
Abstract
Adult adipose-derived stem cells (ASCs) are considered to be an alternative cell source for cell-based cartilage repair because of their multiple differentiation potentials. This article addresses the chondrogenic differentiation of ASCs seeded into poly-lactide-co-glycolide (PLGA) scaffolds after implantation in a subcutaneous pocket of nude mice. Human ASCs were seeded into PLGA (polylactic acid:polyglycolic acid = 90:10) scaffolds and cultured in transforming growth factor beta 1 (TGF-β1)-containing medium for 3 weeks in vitro. Then specimens were implanted into a subcutaneous pocket of severe combined immunodeficiency mice and harvested after 8 weeks. Chondrospecific messenger RNA (mRNA) expression was analyzed using reverse transcriptase polymerase chain reaction. Corresponding extracellular matrix (ECM) synthesis was demonstrated using immunohistochemical staining. Chondrospecific marker molecules such as collagen type II and type X, cartilage oligomeric matrix protein, and aggrecan subsequently increased during the 3 weeks period in vitro. After a further 8 weeks, in vivo samples pretreated with TGF-β1 continued expressing collagen type II and aggrecan mRNA, and collagen type II was found within the ECM using immunohistochemistry. Chondrospecific mRNA was not detected in control samples. ASC-seeded PLGA scaffolds express a stable chondrogenic phenotype in a heterotopic model of cartilage transplantation and represent a suitable tool for tissue engineering of cartilage.
Introduction
Because of the poor biomechanical properties and the fast degradation of fibrin and alginate, polymer scaffolds with a stable, biodegradable, permeable pore network were introduced to support cell attachment and proliferation and nutrient exchange and to provide stability. The most common of such polymers are polylactic acid (PLA), polyglycolic acid (PGA), and poly-lactide-co-glycolide (PLGA), often in the form of fibers 15 and sponges. 16 These biomaterials are biocompatible because random hydrolysis of the ester bonds in the polymer chain leads to bulk degradation, and are approved by the Food and Drug Administration for clinical use.17–19
Because we recently demonstrated that our manufactured PLGA copolymer scaffolds are suitable cell carriers for chondrocytes in vivo, 20 it was suggested that ASCs could differentiate into chondrocyte-like cells in such PLGA scaffolds and maintain the chondrogenic phenotype after transplantation in vivo. Until now, ASCs have been transplanted in vivo using fibrin or alginate, which have minor biomechanical drawbacks such as high deformability, undue elasticity, and low volume stability, as a cell carrier. Therefore, non-woven PLGA scaffolds were seeded with ASCs and transplanted into subcutaneous pockets of severe combined immunodeficiency (SCID) mice in this study. We hypothesized that such cell-seeded constructs would demonstrate a cartilage-like morphology with expression of chondrospecific molecules while conserving sufficient biomechanical characteristics such as volume stability and elasticity at the same time.
Materials and Methods
Isolation of human ASCs
ASCs were isolated from subcutaneous adipose tissue from healthy young donors (n = 5, mean age 26 ± 1.5, m:f = 2:3) undergoing abdominoplastic surgery, as previously described. 21 The ethical committee of the University of Freiburg approved the donor program. Fat tissue was minced into small pieces (1-2 mm3), washed with phosphate buffered saline (PBS), and digested with 2 mg/mL collagenase type I (Biochrom, Berlin, Germany) for 90 min at 37°C with continuous shaking. The floating adipocytes were separated from the stromal cell fraction using multiple centrifugation and washing steps. The stromal cells were plated at 4000 cells/ cm2 in 175-cm2 tissue culture flasks filled with 25 mL of Dulbecco's modified Eagle medium (DMEM)/F12, 10% fetal bovine serum (FBS), 10 ng/mL basic fibroblast growth factor (bFGF), 100 units/mL of penicillin, and 100 μg/mL of streptomycin. The medium was changed completely every third day, which washed out all nonadherent cells. Once adherent cells had grown to confluence, they were detached with trypsin–ethylenediaminetetraacetic acid (EDTA; Sigma, Steinheim, Germany), re-plated at a density of approximately 2000 cells/cm2 and cultured for two more passages.
Characterization of cells
For flow cytometric analysis of in vitro expanded ASCs, cells were detached with trypsin-EDTA after reaching confluence two times (Passage 2), washed with PBS, and stained with fluorescent antibodies against human CD34, CD45, CD73 (Becton Dickinson, San Jose, CA), CD90, and CD105 (Pharmingen, San Diego, CA) and corresponding isotype controls.
Polymer scaffolds
A nonwoven copolymer scaffold of L-lactide and glycolide (90/10, PLGA) in the form of fibers were generously provided from the Insitut für Textil- und Verfahrenstechnologie (ITV, Denkendorf, Germany). The scaffolds were round, with a diameter of 8 mm and a height of 2 mm. The pore sizes of the nonwoven fibers were on average 75 μm, the pore volume accounts for 97% of the total volume, and the filament diameter was 13 μm.
The PLGA constructs were treated using the low-pressure plasma technique at the end of the production process. A partially ionized gas reacted with the surface of the scaffolds and formed reactive particles. Replacing weak bonding with highly reactive carbonyl and hydroxyl groups increases the adhesion of fluids.
Degradation time of polymer scaffolds
The degradation test of the nonwoven fibers was performed in accordance with International Standards Organization (ISO) Draft International Standard 13781-1995 at 37°C in PBS pH 7.4, as recently described. 22 Samples were retrieved after 1, 3, 8, and 11 weeks of incubation and evaluated regarding loss of strength (tensile test according to ISO 9073-3). A sample (n = 5) 150 mm long (length between the clamps) and 60 mm wide was fixed in the universal tensile tester, folded in the form of a tube, with the long axis (150 mm) in the testing direction. The testing speed was 100 mm/min. Breaking strength and elongation were determined from the recorded force-elongation curve. The results were reported as strength in percentage of initial strength of the undegraded material.
Three-dimensional adhesion assay using ASCs
PLGA scaffolds (n = 4) were resized using a biopsy punch (5 mm) and put into a 1-mL insulin syringe. The scaffolds were pushed to the bottom, and the syringes were used as columns in a specially designed rack. ASCs were stained before the assay using 2',7'-Bis(2-carboxyethyl)-5(6)-carboxyfluorescein acetoxymethyl ester.23,24 After calibrating the columns with assay buffer (PBS with 0.1% FBS), 1.25 × 106 cells were applied to the column and incubated for 0, 45, and 90 min at 37°C in a humidified chamber with 5% carbon dioxide (CO2). Afterwards, the columns were washed with 1.25 mL of assay buffer, and the cells collected in the rinsing fluid were counted using an enzyme-linked immunosorbent assay reader. Quantification of ASCs has been achieved by running a standard curve within each assay so that differences after the staining procedure can be excluded using a direct correlation of fluorescence and cell number. Calculating the difference of total cell number and cells that were washed out resulted in the number of immobilized cells within the scaffold.
Chondrogenesis in polymer scaffolds
ASCs were harvested using trypsin-EDTA and counted. The dry PLGA scaffolds (n = 4) were seeded with a suspension of 2 × 106 ASCs in 50 μL of medium for 45 min in a CO2 incubator to allow the cells to adhere to the polymer fibers, as recently described. 20 Scaffolds were covered with the cell suspension, which invaded the matrix immediately. An additional treatment to enhance the cellular uptake of the scaffold was not used because the adhesion assay obtained satisfying results using the same cell-loading technique. The scaffolds were transferred to a six-well plate, and 3 mL of medium was added. Cell-seeded scaffolds were cultured in control medium containing high-glucose DMEM with sodium pyruvate (100 mg/L), L-glutamine (100 mg/L), pyridoxine hydrochloride (100 mg/L), 1% penicillin/streptomycin, l-ascorbate 2-phosphate (37.5 μg/mL), and insulin, transferrin, selenium+ Premix (6.25 mg/mL; all from Gibco, Karlsruhe, Germany) or in differentiation medium. Differentiation medium differed in that it contained 10 ng/mL of TGF-β1 (R&D Systems, Minneapolis, MN) in addition to the control medium. For chondrogenic differentiation assays, all samples were cultured for 21 days in total, and the medium was changed every third day. Additionally, cell-free constructs were cultured in control medium under identical culture conditions as cell-loaded scaffolds.
Animals
Adult male SCID mice (C.B. 17-scid/leRCrl; Charles River Deutschland GmbH, Sulzfeld, Germany) were used (n = 12). Each mouse weighed approximately 30 g and was fed a special diet for mice (V1185-300 ssniff M-Z, Fa. ssniff Spezialdiäten GmbH, Soest, Germany). They were kept in Macrolon cages size MII (Fa. Tecniplast Deutschland GmbH, Hohenpeiβenberg, Germany). To exclude variability between the SCID mice, every mouse received two scaffolds (one composite of each group). No animal died during the experimental period.
Surgical procedure
Under general intraperitoneal anesthesia and after disinfection of the back of the mice, two subcutaneous pockets were bluntly created through a 1.5-cm incision in the back. A composite (cell-loaded TGF-β1-treated, cell-loaded control or cell-free construct) was inserted in each subcutaneous pocket. The wound was closed using a single interrupted suture. The animals were killed; the biomaterial–cell composites were harvested at 8 weeks; and specimens were retrieved for histological, biomechanical, and biochemical evaluation.
Semi-quantitative reverse transcriptase polymerase chain reaction
Messenger RNA (mRNA) samples were taken at days 9, 14, and 21 and after 8 weeks in vitro (n = 4), transcribed into complementary DNA (cDNA) and reverse transcriptase polymerase chain reaction (RT-PCR) analysis for gene expression of integrin subunit-α1, -α2, -α5, -αV, -α10; aggrecan; α1-collagen type II (col2a1); runt-related transcription factor 2 (Runx-2), and alkaline phosphatase (ALP) was carried out as described. 21 Total mRNA was prepared using TRIzol reagent according to the manufacturer's instructions (Invitrogen, Life Technologies, Karlsruhe, Germany). Total RNA (1 μg) was treated with 1 U of deoxyribonuclease I (DNase I; Invitrogen, Life Technologies) to digest genomic DNA contamination. Random-primed cDNA synthesis was performed using 1 μg of DNase I–treated total RNA and 50 U of Stratascript reverse transcriptase according to the manufacturer's instructions (Stratagene, La Jolla, CA). TaqMan PCR assays were performed in 96-well optical plates on an ABI Prism 7700 Sequence Detection system (Applied Biosystems, Forster City, CA) using Absolute QPCR ROX Mix (Abgene, Hamburg, Germany) according to the manufacturer's instructions. Oligonucleotide primers and TaqMan probes were designed using Primer Express (Applied Biosystems, Forster City, CA) according to company guidelines (Table 1).
The thermal cycling conditions were 95°C for 15 min followed by 40 cycles at 95°C for 15 s and 60°C for 1 min. Data were analyzed using the relative standard curve method, with each sample being normalized to glyceraldehyde 3-phosphate dehydrogenase to correct for differences in mRNA quality and quantity. For the standard curve, Universal Mix RNA (Stratagene) and RNA from human articular chondrocytes were transcribed into cDNA. Data are expressed as arbitrary units. RNA samples of human fibroblasts were used as negative controls for chondrogenic marker genes.
Histomorphological and immunohistological evaluation
For paraffin sections, cell-seeded polymer scaffolds were explanted after 8 weeks in vivo, fixed in 4% phosphate buffered formalin, dehydrated, and embedded in paraffin. Sections were cut dry (2.5 μm) on a Leica RM 2165 microtome (Leica, Wetzlar, Germany). For collagen type I and type II immunohistochemistry, sections were incubated for 30 min with 5% normal goat serum, followed by incubation with a 1:50 monoclonal mouse anti-collagen type I antibody (MAB3391, Clone 5D8-G9, Chemicon, Hofheim, Germany) or a 1:50 monoclonal mouse anti-collagen type II antibody (MAB8887, Clone 6B3, Chemicon) for 12 h, three washings with PBS, and incubation with a biotin-labelled goat anti-mouse immunoglobulin for 30 min (Acris, Herford, Germany). Afterwards, sections were incubated with avidin for 30 min and with 3-amino-9-ethylcarbazole (AEC) substrate for 10 min.
To show glycosaminoglycan synthesis, sections were stained with Alcian blue (pH 2.5). Anti-vimentin staining (M0725, DAKO, Hamburg, Germany) was used to detect cells of human origin in specimens explanted after 8 weeks. The antibody used was highly specific for human vimentin as tested and confirmed by the manufacturer. No cross-reactivity could be shown staining mouse and goat tissue samples. After heating the slides in citrate buffer (microwave oven, 3 × 3 min), the sections were washed in PBS and blocked for 20 min with blocking solution (5% goat serum, Sigma, Deisendorf, Germany). The samples were incubated with the primary anti-vimentin antibody in a humid chamber at room temperature for 1 h. A corresponding secondary horseradish peroxidase–conjugated goat anti-mouse antibody (P0447, DAKO,) was applied, and sections were incubated for another 30 min. Thereafter, sections were exposed to AEC chromogen substrate (K3469, DAKO,) and counterstained with hematoxylin.
Statistical analysis
Numerical data were analyzed using SPSS statistical program, version 11.5 (SPSS, Inc., Chicago, IL). All values are reported as means ± standard deviations of the mean. Statistical significance was determined using the Wilcoxon-(Mann-Whitney) test at a confidence level of 95% (p < 0.05).
Results
Flow cytometry
ASCs (passage 2) were characterized according to the expression of surface antigens. The expression of all surface antigens studied was similar for the entire expansion period and for all samples used in the experiment: Similar fractions of both cell populations were positive for CD90 (Thy-1) (60–70%), CD73 (ecto-5-nucleoditase) (70–80%), and CD 105 (35–40%). All samples were negative for CD34 (sialomucin–hematopoietic progenitors) and CD45 (leukocyte common antigen–hematopoietic progenitors) surface antigens.
Degradation time of polymer scaffolds
To assess the degradation behavior of PLGA-polymer scaffolds, the loss of strength during incubation for 1, 3, 8, and 11 weeks in PBS was analyzed using a tensile test according to ISO 5081. After 1 week of incubation, there was no loss of strength; after 3 weeks there was 5% loss, after 8 weeks 50% loss, and after 11 weeks, 80% loss.
Three-dimensional adhesion assay using ASCs
ASCs (n = 3) were seeded into the scaffolds and incubated for 0, 45, and 90 min. Afterwards, the columns were washed with 1.25 mL of assay buffer, and the cells collected in the rinsing fluid were counted. Calculating the difference between total cell number applied (1.25 × 106) and cells that were washed out resulted in the amount of scaffold immobilized cells. Furthermore, the fraction of filtered cells was determined by putting the cells on the column without incubation; cells were immediately washed through the column. The portion of filtered cells reached 6 ± 2%. The difference between totally immobilized and filtered cells was regarded as the number of adherent cells. The fraction of adherent cells inside the scaffold was determined as 69 ± 9% after 45 min of incubation and 72 ± 11% after 90 min of incubation. There was no significant difference in cellular adhesion between samples incubated for 45 min and 90 min. Therefore, cells were incubated for 45 min for all following experiments.
Evaluation of the scaffold gross morphology
All cell-loaded constructs conserved the round shape of the scaffolds and had a compact consistence after 8 weeks of in vivo culture. Cell-free scaffolds were weak and floppy and lost the round shape of the scaffold or were not detectable after 8 weeks of in vivo culture.
Quantitative RT-PCR
To evaluate whether PLGA-seeded ASCs differentiate into chondrocyte-like cells in response to TGF-β1 mRNA expression of the extracellular matrix (ECM) molecules α1-collagen type II (col2a1), aggrecan, Runx-2, and ALP was analyzed using quantitative real-time PCR at days 9, 14, and 21 (n = 4). To examine whether ASCs maintain their chondrogenic properties after transplantation into SCID mice mRNA expression of the markers listed above was measured at day 77 (21 days in vitro plus 8 weeks in vivo).
The mRNA expression of the cartilage marker col2a1 was detected in samples treated with TGF-β1 for 9 days at least, increased 8-fold between day 9 and day 14 and was maintained up to day 21 at the same level. After implantation for 8 weeks in the subcutaneous pocket, col2a1 mRNA expression was as high as at day 21 before implantation. In control samples, col2a1 was not detected at any time during the experiment (Fig. 1).

Aggrecan and collagen type II (col2a1) messenger RNA (mRNA) expression was detected only in samples treated with transforming growth factor beta 1 (TGF-β1) in vitro (9, 14, and 21 days) and in samples pre-treated with TGF-1 in vivo (21 days and 8 weeks). mRNA expression of samples treated with TGF-β (controls) was measured using quantitative real-time polymerase chain reaction at 9, 14, and 21 days and after transplantation in vivo at 21 days and 8 weeks (n = 4 each). mRNA expression was displayed relative to the housekeeping gene glyceraldehyde 3-phosphate dehydrogenase. Values are reported as means ± standard deviations of the mean. 103 × 71 mm (300 × 300 DPI).
The mRNA expression of the cartilage marker aggrecan was detected in samples treated with TGF-β1 at day 9 and more than doubled by day 21. After implantation for 8 weeks in the subcutaneous pocket, aggrecan mRNA expression was as high as at day 21 before implantation. In control samples, aggrecan was not detected at any time during the experiment (Fig. 1).
The mRNA expression of the osteogenic transcription factor Runx-2 was detected in all samples at day 9 and more than doubled by day 21. After implantation for 8 weeks in the subcutaneous pocket, Runx-2 mRNA expression was more than twice that (p < 0.05) in samples at day 21. There was no difference between TGF-β1–treated and control samples (Fig. 2A).
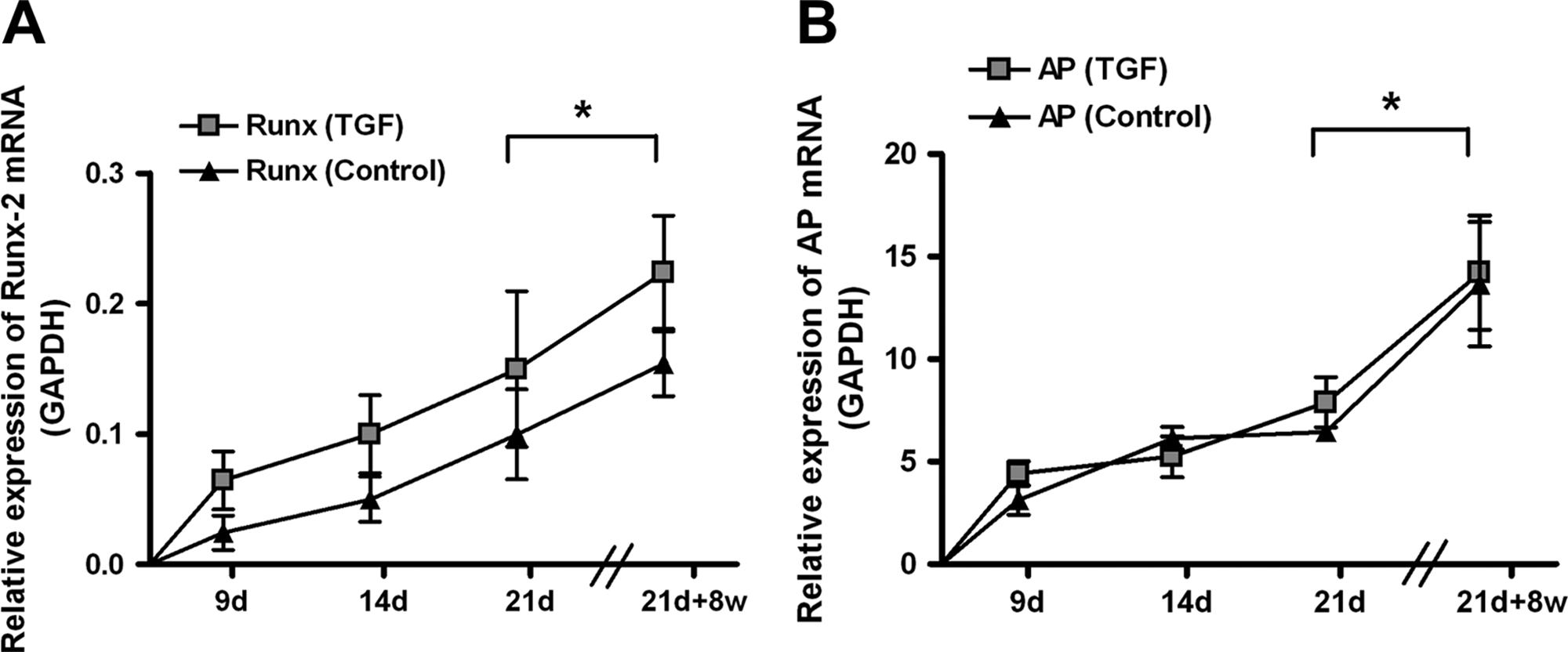
No differences in runt-related transcription factor 2 messenger RNA (mRNA) (
The mRNA expression of the osteogenic marker ALP was detected in all samples at day 9 and was more than 1.5 times as great at day 21. After implantation for 8 weeks in vivo, ALP mRNA expression significantly was twice that (p < 0.05) of samples at day 21. There was no difference between TGF-β1–treated and control samples (Fig. 2B).
To examine the integrin receptor profile before transplantation in vivo, mRNA expression of the integrin subunit-α1, -α2, -α5, -αV, and -α10 was measured in response to TGF-β1 and displayed in percentage up-regulation compared with untreated controls at day 21 (Fig. 3). Integrin-α1 mRNA was 37 ± 21% higher, integrin-α2 mRNA was 39 ± 15% higher, integrin-α5 mRNA was 67 ± 17% higher, integrin-αV mRNA was 93 ± 23% higher, and integrin-α10 mRNA was 10 ± 5% lower than untreated controls.
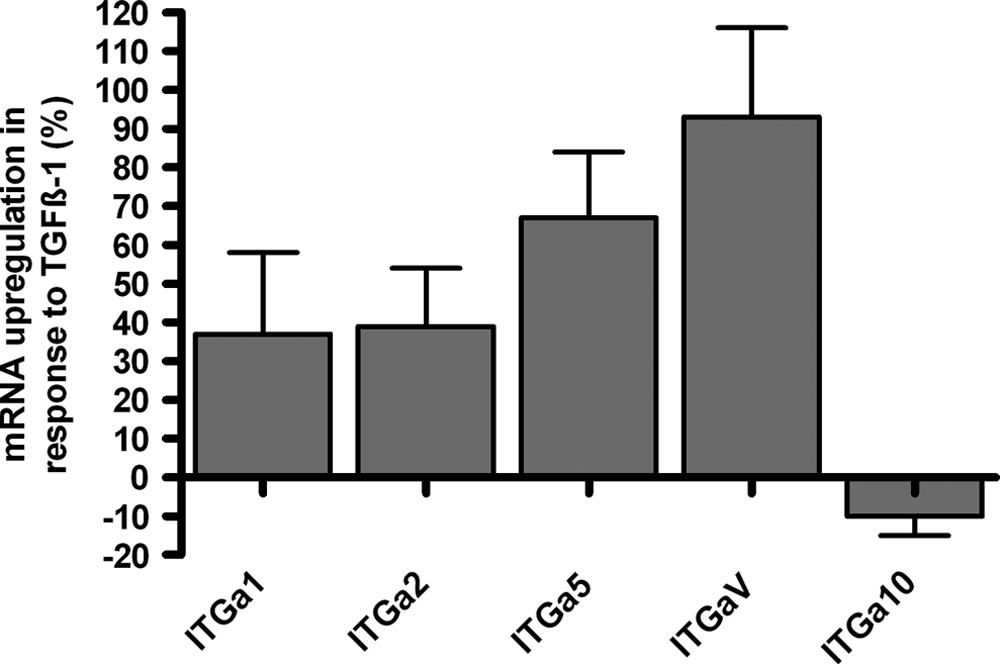
Integrin subunits (α1, α2, α5, αV, α10) were differentially regulated in response to transforming growth factor beta 1 (TGF-β1). Messenger RNA (mRNA) expression of samples treated with TGF-β1 controls was measured using quantitative real-time polymerase chain reaction at day 21 in vitro (n = 4). mRNA expression was displayed relative (up-regulation in percentages) to the housekeeping gene glyceraldehyde 3-phosphate dehydrogenase and corresponding control samples. Values are reported as means ± standard deviations of the mean. 155 × 101 mm (300 × 300 DPI).
Histology
Histomorphological evaluation of the explants revealed a homogenous distribution of round cells within the transplant. The PGLA fibers of the scaffolds were still visible in histological sections. Collagen type I immunohistochemistry revealed the detection of collagen type I in all TGF-β1–treated explants and in all cell-loaded control samples. The intensity of staining for collagen type I was lower in all samples treated with TGF-β1 than in control samples (Fig. 4I). Collagen type II immunohistochemical staining revealed the detection of collagen type II in 67% of TGF-β1–treated explants (8 of 12 explants); collagen type II was not detected in control samples (0 of 12 explants) (Fig. 4II). Alcian blue staining revealed homogenous distribution of proteoglycans in all TGF-β1–treated explants; Alcian blue staining was weak or not detected in control samples (Fig. 4III). To prove that the cells producing cartilage-specific ECM were of human origin, samples were stained for human-specific vimentin. In all cell-loaded samples, vimentin-positive cells could be detected within the scaffold; surrounding mouse tissue was negative for human-specific vimentin (Fig. 4IV).

Transforming growth factor beta (TGF-β)-pre-treated samples (
Discussion
Cartilage defects of the articular joint can be successfully treated using ACT. To gather chondrocytes, a biopsy from the articular joint is taken, which some authors have considered to be a relevant cartilage defect and therefore a pre-arthritic deformity. 4 Thus, mesenchymal stem cells from bone marrow or fat could represent an alternative cell source for cartilage transplantation because they are capable of differentiating into cartilage-like cells.6,25 Chondrogenic phenotype was induced and maintained in ASCs using TGF-β1. 7 For successful chondrogenesis, ASCs must be cultured in a three-dimensional matrix. Alginate, gelatin, and agarose have been shown to have suitable properties for cell culture experiments. 14 Because they have poor biomechanical properties, polymer scaffolds with a stable fiber structure have been developed and used for cartilage tissue engineering.
In this study, we evaluated the suitability of PLGA scaffolds for the chondrogenic differentiation of ASCs in vitro and examined the chondrogenic phenotype of ASCs after transplantation in vivo. A non-woven copolymer scaffold consisting of PLA and PGA (ratio 90:10) fibers was developed and seeded with human ASCs. PLA–PGA has been used in clinical settings as a resorbable suture material and could show satisfactory biocompatibility. Despite the widespread application of PLA–PGA in clinics, we observed foreign body reactions in the surrounding tissue and direct neighborhood of the scaffold after transplantation in vivo. Other researchers26,27 using PGA and PLA scaffolds for tissue-engineered cartilage have made similar observations. To examine the differences of the inflammatory response of both polymers, Sittinger showed that non-woven PLA fibers are more biocompatible during chondrocytes culture than nonwoven PGA fibers. 22
The biocompatibility of polymer scaffolds is related to the affinity and adhesion of cells to its surface. The composition of the scaffold materials gains specific importance when they are being used as a matrix during chondrogenic differentiation of ASCs because chondrogenesis is a multi-step process controlled by adhesive events. During the early phase, cell–cell adhesion through molecules such as N-cadherin and neural cell adhesion molecule is required for chondrogenic differentiation. At later stages, adhesion signaling from ECM proteins through integrins is necessary for normal proliferation, differentiation, and hypertrophy of mesenchymal precursor cells.28,29 The blocking of α2- or β1-integrin function with specific antibodies significantly impeded the in vitro and in vivo chondrogenesis of mesenchymal stem cells.30,31 Moreover, lower cartilage synthesis was observed in α1β1-integrin-deficient mice. 32 In our study, mRNA expression of the integrin subunits α1, α2, α5, and αV was detected in all cells grown on PLGA scaffolds and was higher in response to TGF-β1. The integrin subunit α10 was expressed in PLGA-seeded ASCs but was not greater in the presence of TGF-β1. The integrin subunits α1, α2, α5, α10, and αV are components of collagen receptors (α1β1, α2β1, α10β1) and fibronectin receptors (α5β1, αVβ3, αVβ5) and are responsible for adhesion of ASCs to the cartilage-specific ECM.33,34 We conclude that PLGA scaffolds support the expression of cartilage-specific integrin receptors. An additional treatment of cell-seeded PLGA transplants with TGF-β1 enhances the specific adhesion of ASCs to ECM components, which promotes the differentiation of ASCs into chondrocyte-like cells.
To assess the degradation time of the PLGA scaffolds, we performed tensile testing of the polymers. We calculated 80% less strength after 11 weeks of in vitro culture than in uncultured PLGA scaffolds. Compatible with these results, gross morphology evaluation revealed that cell-free controls were floppy and underwent a loss of shape or were not detectable after 11 weeks of consecutive in vivo and in vitro culture. In contrast, all cell-loaded constructs conserved the initial shape of the scaffold and were compact. We suggest that the ECM produced by ASCs protects the PLGA fibers from degradation. Moreover, ASCs have been found to suppress immunological reactions because they inhibit the migration and proliferation of lymphocytes and macrophages by the production of soluble immunomodulatory factors such as interleukin 10 and tumor necrosis factor α.35,36 Thus, reasons for the delayed degradation of the cell-loaded scaffolds seemed to be the ability of ASCs to produce immunosuppressive factors and to release a shielding ECM.
Biopolymers have been shown not only to be responsible for sufficient tissue integration and compatibility, but also to modulate the phenotype of the scaffold-seeded cells. In the present study, a mixture of non-woven PLA and PGA was developed as a scaffold because PLA has the more advantageous biomechanical characteristics, and PGA possesses the more-favorable biochemical characteristics. Chondrocytes grown on PGA scaffolds produced more chondrospecific ECM than those grown on PLA. 15 This difference has been attributed to the polymer geometry and biodegradation rate of both polymers. Despite the difference in cellular reaction of scaffold materials between the cell types, growth factor–stimulated bone marrow-derived mesenchymal stem cells were shown to differentiate into chondrocyte-like cells producing chondrospecific ECM independent of scaffold material.37,38
It has been shown that TGF-β1 induces a chondrogenic phenotype in ASC-seeded PGLA scaffolds in vitro. When cell-seeded and TGF-β1–preatreated scaffolds were implanted subcutaneously in vivo, the chondrogenic phenotype was maintained for at least 8 weeks. This observation confirmed and completed results of former studies that mesenchymal stem cells maintain their chondrogenic phenotype even in the presence of non-chondroinductive (e.g., osteogenic or adipogenic) cues if they are pre-incubated with TGF-β1 long enough. 39 The pre-incubation time with TGF-β seems to be crucial because Pelletari showed that, after ectopic transplantation in SCID mice, bone marrow-derived mesenchymal stem cells pre-treated with TGF-β1 for 21 days or longer continued to produce proteoglycan and type II collagen in vivo but started to build mineralized ECM. 40
In our samples, osteospecific mRNA—the osteogenic transcription factor Runx-2 and ALP—was detected during in vitro culture and increased after transplantation in vivo. A significant difference between TGF-β1–treated samples and control samples was not detected. In contrast, we recently showed that TGF-β1 inhibits bone morphogenetic protein-2–mediated osteogenesis in alginate-encapsulated ASCs. 41 We explain this conflicting observation by the different type of scaffold used in both studies and by the heterotopic transplantation site. Alginate favors the chondrogenic phenotype, whereas non-woven PLGA seems to favor both osteogenic and chondrogenic differentiation in ASCs. 15 Subcutaneous implantation of the cell-seeded scaffolds increased the expression of the osteogenic markers because ALP and Runx-2 were higher after 8 weeks of culture in vivo than in corresponding samples before transplantation. Adipose tissue–localized growth factors of the transplantation site (e.g., leptin) might be responsible for the expression of a fibro- or osteoblastic phenotype in the transplanted cells. Leptin was recently shown to regulate bone remodelling and bone mass in mammalians. 42 The fibroblastic marker collagen type I was detected in histologic sections of treatment and control groups, but control samples showed overall stronger staining than TGF-β1–treated samples. We suggest that TGF-β1 pre-treatment inhibits the conversion to a fibroblastic phenotype in ASC-seeded PLGA scaffolds but not the expression of osteogenic marker genes after subcutaneous implantation.
In summary, we showed that PLGA scaffolds are suitable for TGF-β1–mediated chondrogenesis in ASCs. Pre-treatment with TGF-β1 induced and maintained chondrogenic phenotype of ASCs but did not prevent the expression of osteogenic markers in a heterotopic animal model. Despite this, TGF-β1 pre-treatment reduced the fibroblastic conversion of ASCs after subcutaneous transplantation. The cell-seeded PLGA scaffolds had high biodegradability, excellent volume stability, and sufficient elasticity and provided ASCs with a similar chondrogenic environment as alginate or fibrin. In future, other culture conditions need to be developed that can enhance the biomechanical and biochemical properties of tissue-engineered cartilage transplants. Recently, Hennig et al. and Estes et al. introduced bone morphogenic protein 6 for treatment of ASCs, which significantly enhances the ECM production in vitro and gives hope for further advances in cartilage tissue engineering with ASCs.43,44