
Abstract
Human mesenchymal stem cells (MSCs) from bone marrow stroma can home to and repair injured tissue, but the rate of engraftment is generally low. Regulating migration-related signaling of MSCs may be a powerful strategy to enhance this process. To gain insight into the molecular mechanisms governing homing, we identified negative factors affecting MSC migration using an in vitro model of injured lung. Heat-labile factors in bovine pituitary extract, a component of serum-free epithelial medium, inhibited more than 97% of MSC migration. This was partly due to a dose-dependent response to macrophage migration inhibitory factor (MIF). Eighty-five ng/mL recombinant MIF, the concentration found in the epithelial medium, inhibited about 50% of MSC migration. Media conditioning by uninjured or bleomycin-injured bronchial epithelial cells partially attenuated this suppressive effect. Additionally, the anti-inflammatory agent ISO-1, a small-molecule MIF antagonist, further increased MSC migration by nearly fourfold in conditioned epithelial media. This is the first report of the effect of MIF and ISO-1 on MSC migration, and the data suggest that MIF and its antagonists may have therapeutic applications in controlling MSC homing during repair of injured lung and in other clinically relevant systems.
Introduction
The focus of this research is to develop strategies to enhance migration of MSCs to lung by regulating migration-related signaling. Many positive chemotactic factors are known to induce MSC migration, including growth factors like fibroblast growth factor-2 and platelet-derived growth factors AB and BB, 13 and chemokines like stromal-derived factor-1 and RANTES. 14 Negative or inhibitory factors affecting MSC chemotaxis have received less attention, although inhibition of migration by the phospholipids sphingosine-1-phosphate and lysophosphatidic acid has been observed recently. 15 Additionally, factors that suppress migration in other cell types are known, as with the effect of all-trans retinoic acid on peripheral blood mononuclear cells. 16 Suppressing this type of negative signaling may be an effective method to enhance the innate homing response of MSCs.
One potential inhibitor of MSC migration is the tautomerase macrophage migration inhibitory factor (MIF) because two of its known receptors, CD74 and CXCR2, 17 are expressed on MSCs.18,19 This cytokine is involved in bone-specific processes in both homeostasis and pathogenesis, including bone remodeling and osteoporosis. 20 MIF is a critical mediator of inflammation and, in this capacity, is known to activate monocytes and arrest their migration. 21 It is likely that the concentration of this proinflammatory cytokine is elevated in injured tissues targeted by MSC therapy. 22 Given the importance of MIF to inflammation, it is surprising that its effect on MSCs has not yet been described. We hypothesized that the MIF small-molecule antagonist (S,R)-3-(4-hydroxyphenyl)-4,5-dihydro-5-isoxazole acetic acid methyl ester (ISO-1), which inhibits the tautomerization and proinflammatory functions of MIF, 23 could enhance migration of MSCs.
To assess the migratory response, we used a well-defined system consisting of human MSCs stably transfected with green fluorescent protein (GFP) and differentiated air–liquid interface cultures of human bronchial epithelial cells (BECs). Interfacial BECs differentiate to a greater extent than when the cells are submerged in culture medium and, consequently, are a popular in vitro model of native bronchial epithelium. 24 With this culture system, we observed that bovine pituitary extract (BPE), a component of serum-free (SF) BEC medium, inhibited MSC migration, partly due to a dose-dependent response to MIF. BEC conditioning mitigated this suppressive effect. Additionally, the MIF antagonist ISO-1 further increased MSC migration in conditioned BEC media. This is the first reported use of ISO-1 with MSCs, and it suggests a possible application of ISO-1 and related small-molecule anti-inflammatory agents as pharmacological adjuvants to MSC therapy.
Materials and Methods
MSC culture
Primary MSCs were harvested from 2 mL of iliac crest bone marrow aspirate from healthy adult volunteers as previously described, 25 under a protocol approved by the Tulane Institutional Review Board. Cell culture supplies were obtained from Invitrogen (Carlsbad, CA) except where noted. Plastic-adherent MSCs were inoculated at 100 cells/cm2 into 150 cm2 T-flasks in stem cell growth medium (SGM) consisting of α-MEM supplemented with 2 mM L-glutamine, 17% fetal bovine serum (FBS; Hyclone, Logan, UT), 100 U/mL penicillin, and 100 μg/mL streptomycin. The medium was replaced every 3–4 days. MSCs were maintained at <50% confluence and subcultured with 0.25% trypsin/1 mM EDTA. Research described herein was conducted with MSCs at passages 2 to 4 and cells cultivated in a 37°C humidified incubator at 5% CO2.
Lentiviral gene transfer
Lentivirus containing the pLenti6.2-GW/EmGFP vector was produced in 293FT embryonal kidney cells using the ViraPower lentiviral expression system according to the supplier's instructions (Invitrogen). Passage 2 MSCs were plated at 500 cells/cm2 in 150 cm2 T-flasks and transduced overnight with lentivirus at multiplicity of infection = 2 in 10 mL SGM containing 6 μg/mL polybrene (Sigma-Aldrich, St. Louis, MO) to provide 20–40% GFP+ cells. Three days after transduction, cells stably expressing EmGFP (λex/λem =487 nm/509 nm) were isolated using a FACS Vantage SE flow cytometer (BD Biosciences, San Jose, CA). GFP+ MSCs were employed for coculture at passage 4.
BEC culture
Passage 1 human BECs (Lonza, Walkersville, MD) were plated at 500 cells/cm2 and expanded according to Gray et al. 26 in a basal medium supplemented with insulin, transferrin, triiodothyronine, epinephrine, hydrocortisone, retinoic acid, recombinant human epidermal growth factor with bovine serum albumin, BPE, gentamycin, and amphotericin-B in a proprietary formulation purchased from Lonza. For air–liquid interfacial culture, BECs were cultivated on polyester Transwell membranes (0.4 μm pores; Corning Costar, Cambridge, MA) coated with 60 μg/mL collagen VI (Sigma-Aldrich) as described by Karp et al. 24 Collagen coating was verified by staining representative membranes with 0.25% Coomassie brilliant blue R-250 in 10% acetic acid/25% isopropanol for 5 min. 27 Passage 2 BECs were inoculated onto the membranes at 2 × 105 cells/cm2 in epithelial differentiation medium (EDM) consisting of epithelial basal medium (EBM, a 50/50 mix of the Lonza basal medium and high-glucose DMEM) with the same supplements as above. 26 Apical medium was removed upon cell confluence, and medium in the bottom Transwell chamber was replaced every 2 days. Interfacial BEC cultures were injured by exposure to 100 mU/mL BM (Sigma-Aldrich) in EDM for 24 h, followed by three 5 min washes with phosphate-buffered saline (PBS).
MSC/BEC coculture
MSCs and BECs were separated by a Transwell insert in coculture, with MSCs in the bottom chamber and an interfacial BEC culture in the upper chamber. First, GFP+ MSCs were plated at 100 cells/cm2 in SGM in 12-well plates. Coculture was initiated 1 h after removing BM from BECs and 24 h after plating the MSCs by replacing SGM with EDM and by sterilely transferring 14-day interfacial BEC cultures on Transwell inserts into wells containing GFP+ MSCs. Half of the EDM in the bottom chamber was replaced daily, while BECs remained at the air–liquid interface.
Cell concentration and viability
For MSC and submerged BEC cultures, cell concentration and viability were determined by trypan blue exclusion and hemocytometer counting. To quantify proliferation of adhesive interfacial cultures, BECs were washed with PBS and stained with 0.1% crystal violet in 0.1 M citric acid for 2 h at 37°C, after which nuclei from the lysed cells were counted with a hemocytometer. 28 Viability of interfacial cultures was examined in situ by staining BECs with 5 μg/mL Hoechst 33342 (λex/λem = 350 nm/460 nm) for 30 min, followed by staining with 100 μg/mL ethidium bromide (λex/λem = 518 nm/605 nm) for 1 min, washing with PBS before and after each step. 29 Stained cultures were imaged with an IX-50 fluorescent microscope (Olympus, Center Valley, PA). BECs exposed to 1 μg/mL actinomycin D (Sigma-Aldrich) served as the positive control. 30 Viability of interfacial BEC cultures was quantified with the CellTiter-Glo luminescent viability assay (Promega, Madison, WI) according to the manufacturer's instructions. The lysed cell suspension was transferred to an opaque 96-well Cliniplate (Thermo Fisher Scientific, Waltham, MA) to read the luminescence on a SpectraMax Gemini EM plate reader (Molecular Devices, Sunnyvale, CA). Loss of membrane integrity of interfacial BEC cultures was detected by release of lactate dehydrogenase into culture medium (kit TOX-7; Sigma-Aldrich).
Colony-forming efficiency, immunophenotype, and differentiation potential
To determine colony-forming efficiency, MSCs were inoculated at 100 cells/plate in 10 cm tissue culture plates and cultivated in SGM for 14 days. Colonies were washed with PBS and stained for 5 min with 3% crystal violet in methanol. 31 The immunophenotype of MSCs was assessed by flow cytometry for expression of hematopoietic and stromal markers as previously described, 31 using antibodies listed in Table 1. Chondrogenesis was induced by plating 105 MSCs in 10 μL SGM to form a micromass in a 12-well plate. 32 After the cells were allowed to adhere for 2 h, 1 mL of chondrogenic medium was added, consisting of high-glucose DMEM supplemented with 500 ng/mL BMP-2, 10 ng/mL TGF-β3 (R&D Systems, Minneapolis, MN), 100 nM dexamethasone, 50 μg/mL ascorbate-2-phosphate, 40 μg/mL proline, 100 μg/mL pyruvate (all from Sigma-Aldrich), 50 mg/mL ITS + (BD Biosciences), 100 U/mL penicillin, and 100 μg/mL streptomycin. 33 The micromass was incubated for 21 days with biweekly medium exchange. Differentiated cultures were washed with PBS, fixed for 15 min with 4% paraformaldehyde, and stained overnight at 25°C with 1% alcian blue 8-GX (pH 1; Sigma-Aldrich) in 0.1 N HCl. 32 The negative control was MSC monolayers in SGM, and micromasses of SW1353 chondrosarcoma cells (ATCC, Manassas, VA) served as the positive control. Osteogenesis and adipogenesis were induced in MSCs and detected according to a protocol by Colter et al. 31
Antibodies and isotype controls were purchased from Beckman Coulter (Fullerton, CA).
Scanning and transmission electron microscopy
Cultures were prepared for ultrastructure analysis by fixation with 1.25% glutaraldehyde and 2% formaldehyde in 0.1 M sodium cacodylate buffer (pH 7.4), followed by three 10 min washes with 5% sucrose in the sodium cacodylate buffer and postfixation with 1% osmium tetroxide in the same buffer for 1 h. Scanning electron microscopy (SEM) samples were dehydrated with a graded ethanol series, critical-point dried, mounted, and sputter-coated as previously described, 34 and examined on a Quanta-200 SEM (FEI, Hillsboro, OR). Transmission electron microscopy (TEM) samples were stained with 2% uranyl acetate in 0.2 M sodium acetate buffer (pH 3.5), dehydrated, infiltrated with LR White resin, polymerized, and sectioned as in Krunkosky et al., 35 and then viewed on a JEM-1011 TEM (JEOL, Tokyo, Japan).
Transepithelial electrical resistance and mucin staining
Transepithelial electrical resistance (TEER) across interfacial BEC cultures was measured with an EVOM volt-ohmmeter equipped with a chopstick electrode (World Precision Instruments, Sarasota, FL) according to the manufacturer's instructions. 24 To detect mucin production, interfacial BEC cultures were fixed with 4% paraformaldehyde for 15 min and stained with 1% alcian blue in 3% acetic acid (pH 2.5) and then with periodic acid/Schiff's reagent (kit 395B-1; Sigma-Aldrich) following protocols by Bancroft and Gamble. 36 Cultures were imaged by phase-contrast microscopy. Confluent MSCs cultured in SGM were used as the negative control and sectioned human intestine as the positive control.
Migration assay
Migration of MSCs was evaluated with membrane inserts (8-μm pore) in 24-well plates 37 (Falcon; BD, Franklin Lakes, NJ). MSCs were cultivated in serum-free SGM (SF-SGM) for 24 h, trypsinized, and inoculated into the upper chamber at 2 × 104 cells/well in 300 μL SF-SGM. The lower chamber contained 900 μL of SGM, EBM, or EDM. After 6 h at 37°C, MSCs were stained with 3% crystal violet in methanol as above. Cells remaining in the upper chamber were removed with a cotton swab, and MSCs that migrated across the insert were counted by phase-contrast microscopy at 10 × as the average of ≥30 fields of view. Conditioned EDM was obtained 24 h after complete medium exchange from interfacial BECs. Heat-treated (HT) BPE was exposed to 71°C for 10 min. 38 Negative and positive controls were SF-SGM and SGM, respectively.
Tautomerization assay
MIF concentration in medium was quantified by its kinetic activity to catalyze the keto–enol tautomerization of 4-hydroxyphenylpyruvate 39 (HPP; Sigma-Aldrich). A 5 mM solution of HPP in 50 mM ammonium acetate (pH 6.0) was allowed to equilibrate overnight at room temperature before use. The absorbance increase at 330 nm was monitored spectrophotometrically for 5 min at 25°C in a 0.5 mL quartz cuvette containing 200 μL of HPP stock, 420 μL of 0.5 M boric acid in 0.2 M sodium phosphate buffer (pH 6.2), and 50 μL of sample. PBS was used as the negative control with recombinant mouse MIF (R&D Systems) as the positive control. The MIF antagonist ISO-140 was purchased from EMD (San Diego, CA) and dissolved in DMSO at 10 mg/mL. All ISO-1 experiments included DMSO-only controls.
Statistical analysis
All experiments were performed at least in triplicate, and results were analyzed using the Student's t-test in Microsoft Excel. The criterion for significance was p ≤ 0.05. Numerical data are reported as mean ± standard deviation.
Results
Characterization of wild-type and lentiviral-transduced MSCs
Immunophenotyping demonstrated that MSCs expressed cell surface molecules (CD44, CD90, and CD166) characteristic of stromal cells and were negative for hematopoietic markers (CD34, CD36, and CD45), in accordance with previous reports14,19 (Table 1). MSCs were transduced with a lentivirus encoding GFP to identify the cells in coculture. After transduction, GFP+ MSCs were robust in terms of their scatter properties, potency, and colony-forming efficiency. Forward versus side scatter plots from before (Fig. 1A) and after (Fig. 1B) sorting for GFP expression showed that GFP+ MSCs exhibited the small, agranular phenotype of healthy cells. Multipotency of the GFP+ cells was examined by induction of osteogenesis (Fig. 1C), adipogenesis (Fig. 1D), and chondrogenesis (Fig. 1E) over 21 days, which revealed extensive trilineage differentiation as compared with controls cultivated in SGM (Fig. 1F). Additionally, the cells retained strong GFP expression after differentiation, as confirmed by fluorescent microscopy (Fig. 1G). Wild-type and GFP+ MSCs had comparable colony-forming efficiencies, ∼50% (Fig. 1H). GFP expression was stable after lentiviral transduction with <10% change in the percentage of GFP+ MSCs over 21 days (Fig. 1I).
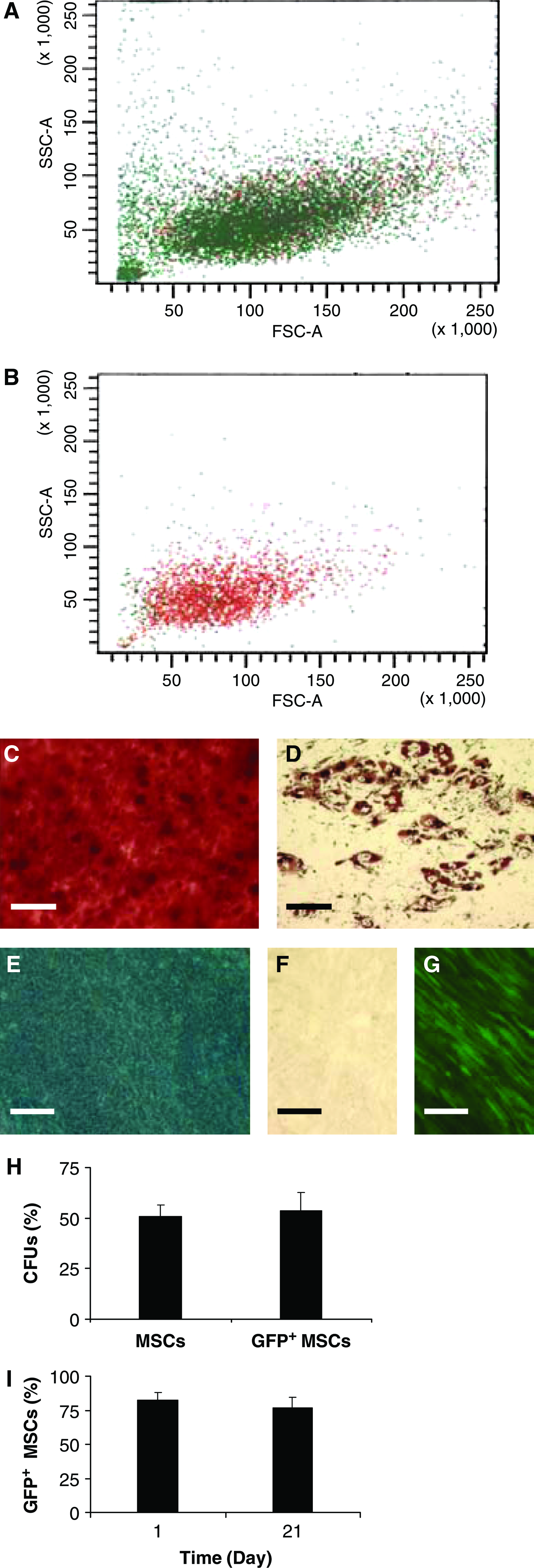
Characterization of mesenchymal stem cells (MSCs) transduced with green fluorescent protein (GFP) lentivirus. Forward versus side scatter plots of (
Differentiation and BM-induced injury of air–liquid BEC culture
Air–liquid interfacial cultures of BECs were employed in this study because their differentiation resembles that of native bronchial epithelium. 24 BEC cell density increased during the first week of interfacial culture to, on average, 6 × 105 cells/cm2 and then remained stable during the second week (Fig. 2A). Interfacial cultures were highly viable with dying cells constituting <1% of the total population (Fig. 2B). In comparison, the apoptotic positive control exhibited extensive loss of membrane integrity and chromatin condensation (Fig. 2C). BEC differentiation at the air–liquid interface was evident in mucin secretion and formation of a paracellular transport barrier. For the latter, TEER stabilized at 950 ± 200 ohm-cm2 after 7 days of culture, a resistance significantly greater than that of the negative control and consistent with other work 9 (Fig. 2D). Staining for acidic and neutral mucins revealed heterogeneous expression in 14-day interfacial BECs (Fig. 2E) relative to the negative control (Fig. 2F), in agreement with previous findings. 35 These results were confirmed by ultrastructure analysis, which detected secretory granules on the apical cell surface of the interfacial BEC culture (Fig. 2G) and desmosomes joining lateral cell surfaces (Fig. 2H).

Establishment of air–liquid interfacial bronchial epithelial cell (BEC) culture. (
BM exposure is a clinically relevant mode of lung injury. 7 Cell density decreased by one-third during the 5 days after interfacial BEC cultures were exposed to 100 mU/mL BM for 24 h, whereas there was no cell loss in mock-injured cultures over the same period (Fig. 3A). While injury from this chemotherapy drug was substantial, viable cells were detected on day 5 by ATP-induced luminescence, albeit at only ∼20% of the initial value (Fig. 3B). Concordantly, BM exposure caused a significant release of intracellular lactate dehydrogenase into culture medium as BECs became more permeable than the control (Fig. 3C). A 10-fold reduction in TEER after injury (Fig. 3D) indicates that this toxin compromised the paracellular barrier.

Bleomycin (BM)-induced injury of interfacial BEC cultures. Cultures were exposed to 100 mU/mL BM for 24 h. (
Migratory and proliferative response of MSCs to BEC cultures
As indicated in Figure 4A, EDM suppressed MSC migration by >97% as compared with chemokinesis in SF-SGM (p < 0.001) and by >200-fold relative to chemotaxis to SGM. Conditioned EDM from interfacial BEC cultures partially restored migration; the concentration of MSCs that migrated over a 6-h period to the spent medium was, on average, 4.3 × 102 cells/cm2 versus 60 ± 15 cells/cm2 for fresh EDM (p = 0.01). Exposing the epithelial cells to BM had no effect on the ability of MSCs to migrate to spent EDM. While BEC conditioning stimulated significant migration, it was not mitogenic. Proliferation of GFP+ MSCs in EDM as a monoculture and coculture with BECs was comparable after 48 h and 5 days, but the cell density was less than in SGM, the preferred growth medium for MSCs (Fig. 4B).

Migratory and proliferative response of MSCs to conditioned medium from uninjured and BM-exposed interfacial BEC cultures. (
Additional research was performed to assess the impact of medium supplements and BEC conditioning on MSC migration. BPE was found to be the migration-inhibiting component of EDM (Fig. 5A). Adding BPE to SF-SGM inhibited MSC migration by >97%, equivalent to that for complete EDM. The inhibitory factor(s) in BPE are likely to be proteinaceous because heat treatment of BPE at 71°C, which is sufficient to induce denaturation, restored migration to the same level as SF-SGM. We questioned whether the inhibitory effect of BPE might be caused by a biphasic response to a chemotactic factor. However, BPE produced dose-dependent inhibition of MSC migration, with no positive chemotaxis observed at any concentration of BPE (data not shown). The eight other supplements contained in EDM were either known MSC chemoattractants that elicited a positive chemotactic response, such as epidermal growth factor, 14 or were factors that did not affect migration, such as all-trans retinoic acid (data not shown). This is contrary to the reported inhibitory effect of retinoic acid on peripheral blood mononuclear cell migration. 16 Omitting BPE from EDM revealed that the eight remaining supplements had a net positive chemotactic effect: the concentration of MSCs migrating to EDM in the absence of BPE, (7.1 ± 0.9) × 103 cells/cm2, was greater than that for the basal medium, EBM (p = 0.02). Last, all supplements were omitted from the epithelial medium to determine whether BECs secrete positive chemotactic factors in addition to mitigating the negative factor(s) contained in BPE. Conditioning had no significant effect on the chemotactic potential of EBM (Fig. 5B). The data suggest that the primary effect of BEC conditioning on MSC migration is to consume or inactivate inhibitory factors rather than to secrete chemotactic factors.
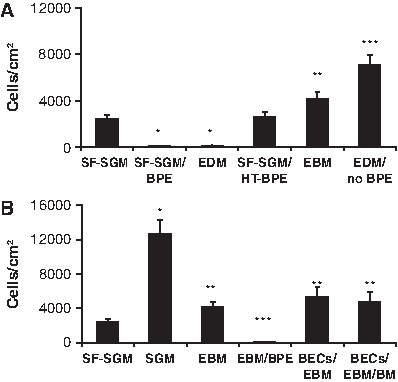
Migration of MSCs toward (
Effect of MIF and ISO-1 on MSC migration
MIF catalyzes the conversion of HPP from a keto to enol tautomer 39 (Fig. 6A). This kinetic activity was utilized to estimate the concentration of the enzyme in BPE and FBS, using recombinant MIF (rMIF) between 25 and 125 ng for calibration (Fig. 6B). The highest concentration, 85.4 ± 3.5 ng/mL, was found in EDM, followed by SGM with 75.8 ±4.3 ng/mL (Table 2). Conditioning by uninjured or BM-exposed BECs reduced the level of MIF in EDM by an average of 20% and 57%, respectively (p < 0.001). rMIF elicited a dose-dependent inhibition of MSC migration over the range of 1–1000 ng/mL (Fig. 6C). The quantity of MIF found in EDM and SGM was sufficient to inhibit MSC migration by ∼50%. EDM and SGM contain similar concentrations of MIF, yet produce divergent migratory behavior, which demonstrates that it is possible to counteract MIF inhibition.

Migration inhibitory factor (MIF) quantitation and effect on MSC migration. (
All groups are statistically different (p < 0.001). EDM, epithelial differentiation medium; SGM, stem cell growth medium; BECs, bronchial epithelial cells; BM, bleomycin; MIF, migration inhibitory factor.
The small-molecule antagonist ISO-1 (Fig. 7A) inhibits MIF tautomerase activity in a dose-dependent manner (Fig. 7B), consistent with previous findings. 40 While ISO-1 is recognized as an inhibitor of the MIF active site, 23 its effect on MSC migration was unknown. Based on the dose-response curve (Fig. 7B), we selected two concentrations, 500 and 1000 μg ISO-1/μg MIF, for migration studies. ISO-1 restored MSC migration equally well at both tested concentrations, insensitive to the exact quantity of antagonist added in this range (Fig. 7C). Specifically, these concentrations restored >90% of MSC migration in the presence of 85 ng/mL MIF in SF-SGM (p < 0.001), the same concentration found in EDM (Table 2). ISO-1 by itself is not a chemoattractant; migration in SF-SGM was unaltered by the antagonist in the absence of MIF (Fig. 7C).
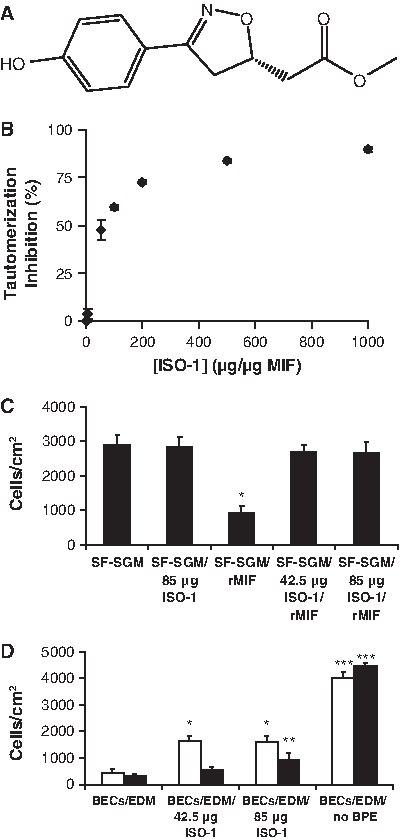
Antagonization of rMIF by (
The antagonist was then applied to conditioned media from interfacial BEC cultures to determine whether MIF inhibition in an in vivo–like system could enhance MSC migration. Use of ISO-1 at either 500 or 1000 μg/μg MIF produced nearly a fourfold increase in migrating MSCs (p < 0.001) compared with conditioned medium from uninjured BECs alone (Fig. 7D). Conditioned EDM from BM-exposed BECs, in contrast, required a larger dose of ISO-1, 1000 μg/μg MIF, to produce approximately a threefold increase in migrating MSCs (p < 0.05). No tested condition restored migration to the level found in conditioned EDM without BPE, suggesting that BPE contains inhibitory factors other than MIF. These findings suggest the intriguing possibility that small-molecule MIF antagonists, such as ISO-1, may have value as pharmacological adjuvants to MSC therapy for damaged lung tissue by improving stem cell migration.
Discussion
Regulating migration-related signaling has the potential to be a powerful strategy to enhance homing of MSCs to damaged tissue. To gain insight into the molecular mechanisms governing migration, we identified negative factors affecting MSC migration in an in vitro model of injured lung. BPE was found to contain proteins that cause dose-dependent inhibition of MSC migration. We identified one of these proteins as MIF and showed that the quantity of MIF in EDM, 85 ng/mL, causes ∼50% inhibition of MSC migration in our study. This discovery presents a new drug target to regulate MSC migration. The MIF antagonist ISO-1 attenuates the effect of MIF by restoring MSC migration in the presence of rMIF and by partly restoring migration in BPE-containing conditioned media from uninjured and BM-exposed BEC cultures. MIF and its antagonists may have therapeutic applications to control MSC homing during repair of injured lung and in other clinically relevant systems.
BPE proteins inhibit MSC migration
The anterior pituitary secretes endocrine hormones, such as growth hormone and luteinizing hormone, for homeostasis and in response to various forms of stress, including injury. 41 This gland also produces more than 100 compounds that may participate in autocrine and paracrine signaling, including neurotransmitters and cytokines like MIF. 41 BPE is widely used as a supplement in SF media for epithelial cells,26,35,38 but its composition is complex and not well defined. 38 We found that BPE strongly inhibits migration of MSCs at all tested concentrations. Given the prevalence of BPE as a media supplement, this inhibition should be considered in situations such as coculture, where MSCs may be exposed to epithelial media.
Several components present in BPE are known to influence migration, including epidermal growth factor and platelet-derived growth factor. 38 However, these factors all elicit positive chemotaxis in MSCs, 13 suggesting that they are not responsible for the observed inhibitory effect. Two other potential components of BPE, the lipid messengers lysophosphatidic acid and sphingosine-1-phosphate, have been reported to halt MSC migration by activating Rho GTPase, 15 but they, too, are unlikely to be the source of the observed inhibition because these lipids are heat-stable.42,43 Proteins, not lipids, were probably the cause of migration inhibition in our study because heat treatment at 71°C, sufficient to induce denaturation, eliminated the inhibitory property of BPE. Among the proteins known to be components of BPE, we selected MIF as a potential inhibitory cytokine due to its ability to arrest macrophage migration. 21 In the future, BPE may be a good source to discover other heat-labile, inhibitory factors from among the many proteins in the extract that have likely not yet been identified.
MIF inhibits MSC migration
This research demonstrated that MIF contributes to BPE inhibition of MSC migration. MIF is a 37.5 kDa homotrimer 21 rapidly secreted from the pituitary into the bloodstream in response to injury and other forms of stress. 44 This is the first report of MIF regulating MSC migration. A potent cytokine, MIF inhibited MSC migration by ∼50% at concentrations found in culture medium and nearly suppressed migration entirely at higher levels in this study. MIF signaling is mediated though three putative receptors, all of which are expressed on MSCs.18,19 CD74, the major histocompatibility class II–associated invariant chain, transduces the signal by recruiting the cell-surface glycoprotein CD44, 21 resulting in ERK 1/2 activation. 45 MIF has also been observed to signal through CXCR2. 17
In the native environment of bone marrow, MIF regulation of MSCs may contribute to bone remodeling. MIF is upregulated at the fracture site during bone healing 46 and stimulates osteoblasts to express metalloproteinases that degrade bone matrix. 47 Transgenic mice overexpressing MIF exhibit increased bone formation rate, higher metalloproteinase expression, and high-turnover osteoporosis, without affecting osteoclast formation. 20 As precursors to osteoblasts, MSCs may participate in MIF-induced bone remodeling. The effect of MIF on MSC proliferation, lineage commitment, and other relevant processes remains to be determined.
When MSCs are employed in therapies to repair tissue beyond the confines of the bone marrow, they encounter MIF in the context of inflammation. For example, circulating concentrations of MIF in the serum of pediatric patients can approach 100 ng/mL after cardiopulmonary bypass surgery, as compared with ∼20 ng/mL in healthy children. 48 During sepsis, bronchoalveolar lavage fluid level of this proinflammatory cytokine increases from <5 ng/mL to >40 ng/mL in an animal model. 22 In patients with severe sepsis, plasma MIF can reach 3200 ng/mL, with a median of >100 ng/mL. 49 Inflammation and therefore elevated MIF levels are likely to be present in damaged tissues targeted by stem cell therapy. MIF suppressed migration of MSCs in our in vitro assay at clinically relevant concentrations; hence it may also impact homing of MSCs in vivo. Future research in our lab will determine the extent to which MIF signaling can influence MSC homing in the complex mixture of cytokines and chemokines found in vivo that direct cell recruitment to the site of injury.
ISO-1 antagonizes MIF inhibition of MSCs
The MIF antagonist ISO-1 binds to a hydrophobic pocket in the MIF active site and functions as an inhibitor of tautomerization. 40 The tautomerase function is unnecessary for MIF to participate in cell signaling because a catalytically inactive mutant retains the ability to inhibit monocyte migration. 50 ISO-1, however, inhibits not only the catalytic activity but also other biological functions of MIF. The anti-inflammatory effects of ISO-1 include reversing the glucocorticoid-regulating activity of MIF both in vitro 40 and in vivo, 23 and attenuating MIF-induced arachidonic acid release. 40 ISO-1 also inhibits MIF-induced proliferation of DU 145 prostate cancer cells. 45 Our experiments show that ISO-1 attenuates the migration-inhibitory function of MIF in MSCs. It remains to be determined whether ISO-1 affects other biological activities of MIF that may be relevant to MSCs.
To enhance MSC homing to injured tissue, it would be beneficial to attenuate signaling that inhibits migration. We report that ISO-1 partly restores MSC migration in BEC-conditioned media containing BPE, suggesting that inhibitory signals from MIF are relevant in a complex milieu of signaling factors. Although ISO-1 has been shown to negate the effect of MIF on monocyte migration, 51 this is the first report of the effect of ISO-1 on MSCs. While our research was conducted with an in vitro model of bronchial epithelium, it is likely that the antagonist activity of ISO-1 would be germane to MSC therapies for epithelial and nonepithelial tissues alike because MIF expression is seminal to the inflammatory response induced by tissue injury. 21 Further, our findings should be applicable to in vitro cultures of MSCs with other types of epithelial cells as BPE is a supplement in many SF epithelial media,35,38 and MIF is consumed by a variety of cells, including those of epithelial origin. 52 We also observed that the beneficial effects of ISO-1 on MSC migration were relevant in conditioned media from both uninjured and BM-exposed BECs. A difference that we observed between these two cultures was a lower MIF concentration in the latter. Injured BECs may have increased consumption of MIF, which is involved in prosurvival signaling in both epithelial and nonepithelial cells.53,54
In addition to ISO-1, antibodies specific to MIF or its receptor have been used successfully to reverse MIF activity. 45 However, ISO-1 has several advantages over antibodies as an MIF antagonist. Small molecules like ISO-1 are generally more stable, less expensive to produce, and easier to deliver. Moreover, rational design strategies can more readily improve the efficacy of small-molecule drugs. To that end, a more potent variant of ISO-1 has recently been reported. 55
Therapeutic applications
MSCs have a wide variety of potential applications in regenerative medicine. Their use, however, is currently hampered by low engraftment rates. 6 To provide maximum benefit, MSCs must be introduced soon after injury, 7 at the same time that MIF concentrations are elevated. 22 Altering migration-related signaling, using small-molecule MIF antagonists like ISO-1 as a pharmacological adjuvant to MSC therapy, could allow a greater fraction of the injected cells to reach injured tissue and participate in the healing process. Conversely, migration of MSCs can also be an undesired outcome, as when the cells migrate spontaneously out of ex vivo cartilage constructs. 56 Cases exist, then, where inhibiting migration, perhaps by enhancing MIF signaling, could increase the fraction of MSCs remaining in the target tissue.
In some situations, there may be synergy between anti-inflammatory drugs, like ISO-1, and the innate anti-inflammatory characteristics of MSCs. Anti-MIF antibodies reduce mortality and lung inflammation in BM-exposed mice, 57 indicating that MIF is involved in the acute phase of lung injury where MSCs have also proven beneficial. 7 Similarly, MIF suppression and MSC therapy both show promise in treating rheumatoid arthritis. An MIF-targeting DNA vaccine reduces joint inflammation in a murine arthritis model, 58 while MSCs suppress proliferation and activation-antigen expression of collagen II–stimulated T cells from arthritis patients. 59 The potential synergy achieved by combining MSC and ISO-1 therapies may be relevant to the treatment of these and other disorders where inflammation contributes to disease progression.
In addition to utilizing innate properties of MSCs to treat disease, the cells can also be used as vectors for drug delivery. For example, MSCs have been used to deliver gene therapy to tumors, 60 while MIF has been implicated in cancer progression. In lung adenocarcinoma cells, autocrine MIF induces cell cycle progression and promotes migration and invasion through activation of RhoA GTPase. 61 ISO-1 has been used to decrease tumor volume in prostate cancer xenografts by decreasing cell proliferation and invasion. 45 If this treatment is combined with MSC therapy, inhibition of MIF activity may provide the dual benefit of slowing tumor development while enhancing homing of MSCs carrying a suicide gene to the tumor.
Conclusion
This research presents a new drug target to regulate MSC migration. We identified the proinflammatory cytokine MIF as a potent inhibitor of MSC migration using an in vitro model of injured lung. MIF in BPE contributes to suppression of MSC migration, but BPE also likely contains other heat-labile, inhibitory factors that have not yet been identified. Further, we demonstrated that it is possible to augment MSC migration by targeting negative migration signaling with the small-molecule MIF antagonist ISO-1. This study suggests that MIF and its antagonists are relevant to the control of MSC homing and efficacy of stem cell therapy in a variety of clinical scenarios. Future work in our lab will extend these studies from a well-defined in vitro system to an in vivo model of injured lung.
Footnotes
Acknowledgments
We thank Alan Tucker for his assistance with flow cytometry, Wen-Tzu Lai and Iryna Isakova for their help in producing lentivirus, and Olga Borkhsenious for performing the electron microscopy. This work was supported by a grant from the National Science Foundation (BES-0514242).
Disclosure Statement
No competing financial interests exist.
This article was presented in part at the 2008 Annual Meeting of the American Institute of Chemical Engineers in Philadelphia, Pennsylvania.