL-B cells) were positive for all genes of interest when compared to the cell lines or tissue controls listed in Table 2. RNA was extracted and reverse transcribed for (
Primers for Tbx5, Dlk/Pref-1 (Dlk), Msx1, Thrombospondin (THBS), Tenascin-C (TnC), Thy1, and glyceraldehyde 3-phosphate dehydrogenase (GAPDH) were purchased from Operon Biotechnologies (Huntsville, AL).
Primers that were designed using the PrimerQuest software from Integrated DNA Technologies (Coralville, IA) and verified by BLAST sequencing. All primer sets span intronic sequences as an added quality control step. Cell lines used as controls included the mouse fibroblast cell line NIH-3T3 and the human breast cancer cell line MCF-7.
Human aortic endothelial cells (HAEC), human microvascular endothelial cells from bladder (HMVEC-bladder), and human microvascular endothelial cells from lung (HMVEC-lung) were purchased from Lonza (Walkersville, MD). HAEC were cultured in EGM2 medium (Lonza) under a humidified atmosphere in 95% air/5% CO2 at 37°C; HMVEC-bladder and HMVEC-lung were cultured in EGM2-MV medium (Lonza) under a humidified atmosphere in 95% air/5% CO2 at 37°C.
Capillary endothelial cells from a primary cell line of bovine adrenal cortex (bovine endothelial cells, or BEC) were purchased from Lonza and cultured in EGM2-MV medium with a bullet kit CC-3202 (Lonza) and a final FCS concentration of 5%. For proliferation assays, cells were plated in 48-well plates at 1000 cells/well in EGM2-MV medium with 10% FCS (JRH Biosciences, Lenexa, KS) supplemented with 2 mM glutamine, 100 U/mL penicillin, 100 μg/mL streptomycin (GPS; Irvine Scientific, Santa Ana, CA), and 2 μg/mL Fungizone (Fisher Scientific, Pittsburgh, PA), without added growth factors under a humidified atmosphere of 95% air/5% CO2 at 37°C.
RNA isolation and analysis
Mouse MRL-B cells and human MLPC were grown to at least 50% confluency in 75 cm2 flasks, collected, and lysed. Total RNA was extracted using the Qiagen RNeasy Kit (Qiagen, Valencia, CA) according to the manufacturer's instructions with the recommended DNase treatment for removal of genomic DNA. RNA concentrations were determined by measuring absorbance at 260 nm on a BioMate3 spectrophotometer (Thermo Spectronic, Rochester, NY). Control RNA samples from human and mouse tissues were prepared by homogenization in TriReagent and chloroform extraction; RNA was extracted from aqueous phase using Qiagen RNeasy Kit as detailed above. First-strand cDNA was synthesized from 1 μg of RNA using the Superscript First-Strand Synthesis System (Invitrogen, Carlsbad, CA) according to the manufacturer's instructions. PCR was performed with primers specific for Tbx5, Dlk/Pref-1 (Dlk), Msx1, Thy1, Thrombospondin (THBS), Tenascin-C (TnC), and glyceraldehyde 3-phosphate dehydrogenase (GAPDH) (Table 2). cDNAs were amplified in EasyStart Micro100 tubes (Molecular BioProducts, San Diego, CA) with 100 nM of primer in a Mastercycler (Eppendorf, Westbury, NY). All PCR runs were 35 cycles. PCR products were separated on 2% agarose gels, stained with ethidium bromide, and visualized on Kodak Image Station 2000R (Kodak, Rochester, NY).
Neuro Probe chamber chemotaxis assay
Responses of MRL-B cells and endothelial cells to ECM degradation products were quantitatively evaluated utilizing the Neuro Probe 48-well microchemotaxis chamber (Neuro Probe, Gaithersburg, MD). Cells to be assayed for migration response were starved for 14–17 h in media with no added growth factors containing 0.5% heat-inactivated FCS. The starved cells were harvested with trypsin, resuspended in serum-free media at a concentration of 6 × 105 cells/mL, and preincubated for 1 h in a humidified 95% air/5% CO2 37°C incubator. Polycarbonate chemotaxis filters (Neuro Probe, Gaithersburg, MD), 8 μm pore size filters (Neuro Probe PFB8) for endothelial cells and 12 μm pore size filters (Neuro Probe PFB12) for MRL-B cells, were coated equally on both sides (by immersion) with 0.05 mg/mL collagen 1 (BD Biosciences, San Jose, CA). The ECM degradation products collected from the pepsin or papain digestion were added to the bottom chamber wells (see Table 1 for optimum concentrations), the filter was placed over the bottom chamber, and the apparatus was assembled according to the manufacturer's instructions. Approximately 30,000 cells were then added to each upper chamber well of the apparatus, and the chamber was incubated for 3 h at 37°C under a humidified atmosphere in 95% air/5% CO2. Cells remaining on the topside of the membrane (i.e., nonmigrated cells) were removed, and then cells on the bottom side of the membrane (i.e., migrated cells) were stained with Diff Quik (Dade AG, Liederbach, Germany). Three predetermined fields were counted from each well at 20 × magnification. Fields were predetermined by location in the well: cells in the top left, top right, and bottom center fields in each well were counted. Each experimental condition was tested in quadruplicate wells, and the average number of migrated cells was determined for each condition. The Student's t-test was used to test the null hypothesis that there was no difference between the results of cell migration toward each ECM degradation product and the appropriate control buffer. p-values ≤ 0.05 were considered significant.
Pilot studies were conducted to determine optimal concentrations of ECM degradation products for the Neuro Probe chemotaxis assay. A range of concentrations for each ECM preparation was tested: UBM pepsin digest was tested at concentrations ranging from 10 to 200 μg/mL dry weight; UBM papain digest was tested at concentrations ranging from 100 to 500 μg/mL protein.
The appropriate filter pore size for each cell type was determined by conducting pilot studies to measure the migration of the cells toward positive control chemoattractants and negative controls in the Neuro Probe chamber. Filters with pore sizes of 5, 8, and 12 μm were included in these pilot studies.
Figure 2A shows a representative filter from a Neuro Probe chemotaxis assay, and Figures 2B and 2C show photographs of migrated cells from that filter. The photograph of the filter is from the bottom side of the filter after cells have been scraped from the topside. This particular assay measured MRL-B cell migration toward a range of UBM papain digest concentrations.
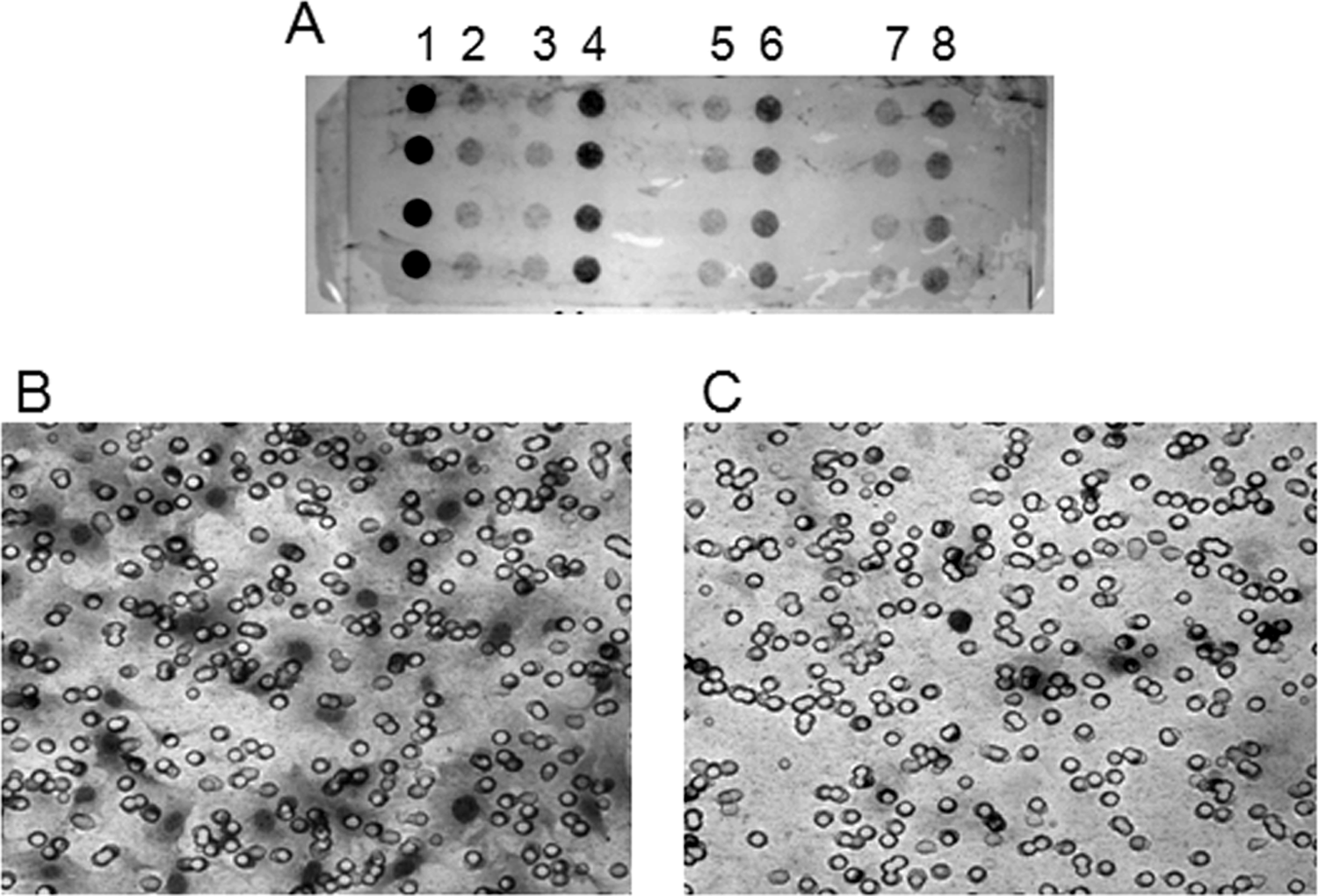
(
Fluorescence-based migration assay
Responses of MRL-B cells and MLPC to UBM pepsin digest were quantitatively evaluated utilizing the 8 μm CytoSelect™ cell migration assay (Cell Biolabs, San Diego, CA). Cells to be assayed for migration response were starved for 14–17 h in media with no added growth factors containing 0.5% heat-inactivated FCS. The starved cells were harvested with trypsin, resuspended in serum-free media at a concentration of 4 × 105 cells/mL, and preincubated for 1 h in a humidified 95% air/5% CO2 37°C incubator. While cells were preincubating, 150 μL of UBM pepsin digest or buffer control was added to each well of the 96-well feeder tray (see Table 1 for optimum concentrations). The 96-well membrane chamber insert was placed onto the feeder tray, and 100 μL of cell suspension was added to each well of the membrane chamber, for a final concentration of 40,000 cells per well. The plate was covered and incubated for 4 h at 37°C under a humidified atmosphere in 95% air/5% CO2. One hundred and fifty microliters of cell detachment solution was added to each well of a clean harvesting tray. The 96-well membrane chamber was separated from the feeder tray, remaining cells on the topside of the membrane chamber were removed by aspiration, and the membrane chamber was placed onto the harvesting tray containing cell detachment solution and incubated in a cell culture incubator for 1 h, rinsing any cells from the bottom of the membranes into the harvesting tray wells. CyQuant GR Dye/cell lysis solution was prepared by diluting the dye in lysis buffer (1:75), the membrane chamber was removed from the harvesting tray, and 50 μL of dye/cell lysis solution was added to each well of the harvesting tray. The tray was incubated at room temperature for 20 min in order to lyse the cells and stain the nucleic acids. One hundred and fifty microliters of the contents of each well was then transferred to a plate suitable for fluorescence measurement. Fluorescence was measured with a SpectraMax M2 Plate Reader (Molecular Devices, Sunnyvale, CA) at 480/520 nm. Each experimental condition was tested in triplicate, and the average number of migrated cells was determined for each condition. The Student's t-test was used to detect significant differences between the results of cell migration toward each ECM degradation product and the appropriate control buffer. p-values ≤ 0.05 were considered significant.
Proliferation assay
Quantitative measurement of MRL-B and capillary endothelial cell proliferation was determined in a cell proliferation assay. Cells were plated in their media without growth factor additions at 1000 cells per well in a 48-well plate. Twenty-four hours later, media was removed, the cells were washed, and media was replenished. At the time of media replenishment, either ECM degradation product or an identical volume of control buffer was added at varying doses to each well in triplicate. Time zero cell counts were determined in three of the wells using trypsinization and electronic cell counting with the Z1 Particle Counter (Beckman Coulter, Fullerton, CA). Three or 6 days after sample additions, cells in triplicate wells for control and each treatment group were trypsinized and counted (Z1 Particle Counter electronic counts). The mean value and standard deviation were determined for each condition. The Student's t-test was used to test the null hypothesis that there was no difference between the results of cell growth for cells grown in the presence of ECM degradation products and those that were grown in the presence of buffer controls.
Results
The UBM papain digest was chemotactic for MRL-B cells (p ≤ 0.01) and inhibited the migration of the endothelial cells HAEC (p ≤ 0.05) and HMVEC-lung (p ≤ 0.05) (Fig. 3). The migration of MRL-B cells toward UBM papain digest was fivefold increased over migration toward papain digest control buffer: the percent increase from control (average of three experiments) was 489% (p ≤ 0.01). Migration of both types of endothelial cells toward papain was decreased compared to migration toward papain digest control buffer. The decrease was 57% for HAEC (average of four experiments) (p ≤ 0.05) and was 42% for HMVEC-lung (average of three experiments) (p ≤ 0.05).
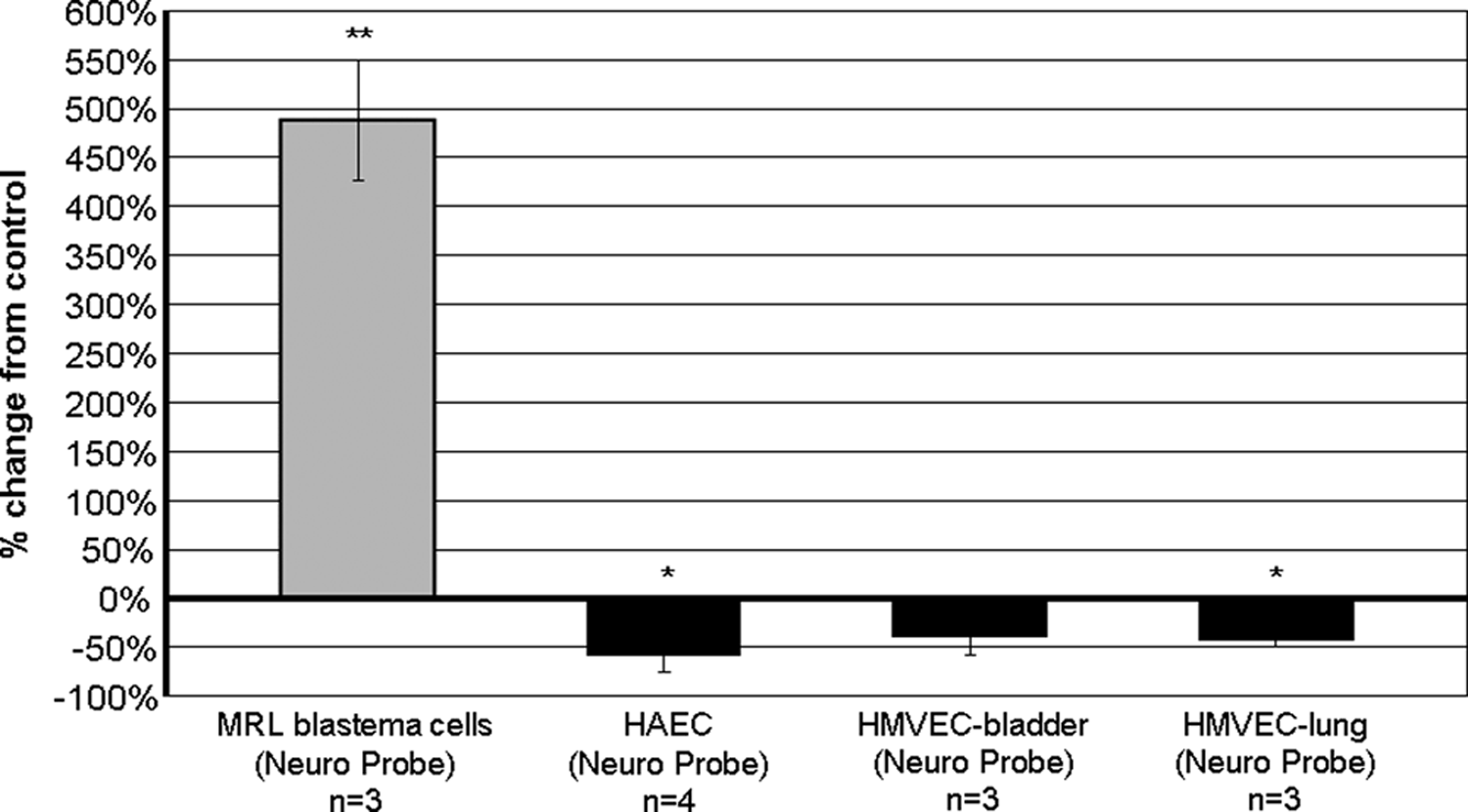
UBM papain digest was chemotactic for MRL blastema cells and was chemoinhibitory for human aortic endothelial cells (HAEC) and human microvascular endothelial cells (HMVEC). Cell migration responses to 500 μg total protein/mL of papain-digested UBM were measured in the Neuro Probe 48-well microchemotaxis chamber. In the comparison of cell migration toward the UBM papain digest and cell migration toward the control buffer, double asterisks (**) signify a p ≤ 0.01 and asterisk (*) signifies a p ≤ 0.05.
In a pattern similar to that observed for papain-digested UBM, the UBM pepsin digest was chemotactic for progenitor cells and inhibited the migration of endothelial cells (Fig. 4). UBM digested with pepsin enhanced the migration of MRL-B cells in both the Neuro Probe microchemotaxis chamber (405% increase from control) (p ≤ 0.01) and fluorescence-based filter migration assays (126% increase from control) (p ≤ 0.01), and enhanced the migration of MLPCs in the fluorescence-based migration assay (104% increase from control) (p ≤ 0.05). The migration response of MLPCs was tested only in the fluorescence-based migration assay because the MLPCs grow slowly and there were not enough cells available for the Neuro Probe chamber assay. This same UBM pepsin digest inhibited the migration of endothelial cells. Migration toward UBM pepsin digest compared to migration toward control buffer for HAEC, HMVEC-bladder, and HMVEC-lung was decreased 44%, 70%, and 73%, respectively (all p-values ≤ 0.05).
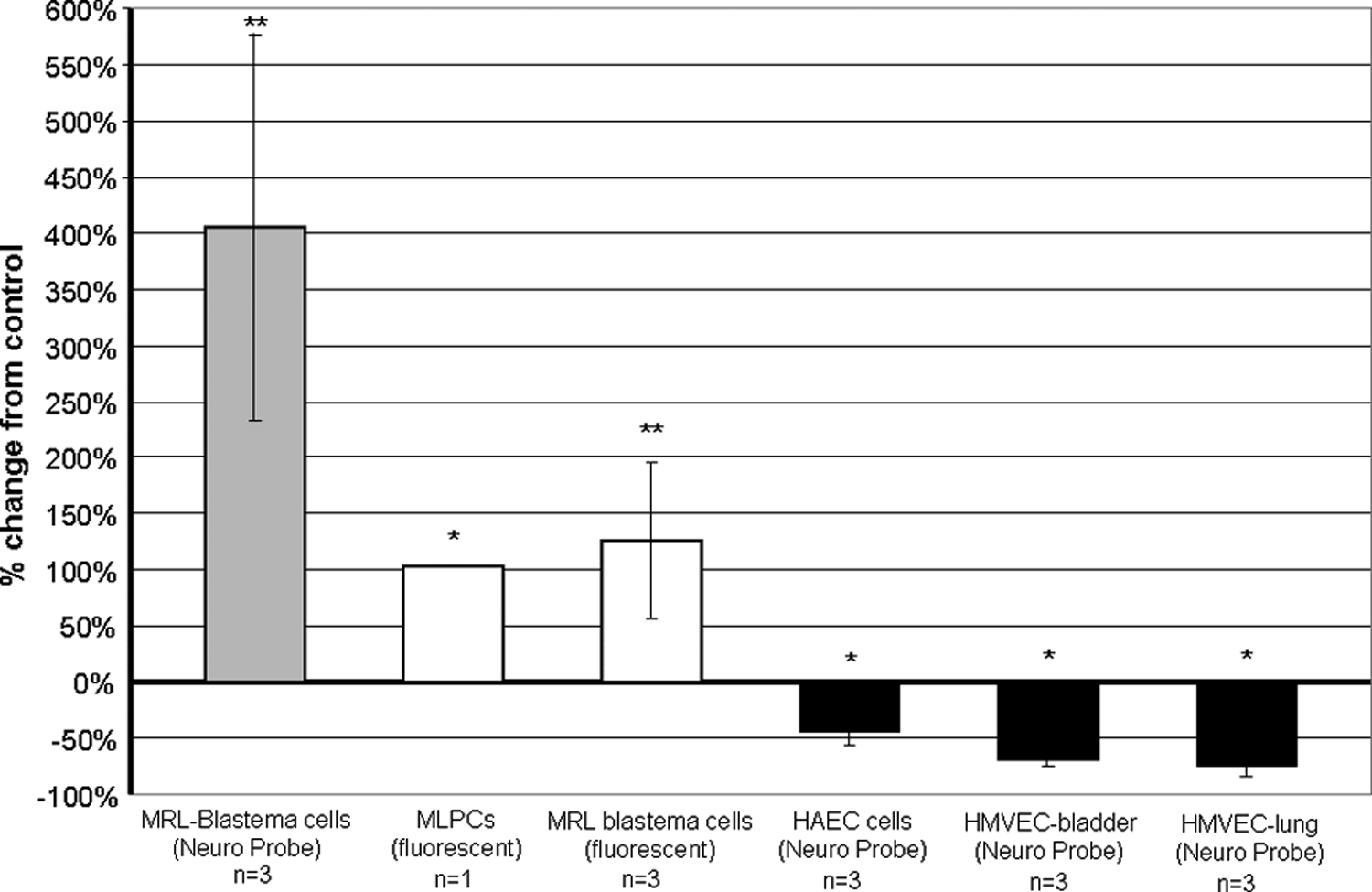
UBM pepsin digest was chemotactic for MRL blastema cells and for multilineage progenitor cells (MLPCs) and was chemoinhibitory for human aortic endothelial cells (HAEC) and for human microvascular endothelial cells (HMVEC-bladder and HMVEC-lung). Cell migration responses to pepsin-digested UBM were measured with either the Neuro Probe 48-well microchemotaxis chamber or the fluorescence-based migration assay. In the comparisons of cell migration toward pepsin digest and cell migration toward control buffer, double asterisks (**) signify a p ≤ 0.01 and asterisk (*) signifies a p ≤ 0.05.
Table 3 provides a summary of the migration experiments performed in the Neuro Probe assay and describes the data both in terms of numbers of migrated cells counted per well and percent change from the control.
Each experiment was set up in either triplicate or quadruplicate wells. n is the number of experiments conducted for each cell type and ECM digest combination. Percent change from control is calculated as the % change in migration of cells toward the pepsin or papain digest compared to migration of cells toward the appropriate buffer control.
p ≤ 0.01; bp ≤ 0.05.
Degradation products of UBM-ECM as a result of pepsin digestion were also evaluated in a quantitative assay of cell proliferation. The UBM pepsin degradation products enhanced proliferation of MRL-B cells and decreased the proliferation of capillary endothelial cells (Fig. 5) (p ≤ 0.05 for MRL-B cells and p ≤ 0.01 for capillary endothelial cells).
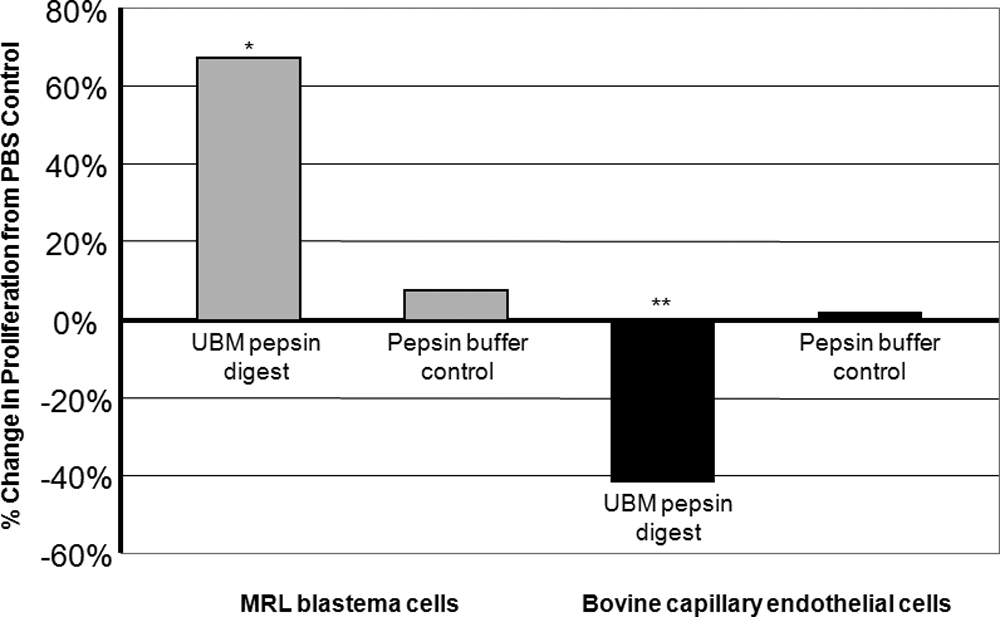
UBM pepsin digest enhanced the proliferation of MRL blastema cells and inhibited the proliferation of bovine capillary endothelial cells. Cell proliferation responses to 78.9 μg protein/mL of pepsin-digested UBM were measured in a 3-day proliferation assay. In the comparison of cell proliferation in the presence of pepsin digest compared to cell proliferation in the presence of PBS alone, asterisk (*) signifies a p ≤ 0.05 and double asterisks (**) signify a p ≤ 0.01.
The proliferation assay with UBM pepsin degradation products was conducted as both a 3-day and a 6-day assay for MRL-B cells. The results for the 3-day assay are shown in Figure 5. The UBM pepsin degradation products enhanced proliferation of MRL-B cells in both assays: in the 3-day proliferation assay, the UBM pepsin degradation products enhanced proliferation of MRL-B cells showing a 67% increase over the PBS control (p ≤ 0.05) (Fig. 5), and in the 6-day proliferation assay, the UBM pepsin degradation products again enhanced the proliferation of MRL-B cells, showing a 50% increase over the PBS control (p ≤ 0.01).
The 3-day proliferation assay with UBM pepsin degradation products was conducted three separate times for the capillary endothelial cells, in either triplicate or sextuplicate for each assay. In all three assays the UBM pepsin degradation products inhibited the proliferation of capillary endothelial cells. For the three assays, the decreases in proliferation from the PBS control for bovine capillary endothelial cells grown in the presence of UBM pepsin digest were −32% (p ≤ 0.05), −42% (p ≤ 0.01), and −66% (p ≤ 0.01). A representative proliferation assay demonstrating the inhibitory effect of UBM pepsin digest on the proliferation of capillary endothelial cells is shown in Figure 5.
Discussion
The results of the present study show that physiologically relevant methods of ECM degradation give rise to products that have clear effects upon the chemotactic and mitogenic responses of both multipotential progenitor cells and differentiated endothelial cells. Of interest, the effects upon multipotential cells and mature differentiated endothelial cells are opposite.
ECM scaffolds have been shown to facilitate the constructive remodeling of tissues in many body systems.16–21 The ECM remodeling process involves complex signaling between the scaffold and host cells, and the mechanisms by which host cell activity is modulated during this process are not completely understood. Growth factors such as transforming growth factor beta (TGF-β), VEGF, and bFGF are present in processed ECM scaffolds, and these growth factors retain most of their biologic activity following processing of the ECM for clinical use.43,44 Additionally, biologic scaffolds composed of ECM have been shown to degrade rapidly after implantation,45,46 and the ECM remodeling continues for days to weeks after the scaffold degradation is complete. Although the present study does not demonstrate a cause–effect relationship between the ECM degradation products and in vivo remodeling, it does suggest a possible mechanism for the constructive remodeling that has been observed in ECM scaffold materials that are subject to in vivo degradation.46,47 These findings may also at least partially explain the recruitment of progenitor cells to in vivo sites of ECM scaffold remodeling.24,25,48
In vitro degradation of SIS-ECM by physical and chemical methods has produced low molecular weight peptides with chemoattractant activity. These peptides range in size from 5 to 16 kDa and have demonstrated chemotactic activity for primary endothelial cells of mouse heart, liver, and kidney origins. 23 While these results indicate that biologically active peptides are produced as a result of ECM degradation, the use of acid and extreme heat to perform the degradation is not a physiologically relevant model. The current study utilized the more physiologically relevant method of enzyme digestion of an ECM bioscaffold—specifically, the use of pepsin and papain, two enzymes found in almost all mammalian systems and which have well-characterized mechanisms of peptide cleavage.
MRL-B cells are isolated from a blastema-like structure following injury in a strain of mice shown to have the capacity to replace numerous tissues and organs without scarring. 34 These mice were first discovered when ear hole punches resulted in the closure of the ear holes and regrowth of cartilage in the ear pinna. 35 The present study showed that this cell population expresses markers commonly found in cells with potential for multilineage differentiation. Since this cell population is only partially characterized,34,37 the significance of the chemotaxis and mitogenesis assay results must be considered with caution. However, combined with the similar results in the MLPC population, the findings suggest a mechanism by which multipotential cells are actively recruited to sites at which ECM is degraded, that is, cell recruitment following tissue injury or during the remodeling of a biologic scaffold composed of ECM.
MLPCs are clonally derived from human umbilical cord blood, express numerous stem cell markers, and are capable of differentiating into cell types from all three embryonic layers.49,50,51 Because it has been demonstrated that stem cells participate in remodeling of tissue in vivo, 25 it was of interest to examine the response of an undifferentiated cell line to the UBM peptides that resulted from UBM-ECM degradation.
The consistent but somewhat unexpected finding that UBM-ECM degradation products inhibit the migration and proliferation of mature endothelial cells is worthy of note. Progenitor cells in an undifferentiated state have been found to exhibit enhanced proliferation and resistance to apoptosis under conditions of low oxygen concentration. 52 We suggest that during early stages of tissue remodeling, blood vessel formation may not be required or even helpful. The in vitro inhibition of mature endothelial cell migration and proliferation by ECM degradation products in the present study may be a reflection of possible in vivo inhibition of blood vessel formation by ECM degradation products during specific (very early) stages of tissue remodeling.53,54
ECM scaffolds, when used in the repair of mammalian tissue, facilitate tissue remodeling and restoration of function. Elucidation of the mechanisms and temporal pattern of this process will optimize the utilization of ECM scaffolds in regenerative medicine. The results of the present study support the concept that molecules produced upon degradation of ECM scaffolds play roles in determining the timing and nature of recruitment and proliferation of appropriate cell types during tissue remodeling.