
Abstract
The design of composite scaffolds with slow degradation kinetics imposes the assessment of the time-course of degradation to predict the long-term in vitro behavior. In this work, the effect of hydroxyapatite (HA) particles on the hydrolytic degradation of poly ɛ-caprolactone composite scaffold was investigated. The study of accelerated degradation mechanisms in alkaline medium enabled analysing comparable degradation profiles at different times. The accurate qualitative and quantitative study of morphology by scanning electron microscopy supported by image analysis demonstrated only a negligible effect on the structural porosity, to be ascribed to the addition of micrometric HA as a filler. Moreover, by comparing the Raman spectra with thermal analysis (thermogravimetry and differential scanning calorimetry) the role of HA on the composite degradation mechanism was defined, by separately quantifying the contribution of HA particles in the bulk and on the surface, on the bone formation as a function of modifications induced in the pore morphology, as well as physical and chemical properties of the polymer matrix. Indeed, HA particles alter the poly ɛ-caprolactone crystallinity inducing a “shielding” effect of the polymer matrix. Meanwhile, the slight reduction of pore size as a function of the increasing HA content and the improvement of the effective hydrophilicity of the scaffolds also influence the degradation by faster mechanisms. Finally, it has been proven that the presence of HA enhances the scaffold bioactivity and human osteoblast cell response, remarking the active role of bioactive signals on the promotion of the surface mineralization and, as a consequence, on the cell–material interaction.
Introduction
In bone tissue engineering, the understanding of bone-forming behavior of cells must be combined with progress in material science, to achieve guided bone regeneration. 2 Today, the scientific community is taking up this challenge through the employment of insoluble signals integrated with the degradable polymer matrix. These act as reinforcement agents as well as osteoconductive signals, offering a valid compromise between the mechanical response and bioactivity of the scaffold. Many papers propose the use of calcium phosphates such as hydroxyapatite (HA) to stimulate a biochemical response from living tissues, to obtain a strong bond between the scaffold and the adjacent tissue with positive results.3,4 However, several aspects remain inadequately explained, particularly regarding the structural changes induced by in vitro degradation mechanisms in the presence of calcium phosphate particles and their effects on scaffold morphology and cell culture are concerned.
Among polyesters, PCL has a long history in tissue engineering, mainly because it was shown to possess sufficient strength and stiffness to function for the period required by bone to heal. However, the need for a mechanically functional material for bone substitution has generated numerous composites of calcium phosphate–reinforced polymers. 5 This combination is well founded, as Ca/P is the inorganic part of bone and has been shown to be osteoconductive. As reported by Khan et al., the combination of calcium phosphate, which on its own is brittle and limited in its applications, and polymer is also well founded as the addition of the polymer can impart beneficial properties such as mechanical toughness, resistance to brittle failure, and formability to the calcium phosphate, as well as expand potential applications of the composite material. 6
It was in this complex scenario that the study on HA-loaded composite scaffolds with bimodal porosity described here was proposed. The matrix made of PCL, a polymer hydrolytically degradable both via bulk and surface erosion, guarantees degradation kinetics slower than other aliphatic polyesters such as poly(glycolic acid) or poly(L-lactic acid), because of its strong hydrophobic nature, 4 offering a valid mechanical support for long-term implantation. However, PCL alone is not adequate as a bone substitute, because of its reduced capability in the retention of osteoblastic phenotype. 4 The integration of HA bioactive particles within the PCL polymeric matrix may serve to improve the osteoconductivity.7,8
A particular aim of this current study is to superimpose the active role of the HA filler on the underlying in vitro degradation mechanisms by the simultaneous assessment of the influence of scaffold morphology and the physicochemical properties of the proposed scaffolds. Several authors9–11 suggested a qualitative investigation of porosity based on images derived from electron microscopy, even if they offer only a qualitative estimation of pore features. Unfortunately, the lack of detailed data prevents an accurate evaluation of the scaffold microstructure and the understanding of the potential effect of pore architecture on the degradation kinetics. In this work, two different methodologies were adopted for porosity feature measurements, based, respectively, on a gravimetry technique and imaging analyses to reach a complete characterization of scaffold architecture (i.e., pore volume fraction, pore size and distribution, and pore orientation). Indeed, the comparative analysis of scaffold porosity by different quantitative methods overcomes the intrinsic limits of each single procedure, giving the opportunity to check the reliability of the result.11,12 Initially, the influence of the HA particles on the in vitro degradation was investigated by testing scaffolds with different PCL/HA ratio in different aqueous media, including a phosphate buffered saline (PBS) solution and a NaOH solution, respectively. Additionally, the ability of nucleating calcium phosphate compounds was assessed by dipping the scaffold in a simulated body fluid (SBF) solution. Vibrational Raman spectroscopy coupled to thermogravimetry (TG) and differential scanning calorimetry (DSC) was efficaciously employed as an investigation tool to follow the progressive scaffold degradation at different times, as already done in previous studies.13–19 Actually, some preliminary data on the degradation of the present scaffolds have been previously reported. 19 The novelty of the present study consists in a more detailed analysis of the degradation mechanism at longer immersion times; more importantly, the same vibrational and thermal techniques were used to comparatively characterize the composites incubated with marrow-derived human osteoblasts (HOB) to evaluate the cell role in degradation. Indeed, Raman spectroscopy gives information on the molecular structure (composition and crystallinity) of the samples, being sensitive to bond vibrations, and its changes during degradation, whereas TGA and DSC analyses were used to gain insight into the architecture of the polymeric component and its changes upon degradation, remarking the key role of the HA particles on the time-evolution of the scaffold properties.
Materials and Methods
Materials and scaffold preparation
HA-loaded composite scaffolds were developed using a combination of phase inversion and salt leaching technique. 20 PCL pellets (MW 65 kDa; Sigma Aldrich, Milan, Italy) were dissolved in N,N-dimethylacetamide (J.T. Baker 06/2007) solution (5 g polymer in 20 mL solvent) by stirring for about 3 h at 58°C. Subsequently, NaCl particles (sieved into specific size range of 212–300 μm) were added up to reach a volume ratio of about 9 to 1, forming a homogeneous mixture that was placed into an antiadhesive mold. Preliminarily, a more uniform distribution of porogen agent into the polymer mixture was reached via static precompression using 1 kg weight for 10 min (equivalent pressure of 0.127 N/mm2). The mixture was then washed in ethanol (3 mL each 20 min for three times) at room temperature and in bidistilled H2O (Carlo Erba, Milan, Italy) for 7 days, with daily changes.
The HA particles were added to the PCL solution before the solvent extraction, once the polymer/solvent system was totally dissolved. In particular, HA particles (HAP205) from Plasma (Biotal, Tideswell, UK) showed particles with spherical morphology and bimodal size distribution (Φrange = 0.4–11 μm, Φ0.5 = 4.02 μm) measured by laser diffractionmeter measurements 21 with a theoretical density of 3.16 g/cm3. Three different scaffolds were prepared by adding a HA relative volume fraction of 13%, 20%, and 26%, respectively. In addition, a PCL scaffold with HA particles unloading was prepared as control. Disk-shaped samples with 5 mm diameter and 3 mm thickness were used for degradation tests (n = 2) and cellular investigations (n = 3).
Morphological investigation by scanning electron microscopy/energy dispersive spectroscopy analysis
The morphology of the scaffold surface was investigated by scanning electron microscopy (SEM Leica Stereoscan mod.420-Oxford Instruments). The samples were gold-coated using a sputter coater set at 15 mA for about 20 min. The porosity was investigated in terms of pore size, shape, and spatial distribution by images at different magnifications, respectively, 150 × (working distance = 23) and 2230 × (working distance = 30). The chemical composition, element by element, with a high degree of precision (∼0.1 wt%) was evaluated through the energy dispersive spectroscopy using a Oxford mod. INCA 200, to confirm the presence of the ceramic particles on the surface up to a depth of about 2 μm. Finally, to examine the distribution and the extent of surface exposure of HA in the scaffolds, a hydrophilic dye (trypan blue) staining was performed. Briefly, the sample was dipped in 1 M aqueous staining solution for 15 min, and then the dye was removed by ethanol washing for 48 h in an orbital shaker. HA exposed at the scaffold surface was observed using an optical microscope (SXZ7; Olympus, Milan, Italy).
Porosity: Gravimetric measurements and image analysis
To evaluate the scaffold porosity, two different methods were used: the gravimetric method (GM) and image analysis (IA) procedures on selected SEM images. The former consists of the evaluation of the structural porosity by measuring the characteristic densities of the scaffold, namely, the effective density related to the porous structure and the apparent density related to the material that comprises the matrix. To do this, the effective density was first determined by measuring the size parameters and the weight of the scaffold as follows:
Then, the porosity, P, was evaluated as:
The apparent density dp is equal to the PCL density (data reported in literature
22
) for pure PCL samples; for the composites, it was obtained from the densities of the single phases, corrected by their relative volume fractions as reported:
The alternative approach to the investigation of the porosity features employs 2D IA procedures, previously used elsewhere. 11 In brief, micrographs obtained by SEM on cross-sectional surfaces were processed using imaging software (ImageJ 1.38b; NIH Freeware; National Institutes of Health, Bethesda, MD). First, the conversion from analogical to digital image, self-performed by the SEM apparatus, was attained. Then, the 8-bit conversion of the image was applied to obtain a monochromatic image (black and white) from an image with 256 gray levels. Meanwhile, the preliminary adjustment of the image contrast and brightness enables to emphasis pore boundaries from the polymer matrix for the calculation of the pore features. Porosity degree, pore size, and spatial distribution were calculated using specific tools contained into ImageJ freeware software pack. The procedure consisted in two steps (Fig. 1): the former provides the tracing of the entire inter-phase boundaries related to the pore walls, whereas the latter provides the count of all pore objects, calculating quantitatively their size and their surface area. The porosity degree was evaluated from the total surface area of counted pores, whereas the pore size was obtained on the basis of the assumption of the pore circularity. Means and standard deviations of pore fraction and size have been determined from measurements on 10 different SEM images.

Illustration of image analysis (IA) procedure applied on cross-sectional scaffold surfaces via imaging software. (
Cell preparation
Human bone–forming cells were obtained from patients at surgery for hip joint replacement. The study was approved by the Institutional Ethics Committee, and informed consent was obtained from donors. At surgery, drilling of the femur was performed before prosthesis insertion, and bone marrow together with bone fragments was collected aseptically. In the lab, bone fragments were removed using a cell strainer, and bone marrow was layered onto Ficoll gradient. Mononuclear cells obtained by centrifugation and sequential washes with PBS were seeded in 25 cm2 flask with the complete Dulbecco's Modified Eagle's Medium (D-MEM). Marrow stromal cells (MSC) adherent to tissue culture plastic were obtained by removal of nonadherent cells; expanded in α-Modified Eagle's Medium (α-MEM) with 10% fetal bovine serum, 1% antibiotic–antimycotic solution, and 50 μg/mL ascorbate-2 phosphate; and used for experiments at passages 1–3. PCL scaffolds were prepared to cell seeding by soaking first in 70% ethanol (1 h), then in 1% antibiotic–antimycotic in PBS (2 h), and prewetting in medium (2 h).
MSC were seeded 4 × 104 per sample in a 10 μL-drop and allowed to settle on the sample surface for 30 min; then, 2 mL of medium was added to the wells. The seeded scaffolds were cultured in the complete medium for 28 days, with medium exchange twice a week, and b-glycerophosphate (5 mM) was added from the third week onward for mineralization.
Each week cell-loaded scaffolds were assayed for cell viability using Alamar blue (data not shown). At 28 days from cell seeding the scaffolds were analyzed using the structural analyses and observed by SEM.
In vitro degradation
To study the in vitro degradation mechanism and kinetics, all samples were weighed and immersed in three different aqueous media, at 37°C: (a) PBS at pH 7.5 (KH2PO4 0.0087 M, Na2HPO4 0.0304 M, and NaCl 0.154 M); (b) 0.01 M NaOH solution; and (c) SBF buffered at pH 7.5 (Tris 50 mM, HCl 45 mM) containing Na+ (142 mM), K+ (4 mM), Ca2+ (2.5 mM), Cl− (148.8 mM), HCO3− (4.2 mM), and HPO42− (1 mM). 23
Pure PCL, PCL/HA 13%, PCL/HA 20%, and PCL/HA 26%, belonging to the same synthesis batch, were used for in vitro degradation tests, whereas only pure PCL, PCL/HA 20%, and PCL/HA 26% were used to assess the degradation in the presence of HOB or into the culture medium. Each degradation test was performed in duplicate under sterile conditions. To this purpose, all samples were kept in 70% ethanol overnight, before their immersion in the degradation media that were renewed every week.
For each degradation time, the samples were recovered, washed with distilled water, vacuum-dried at room temperature, and weighed before being subjected to the various analyses. The weight loss percentage of the samples was calculated according to the equation:
Raman spectroscopy
The Raman spectra of the untreated and in vitro degraded samples were recorded on a Jasco R1100 spectrometer with a 488 nm radiation from a Spectra-Physics argon ion laser source. The spectral resolution was 4 cm−1, and laser power at the sample was ∼40 mW.
Because of the fluorescence of the cell-incubated samples, their Raman spectra were recorded on a Bruker IFS66 spectrometer equipped with an FRA-106 Fourier Transform (FT)-Raman module and a cooled Ge-diode detector. The spectral resolution was 4 cm−1. The excitation source was an Nd3+-YAG laser (1064 nm) in the backscattering (180°) configuration. Since the fluorescence bands typical of apatite 24 were used to evaluate the HA content, all spectra were recorded with the same laser power (180 mW). Raman intensities were evaluated as peak heights. To minimize variability deriving from the possible sample inhomogeneity, three Raman spectra were recorded on three different points of each specimen. The presented spectra are the representative measurements (i.e., average spectra) of the corresponding samples.
Thermal analysis
The thermogravimetry (TG) thermograms of the untreated, in vitro degraded, and cell-incubated samples were performed using a Mettler TA-STAR, TGA/SDTA 851e thermobalance, in air with a heating rate of 2°C/min, from 25 to 1000°C. The PCL content was evaluated through the weight loss in the 200–500°C range.
The DSC thermograms of the untreated, in vitro degraded, and cell-incubated samples were performed with a Mettler TA-STAR, DSC 821e calorimeter, covering 5–120°C. The samples were heated at 2°C/min (first run) and then cooled at −2°C/min (second run). Typically, the first run of a crystalline polymer presents the melting peak of the polymer. From this peak, the crystallinity degree Xc% of the polymer (i.e., its degree of order) was calculated according to the equation:
The crystallization capability of the polymer, once melted in the first run, was evaluated from the crystallization peak observed in the second run; from a quantitative point of view, the crystallizable fraction (CF%) of the samples was calculated according to the following equation:
In the calculation of Xc% and CF% for the composites, ΔHm1 and ΔHc were calculated by taking into account the weight percentage of polymer determined by TG analysis.
All the quantitative results were expressed as mean ±standard deviation for n = 2 or n = 3 (cell-loaded samples). A statistical analysis by a one-way analysis of variance, followed by Bonferroni post hoc test (paired groups), was carried out to determine significant differences in Xc%, CF%, TG, and weight gain data between four groups of samples with different HA content. A value of p < 0.05 was considered to be statistically significant.
Results
Qualitative investigation of morphology
A preliminary evaluation of the morphological features of scaffolds was assessed by SEM analysis of the surface of all samples before degradation. A typical bimodal porosity, induced by the preparation technique, has been clearly evidenced (Fig. 2). A large porosity, or macroporosity, ranging from 20 to 300 μm and a small porosity, or microporosity, ranging from 1 to 20 μm were observed. The former (Fig. 2A) was due to the extraction of the sodium chloride crystals, whereas the latter (Fig. 2B) was ascribed to the solvent/nonsolvent exchange. Moreover, no significant modification of the structure was evidenced by SEM in the HA-loaded composite scaffolds as the HA content varies.
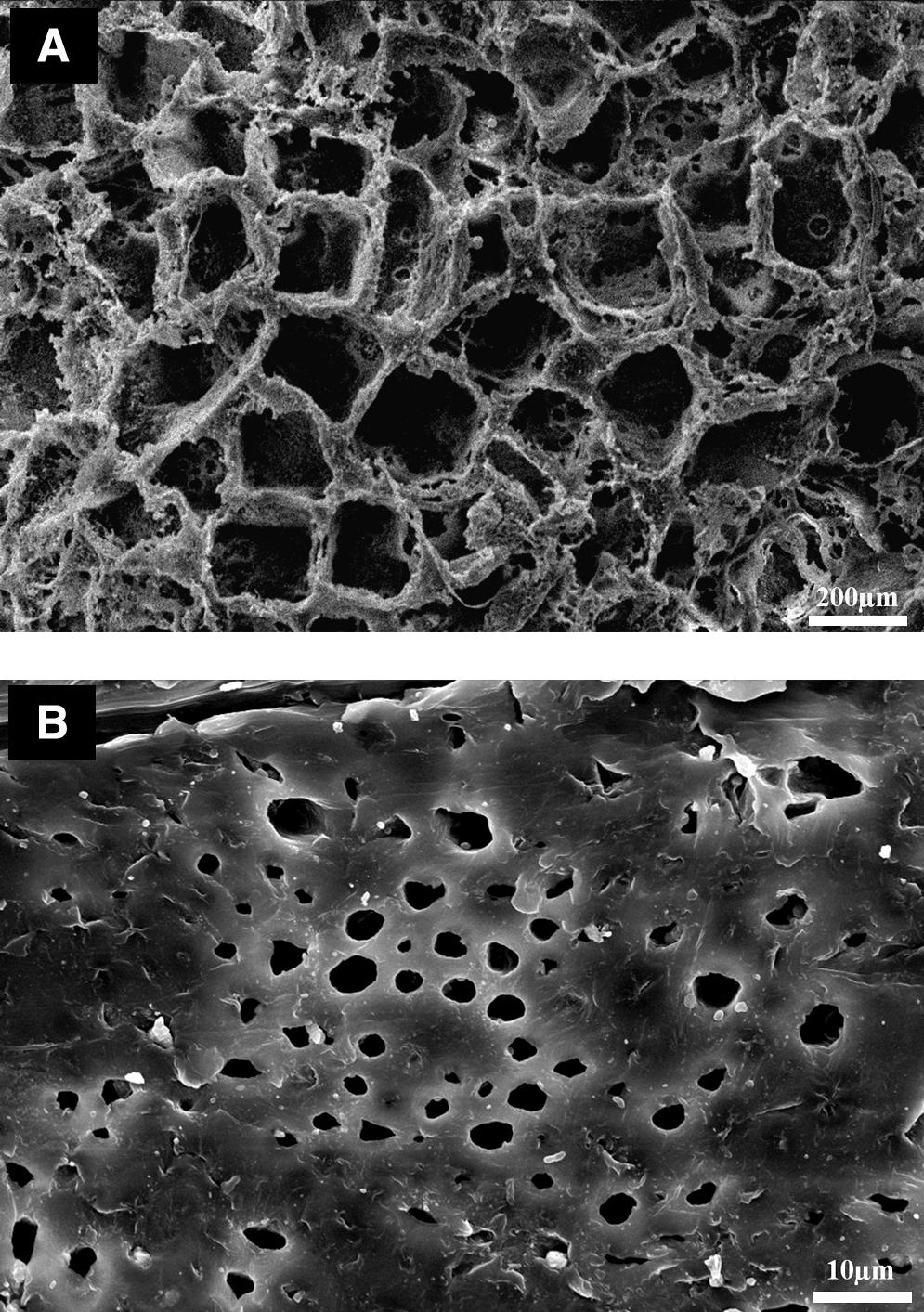
Bimodal porosity of HA-loaded scaffolds (poly ɛ-caprolactone [PCL]/HA 13%) observed on scanning electron microscopy cross-sectional image. (
On the other hand, the energy dispersive spectroscopy analysis performed on the cross section of HA-loaded scaffolds (Fig. 3A–C) confirmed the presence of the apatitic phase: two large peaks due to Ca and P elements were observed, and a Ca/P atomic ratio ranging from 1.62 to 1.76, that is, close to the theoretical value of stoichiometric HA (Ca/P = 1.67), was calculated. Meanwhile, the use of the Trypan Blue hydrophilic dye revealed the exposed HA particles on the pore surfaces, evidencing a more abundant, homogeneous, spatially distributed blue-stained regions as the HA volume fraction increases (Fig. 4A–D).
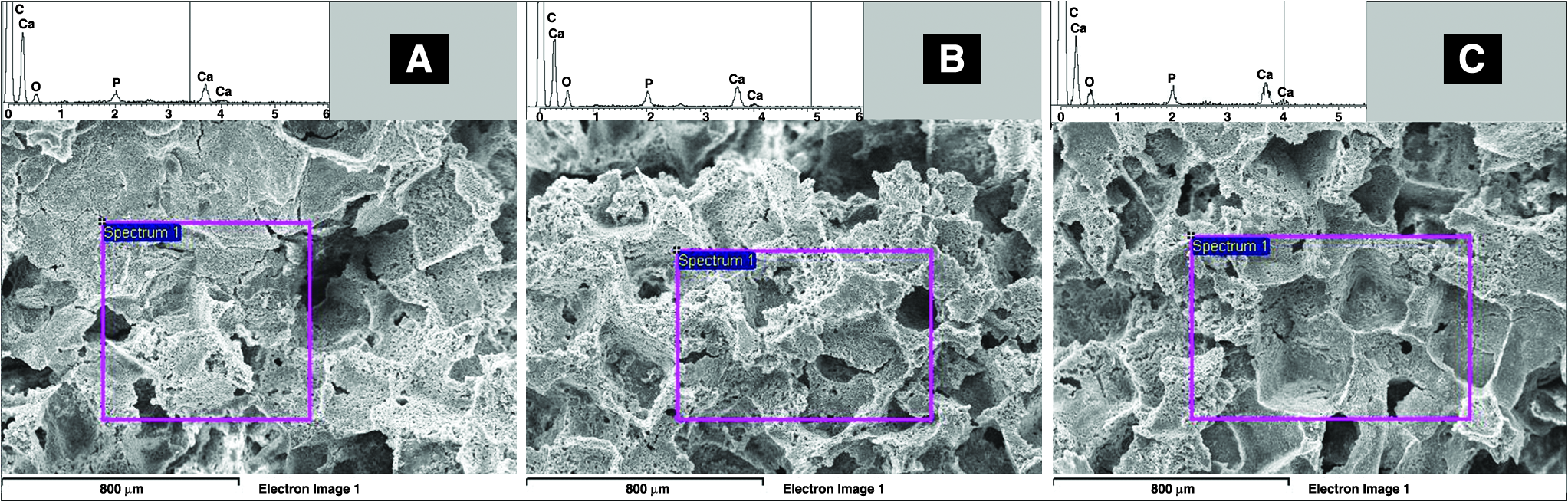
Qualitative evaluation of P and Ca peaks by energy dispersive spectroscopy analysis on the cross section: (
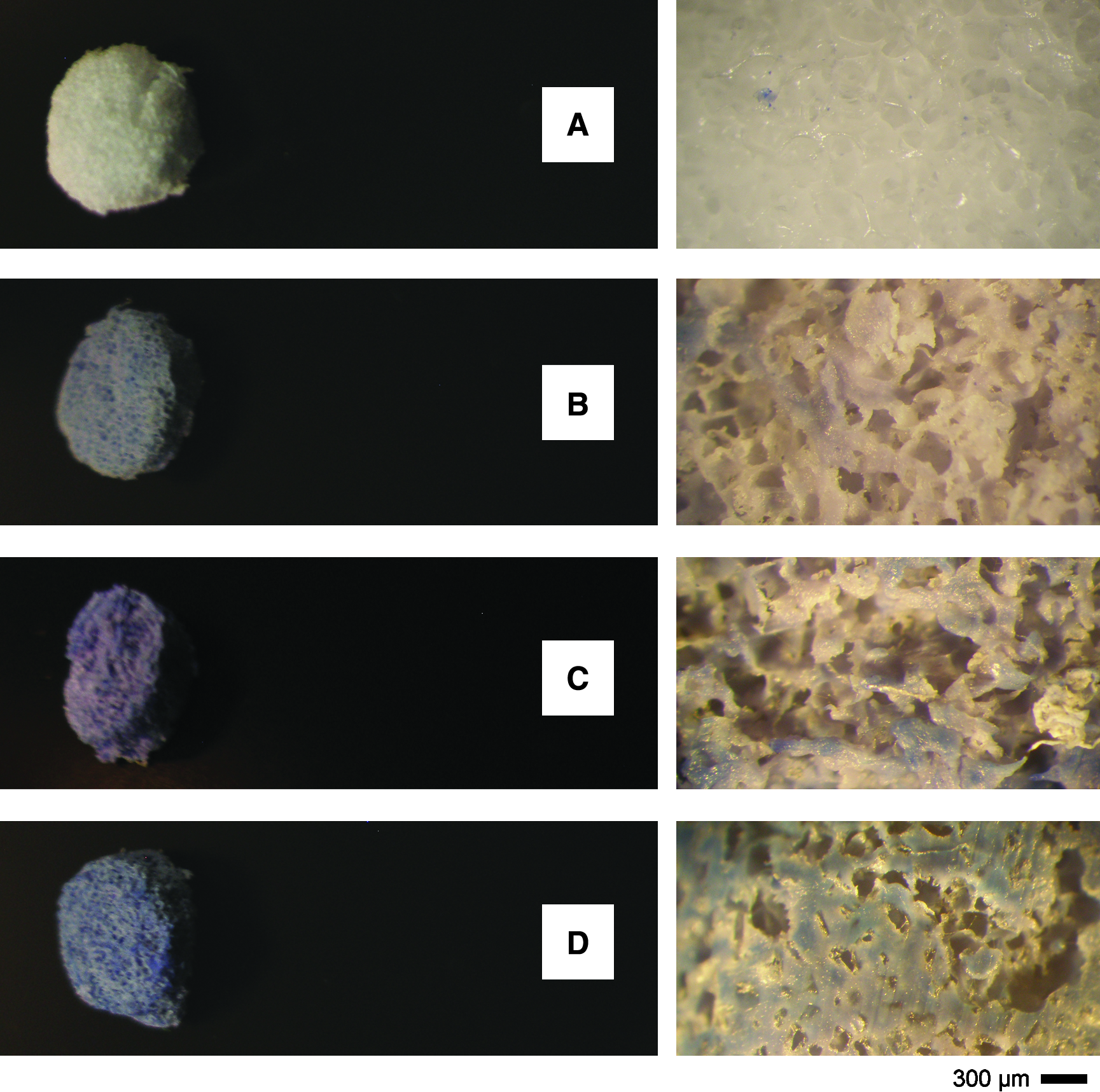
Qualitative evaluation of the HA particles exposure at the surfaces of PCL composite scaffolds, as a function of the HA volume fraction: (
Quantitative estimation of porosity
The porosity was quantified by GM and IA methods. In Figure 5A, the porosity degree calculated by both methods is reported as a function of the HA volume fraction. The results show that the addition of HA does not significantly affect the porosity of the scaffolds at any volume fraction. The employed methods revealed different porosity degrees values ascribable to the different errors associated to the measurements by the two procedures. By weight measurements, porosity degree ranges from 95.1% to 90.6% for the increasing amount of ceramic filler, close to the theoretical values corresponding to the porogen volume fraction used during the preparation (∼90%). Instead, the investigation of porosity by IA showed a lower porosity ranging from 81% to 84%, not strictly dependent on the filler amount (Fig. 5A). The slight differences are likely due to the presence of micropores obtained via phase inversion previously evidenced by SEM investigation. Further, the latter measurement is less reliable because of the intrinsic error of the IA procedure that entails the loss of smaller fundamental details during the image manipulation by working on 2D images representative of 3D objects.
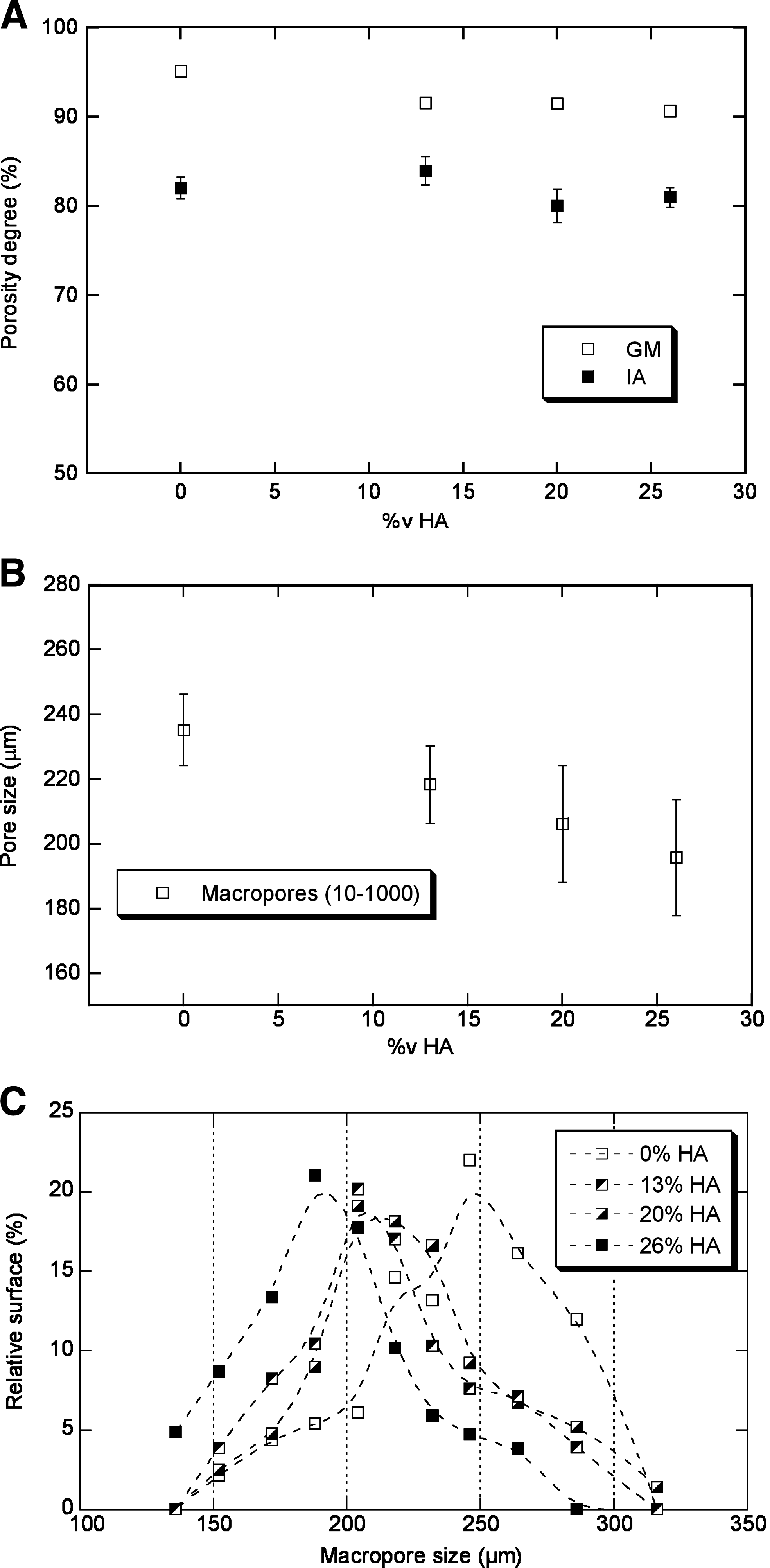
Assessment of the porosity features by different methods. (
However, the procedures of 2D image manipulation represented a valid instrument for the description of additional morphological parameters such as size, distribution, and orientation of pores, not achieved by the GM. In Figure 5B, the average size of macropores was reported as a function of the HA content. Macropore size ranges from 195 ± 18 μm to 235 ± 11 μm coherently with the average size of the used porogen particles (see par. 2.1). The HA content affects the pore size, which decreases for increasing HA volume fraction. These results were also confirmed by the macropore size distribution reported in Figure 5C characterized by statistical modes of 244, 207, 202, and 192, respectively.
Thermal and Raman characterization of untreated samples
Table 1 reports the TG and DSC data obtained from the thermograms of all the samples. TG data on the untreated composites indicated that the HA amount was spanning between 25% and 50% wt/wt, according to the PCL/HA ratios used in the scaffold preparation. In fact, these results completely fit the indicated volume fractions directly calculated by normalizing the weight ratios through the densities of each component. Moreover, all DSC thermograms revealed two different characteristic peaks, that is, an endothermic melting peak at 61–63°C in the first run and an exothermic crystallization peak at 41–37°C in the secondrun.
Calculated according to Equation (2).
Calculated according to Equation (3).
By thermogravimetry (TG) analysis for the composites; nominal for the pure PCL sample.
PCL, poly ɛ-caprolactone; HA, hydroxyapatite; PBS, phosphate buffered saline; SBF, simulated body fluid.
As can be seen from Table 1, the polymer phase in the untreated PCL/HA scaffolds appears more crystalline than in the pure PCL sample. This suggests that the method utilized for preparing the composites affected the macroscopic properties of the polymer: specifically, an increase of crystallinity (Xc%) from 71 to 74–84% was highlighted for increasing HA amounts, while no remarkable variation in the CF% (62–65%) was observed.
These data have been confirmed by the Raman analysis. Figure 6 shows the Raman spectra of untreated PCL (a), PCL/HA 20% (b), and PCL/HA 26% (c). As indicated in the figure, some bands are due to PCL, others to HA, others to both components. The Raman spectrum of PCL reflects polymer morphology; as reported in the figure, some bands can be assigned to bond vibrations of crystalline PCL and others to bond vibrations of amorphous PCL. Therefore, the relative intensity of these bands gives information on the degree of crystallinity of the polymer. For PCL/HA 26%, the components at 1735 cm−1 (C = O stretching) and 1090 cm−1 (skeletal stretching), typical of amorphous PCL, 26 had the lowest intensity, and the intensity ratio between the bands at 1304 cm−1 (CH2 wagging of crystalline and amorphous PCL 26 ) and 1283 cm−1 (CH2 wagging of crystalline PCL 26 ) had the lowest value. These results were in agreement with the previously noticed trends of crystallinity, as determined by thermal analysis: Xc% increased moving from pure PCL to PCL/HA 26%. The Raman analysis also confirmed the relative compositions of the scaffolds. The Raman bands of the HA component at 1065 − 1042 cm−1 (PO4 stretching 27 ), 963 cm−1 (PO4 stretching 27 ), 609–592–581 cm−1 (PO4 bending 27 ), and 487–446–431 cm−1 (PO4 bending 27 ) increased in intensity with respect to those of PCL going from PCL/HA 13% to PCL/HA 20% and PCL/HA 26%, as expected on the basis of the TG data.
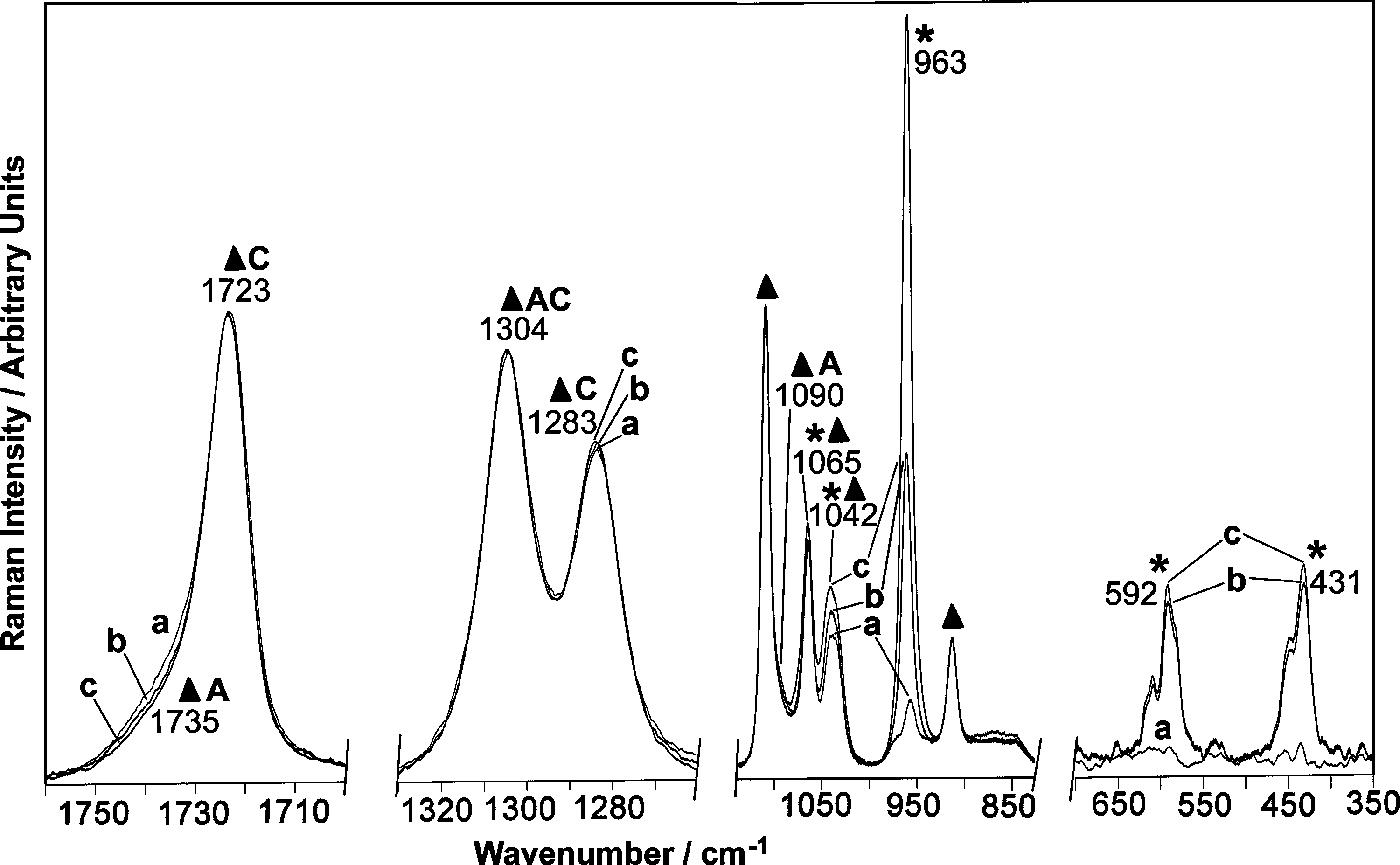
Raman spectra of untreated samples: pure PCL (a), PCL/HA 20% (b), and PCL/HA 26% (c). The bands prevalently due to PCL (▴) and HA (*) are indicated, as well as the bands due to crystalline (C) and amorphous (A) PCL.
In vitro degradation in PBS and NaOH media
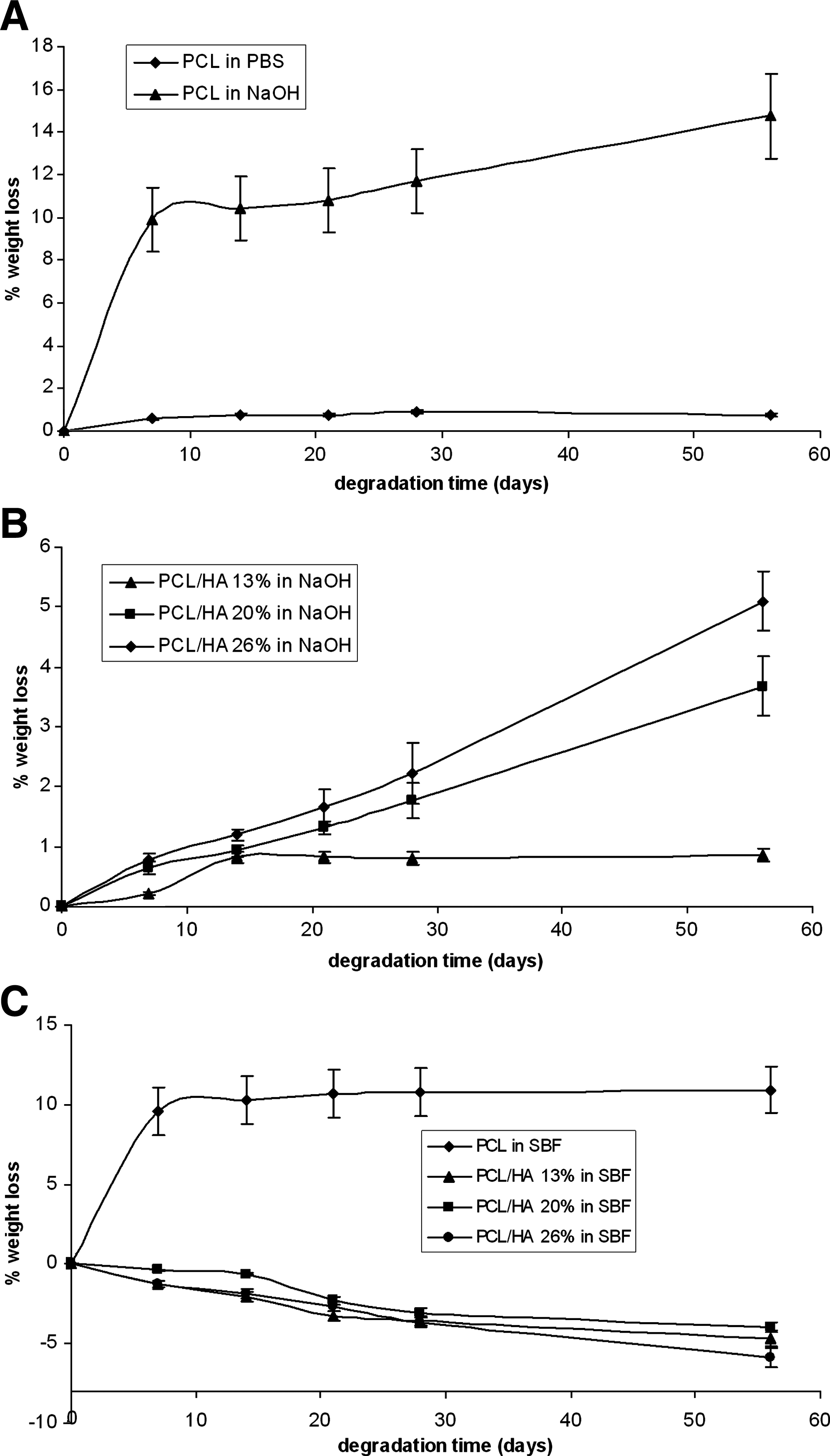
Figure 7A shows the trend of the weight loss percentage of pure PCL samples as a function of degradation time in the different media. As can be easily seen, after 56 days of degradation, the weight loss of the pure PCL samples was higher in NaOH solution than in PBS.
(
As can be seen from Table 1, DSC measurements showed that degradation of pure PCL samples resulted in an increase in crystallinity (Xc%), irrespective of the degradation medium, with the highest increase in NaOH solution (p < 0.01). Further, the samples degraded for 56 days in NaOH solution showed the most significant CF% increase (p < 0.01), which indicates that in this medium the chains are more shortened than in the others.
Accordingly, the latter samples showed also the most pronounced spectroscopic changes with respect to the untreated PCL sample: the above-mentioned marker bands of the amorphous PCL phase at 1735, 1090, and 870 cm−1 decreased in intensity upon degradation, 26 as well as the intensity ratio between the bands at 1304 cm−1 (CH2 wagging of crystalline and amorphous PCL 26 ) and 1283 cm−1 (CH2 wagging of crystalline PCL 26 ).
Interestingly, also the PCL samples treated with PBS for 56 days showed a significant increase in Xc% and CF% (Table 1, p < 0.05), in spite of their only slight weight loss (Fig. 7A). This result indicates that a certain fragmentation of the polymeric chains occurred with a consequent rearrangement toward a more ordered structure characterized by higher Xc% and CF%.
In vitro degradation tests have been also performed on HA-loaded scaffolds. The percent weight loss of the PCL/HA composites upon immersion in PBS medium was as low as that for pure PCL samples (less than 1% after 56 days), independent of relative HA content. As observed for the pure PCL samples, the PCL/HA composites treated with PBS also showed significant changes in Xc% and CF% (p < 0.05), although they displayed only slight weight losses. However, contrary to behavior observed for the pure PCL samples, Xc% decreased upon degradation for 56 days (p < 0.05). This contrasting behavior can be ascribed to the different Xc% of the undegraded samples. The CF% increased upon degradation, as observed for the pure PCL samples (p < 0.05).
The treatment of the PCL/HA composites with the NaOH solution induced more significant weight changes (Fig. 7B) than with PBS. Interestingly, after 56 days of degradation, the PCL/HA samples were characterized by significantly lower weight losses than pure PCL samples (p < 0.01).
To compare the weight loss data referred to the polymeric component, net weight loss data were calculated (Table 2) by taking into account the TG data. According to the data reported in Table 2, it can be seen that the polymeric component present in all the composites underwent significantly lower weight losses than pure PCL, the lowest being that of PCL/HA 26%.
By TG analysis for the composites; nominal for the pure PCL sample.
Calculated according to Equation (1).
DSC analysis confirmed that the polymeric component in the latter sample degraded less than in PCL/HA 13% and PCL/HA 20%. This result may be explained by examining the trend of Xc% during the degradation of the composites in NaOH solution (Table 1). As can be easily seen, Xc% followed the same trend for PCL/HA 13% and PCL/HA 20%, according to their initially similar Xc%. In particular, after 28 days of degradation Xc% decreased (p < 0.05), due to the degradation of the PCL crystalline domains. After 56 days, Xc% increased (p < 0.01), indicating a further degradation: the polymeric chains were shortened to such an extent that they could reorganize into a more ordered state, with a correspondingly higher crystallinity.
The Xc% of the PCL/HA 26% composite showed a different trend as a function of the immersion time in NaOH solution. In particular, it must be stressed that the polymeric component appeared less degraded than in the other composites since the above-discussed Xc% increase was not yet observed at 56 days. The TG curves (data not shown) confirmed that the polymeric component of PCL/HA 26% was the least degraded by reporting a lower temperature–weight loss present neither in the corresponding untreated samples nor in the PCL/HA 26% under the same treatment conditions.
In vitro degradation in SBF medium and bioactivity
Figure 7C reports the trend of the weight loss percentage of the analyzed samples as a function of time of immersion in SBF solution. As can be easily seen, PCL underwent a significant weight loss. More in detail, the percent weight loss followed nearly the same trend as in NaOH solution during the first 28 days of degradation (p > 0.05). This result could be explained according to the literature 28 ; actually, the ionic strength of the degradation medium has been reported to play a significant role on degradation kinetics of aliphatic polyesters.
For the composites, their behavior in SBF solution gave information on their bioactivity, that is, the capability of nucleating an apatitic phase on their surface. Within 56 days of treatment, all the composites underwent a negative weight loss, which corresponds to a weight gain (Fig. 7C). The PCL/HA 26% (i.e., the sample characterized by the highest amount of HA component) showed the highest weight gain (p < 0.001). Accordingly, the TG measurements (Table 1) showed a decrease of the proportion of PCL (wt/wt) for all the composites, with an enrichment in the apatitic component. The Raman spectra recorded on the surface of the composites treated for 56 days in SBF solution confirmed the deposition of an apatitic component. In fact, the above-mentioned marker bands of HA were found to increase in intensity with respect to those of PCL.
HOB incubation tests
HOB were seeded on the prewet scaffolds and cultured as described for 28 days (Fig. 8). Extensive spreading and cell–cell contact on PCL/HA 26% led to the formation of a sheet of cells on the surface. As suggested by Coombes et al. this effect may be ascribed to HA, which is known to enhance cell adhesion and spreading through an inherently high capacity to adsorb proteins. Fourier transform-Raman analysis did not show any significant spectral change in terms of PCL/HA ratio and polymer crystallinity upon HOB incubation of the samples. 29

Scanning electron microscopy image of human osteoblasts grown on PCL/HA 26% at 28 days. The surface is covered by well-spread, interconnected cells, fused together to form a multiplicity of continuous layers. The cells are connected to each other by cytoplasmic filopodia, with the rough PCL/HA surface partially visible beneath the cells (bar = 100 μm).
Thermal analysis provided more insight into the potential composition and crystallinity changes induced by culture with cells (Table 3). As can be easily seen from Table 3, the composition of the samples (expressed as % PCL and % HA) did not undergo marked change upon treatments with either culture medium or cells. On treatment with the culture medium, the PCL/HA 20% composite showed no significant change in Xc% or CF% with respect to the corresponding untreated sample (p > 0.05). The PCL and PCL/HA 26% samples underwent a more pronounced degradation, both showing a significant increase in CF% (i.e., decrease of polymeric chain length, p < 0.01) with respect to the corresponding untreated specimens. Pure PCL samples, in contrast, showed no further degradation induced by culture with cells, as confirmed by the unaltered Xc% and CF% values (p > 0.05). Upon cell incubation, the PCL/HA 20% and PCL/HA 26% composites behaved similarly; for both samples, Xc% decreased with respect to the sample treated with the culture medium (p < 0.05 and p < 0.001, respectively), while the CF% remained nearly the same (p > 0.05). The most significant Xc% decrease was recorded for PCL/HA 26%.
Calculated according to Equation (2).
Calculated according to Equation (3).
By TG analysis.
HOB, human osteoblasts.
Discussion
Polymer-based composites, increasingly applied for engineering bone, show advantages and limitations. 30 Slow-degrading PCL has attracted much interest since its first appearance as bone substitute and has been extensively tested as film, 3D, and nanofibrous scaffold, and for drug delivery.31–35 The employment of different preparation methods, such as blending and grafting, and fabrication procedures, such as solid free-form fabrication and electrospinning, of PCL-based composites broaden the range of combinations of PCL with other materials and consequently the potential applications in bone engineering.36,37
The ability of controlling pore geometry within the scaffold represents a powerful method to optimize mechanical properties and biological interactions. 38 Several papers have shown that there is an intimate correlation between the pore size and biological mechanisms, directly related to the surface/volume ratio, which actively participates in cell adhesion and degradation mechanisms.6,39,40 However, the presence of a high porosity reduces the mechanical properties of the scaffold, suggesting that the employment of reinforcement agents, such as ceramic particles mimicking the natural bone tissue. 41 Recently, it has been demonstrated that the incorporation of insoluble signals, such as HA, promotes cell activity. This is based upon the well-known osteoconductive potential, as well as the improvement of mechanical properties due to their reinforcement action.1,19,20,42
It is critical that the scaffolds maintain the integrity of their framework and 3D architecture until the completion of the tissue ingrowth. This requires a close knowledge of the degradation profile of the composite scaffold, which should match the regenerative rate of the new tissue.16,18,43
The synthetic process, consisting of the combination of phase inversion and salt leaching techniques, seems to meet such specific demands. Indeed, it enables the incorporation of HA solid signals without relevant complications during the preparation procedure, using solvents that do not alter the osteoconductive potential of the HA filler, as confirmed by Ca/P ratios close to that of natural HA (Fig. 3). From a structural point of view, a highly controlled porosity characterized by a bimodal distribution of pore size is achieved: an open macroporous network (Fig. 2A) that assures a uniform cell distribution and tissue regeneration, appears interconnected by micropores (Fig. 2B) that provide an efficient transport of soluble signaling molecules, as well as nutrients and oxygen, and metabolic waste removal. The employment of gravimetric measurements and IA (Fig. 1) guarantees a precise estimation of porosity features, embossing the effect of HA particle addition on the final macropore size. The presence of a network of interconnected pores is clearly remarkable by SEM images (Fig. 2). Previous work has just demonstrated the complete interconnection of pores in the proposed structures by mercury intrusion measurements (data reported elsewhere 20 ). Porosity data obtained via image analyses strictly fit previous experimental measurements and consequently confirm the presence of a percolative pore network independently on the HA particle amount, also proving the validity of imaging analysis as valid instrument for porosity calculation.
Noteworthy, the addition of HA particles was found to modify the polymer morphology; actually, the preliminary study of untreated samples evidenced that the presence of HA phase in the matrix drastically increases the Xc%, promoting the formation of more densely packed PCL crystalline phases within the composite (Table 1) with a lower amount of amorphous regions that are potentially more susceptible to hydrolytic attacks due to a better accessibility of the ester linkage.
To investigate the complex role of HA particles on the scaffold degradation, an experimental approach through the combined use of the vibrational Raman spectroscopy, TG, and DSC performed on untreated and treated scaffolds in different media (PBS, NaOH) has been employed. Slight weight losses have been measured after the treatment of the scaffolds in the low-aggressive medium (i.e., PBS) because of the very slow degradation kinetics of PCL, as previously found by Woodard et al. 44 ; however, DSC analysis showed that also in this medium the polymeric component underwent a certain chain fragmentation, in spite of the slight weight loss.
The employment of a more aggressive medium such as the NaOH solution enabled the resizing of the characteristic time window of degradation, thanks to the catalytic effect of the OH− ion that accelerates the degradation mechanisms of aliphatic polyesters.7–9,45,46 For pure PCL samples, the weight loss curve (Fig. 7A) is characterized by a drastic increase of lost mass during the first stage of degradation (7 days), followed by a noticeably decreased degradation rate according to trends reported in the literature for aliphatic polyesters.3,4,45,46 In the case of PCL/HA composites, degradation proceeds more slowly compared to pure PCL (Table 2), due to the higher values of Xc% (Table 1).
However, the behavior of PCL/HA 13% (Table 1) suggests that crystallinity cannot be the only parameter that determines the degradation kinetics of the composites. As a matter of fact, although the PCL/HA 13% composite was characterized by a Xc% quite similar to pure PCL (Table 1), the net weight loss percentage of the former after 56 days of degradation in NaOH solution was significantly lower than that of the latter under the same conditions, being 0.7% versus 15%, respectively (Table 2). These results lead to the conclusion that the HA component also plays a significant and complex role in the inhibition of degradation, in agreement with other authors.47,48
The staining test with the hydrophilic dye demonstrated that the addition of HA determines an overall increase of the scaffold hydrophilicity, which may promote the diffusion of the medium within the highly porous scaffold. 4 Meanwhile, a higher total surface/volume ratio of pores 20 certainly assures a wider exposure of the hydrophilic particles and, in consequence, an easier access of fluids to the internal porous structure. Therefore, the reduction of macropore size with the increase of the HA content, for the same porosity degree (Fig. 5), doubly contributes to promote the fluid penetration.
It is worthy of note that the increase of crystallinity of the polymer matrix in HA-loaded scaffolds hinders the degradation of the composites, preferentially deflecting the fluids at the polymer/ceramic interface, which are more susceptible to hydrolytic attack. As a consequence, as suggested by the TG data (Table 1), a more frequent escape of HA particles may occur preferentially, at longer times (56 days), with the creation of voids within the polymeric structure.13,14,17
In other words, the mismatch between the rough and net weight loss after 56 days in NaOH solution (Table 2) is explained by the effects of the chemical and structural properties of the scaffold, which combine to modulate HA particle removal during degradation process. However, some fluctuations of net weight loss values may be ascribed to the presence of clusters, which partially alter the decreasing trend with the increasing HA amount.
Regarding bioactivity (Fig. 7C), it has been demonstrated that the inclusion of HA solid signals into the polymer matrix catalyzes the apatite deposition on the surface in a quasi-physiological environment (SBF), with a gain of weight fraction of about 13–15% with respect to the starting amount, detected using thermogravimetric analysis (Table 1). In detail, the measured weight gain represents the sum of two opposite processes, that is, the weight loss due to the degradation of the PCL component and the weight gain due to apatite deposition. Between these two phenomena, the latter generally prevailed, leading to the conclusion that the effective apatite deposition should overcome the weight gain just due to the PCL weight loss. Since the contribution of the PCL component to the weight changes in the various composites is unknown, it is difficult to say which composite nucleated the highest amount of apatitic component upon treatment with SBF. Indeed, the highest weight gain recorded for the PCL/HA 26% sample does not demonstrate that this composite, per se, is the most bioactive, unless it can be demonstrated that the polymeric component in this sample is degraded to the same extent as in the other composites upon treatment with SBF. However, DSC data, reported in Table 1, demonstrated that Xc% and CF% showed a similar trend for the three composites, as a function of the immersion time in SBF, supporting the idea that the polymeric component in all composites should have not been degraded to different extents upon SBF treatment. As a consequence, it can be affirmed that PCL/HA 26% had the highest bioactivity, thanks to the highest HA content, which probably induces an increased calcium phosphate nucleating capability. From this point of view, it does not appear surprising that this sample was found the most degraded in cell cultures, as shown by the data reported in Table 3, supporting the idea that the amount of HA also affects the composite degradation in cell cultures. These results are in agreement with a previous study of our group, which demonstrated a more significant interaction of MSC with HA-loaded PCL substrates than PCL alone. 2
Conclusion
The effect of micrometric HA particles on structure and in vitro degradation of PCL-based highly porous scaffolds obtained via phase inversion and salt leaching technique was examined. The accurate qualitative and quantitative study of 3D architecture by SEM, supported by porosity measurements through different methods (weight measurement, IA), demonstrated only negligible effects on the structural porosity directly referable to the addition of ceramic particles. On the other hand, degradation appears strongly conditioned by the inclusion of micrometric HA, which alters the PCL crystallinity, inducing a shielding effect of the polymer matrix against degradation. Meanwhile, the slight reduction of pore size as a function of the increasing HA content, as well as the effective hydrophilicity of the scaffolds, also influences the degradation mechanisms. Further, the presence of HA as a filler into the polymeric matrix has been proven to enhance the scaffold bioactivity and HOB response. It is notable that Raman spectroscopy coupled to thermal analysis demonstrated that the PCL/HA 26% composite (i.e., the sample containing the highest amount of HA) was characterized by the highest bioactivity in SBF and the highest polymer degradation in HOB cultures, highlighting the active role of bioactive signals on the promotion of the surface mineralization and, as a consequence, on the cell–material interaction.
Footnotes
Acknowledgments
This work was supported by PRIN (Programmi di ricerca scientifica di rilevante interesse nazionale) (ex 40% grants) and FIRB (Composite scaffolds for bone tissue engineering– RBAU01N79B) from the Ministero dell'Università e della Ricerca and Rete nazionale di ricerca TISSUENET (n. RBPR05RSM2).
Disclosure Statement
No competing financial interests exist.