
Abstract
The bone marrow harbors multipotent mesenchymal stromal cells (MSCs) that nurture hematopoietic stem cells (HSCs). The extracellular matrix (ECM) is an integral part of the bone marrow, and the aim of this study was therefore to examine the effect of engineered ECM substrates on MSC gene expression over time and to determine quantitatively the functional ability of ECM-cultured MSCs to support HSCs. ECMs were surface immobilized using thin films of maleic anhydride to covalently immobilize tropocollagen or fibrillar collagen type I to the substrate. Where indicated, collagen type I fibrils were supplemented with heparin or hyaluronic acid. All surfaces maintained MSC viability and supported cell expansion. Microarray analysis of MSCs cultured on engineered ECM substrates revealed that culture time, as well as substrate composition, significantly affected expression levels. Based on these studies, it was possible to predict the effect of these substrates on in vitro HSC clonogenicity and self-renewal. The ability to regulate the expression of stromal factors using engineered ECM is exciting and warrants further studies to identify the ECM components and combinations that maximize the expansion of clonogenic HSCs.
Introduction
Because MSCs are key regulators of HSCs in vitro and vivo, 6 it is critical to maintain and maximize their supportive function. However, in vitro cultivation of MSCs on plasma-treated polystyrene (PTP) substrates (tissue culture plastic) has been associated with senescence and a loss of differentiation capacity,7–11 indicating that standard culture systems do not provide all the signals required for self-renewal and therefore do not fully mimic the in vivo situation. However, the provision of carefully selected ECMs (e.g., MSC-derived ECMs, PYS-2 cell-derived basement membrane, denatured collagen) delays replication senescence and maintains their differentiation potential.12–15 The use of in vitro–generated bone ECM has been shown to augment the osteogenic differentiation of MSCs, 16 which in part could be attributed to laminin-5, collagen type I, and vitronectin.17,18
Because ECM is an integral part of the bone marrow, 1 the aim of this study was to examine the effect of engineered ECM substrates on MSC gene expression over time, complemented by cell viability, proliferation, and differentiation studies, and to determine quantitatively the functional ability of ECM-cultured MSCs to support HSCs in co-cultures. Therefore, we prepared defined collagen type I–based ECM culture substrates supplemented with hyaluronic acid or heparin where indicated. Gene expression analysis of MSCs cultured on engineered ECM substrates revealed that culture time, as well as substrate composition, significantly affected expression levels. Based on these studies, it was possible to predict the effect of these substrates on in vitro HSC clonogenicity and self-renewal.
Materials and Methods
Engineered ECM substrates
Methods for surface immobilization of ECMs and their subsequent characterization have been described in detail elsewhere.19–21 Briefly, freshly oxidized glass coverslips were aminosilanized and spin-coated with a poly(octadece-alt-maleic anhydride) (POMA; molecular weight (MW) 30,000–50,000g/mol) 0.08 wt% tetrahydrofuran solution (Polysciences, Warrington, PA) to allow covalent bonding of thin polymer films (∼5 nm) to glass surfaces. Collagen fibrils were reconstituted from sterile, ice-cold, ultrapure (99.9%) bovine hide collagen in 0.01 M hydrochloric acid (Inamed Biomaterials, Fremont, CA) by mixing eight parts of the collagen stock with one part of 10-fold concentrated phosphate buffered saline (PBS) and one part of 100 mM sodium hydroxide on ice to yield a neutralized collagen solution (1.2 mg/mL). Where indicated, the collagen solution was supplemented with heparin (from porcine intestinal mucosa, MW 4000–6000 g/mol, Sigma-Aldrich, Munich, Germany) or hyaluronic acid (from human umbilical cord MW ∼750,000 g/mol, Sigma-Aldrich) (0.4 mg/mL). ECM preparations were applied to freshly annealed POMA-coated surfaces to achieve covalent bonding of the ECM to the polymer substrate. Reconstituted ECMs were allowed to fibrilize for approximately 24 h at 37°C. Substrates were then washed with PBS, and excess ECM was removed. Tropocollagen surfaces (reconstituted from a 0.01 M HCl solution pH 2.0 containing 0.1 mg/mL collagen) were prepared in a similar fashion but allowed to dry. The amount of immobilized ECM was quantified using high-performance liquid chromatography based on amino acid analysis after acidic hydrolysis, as described elsewhere. 22
Cell culture and treatments
Human bone marrow–derived MSCs were isolated from healthy female/male donors (aged 26–37) after obtaining their informed consent. The cells were expanded and characterized as described previously. 23 The institutional review board of the Medical Faculty at the University Hospital Dresden approved the study. Cells were cultured in low-glucose Dulbecco's modified Eagle medium (DMEM) supplemented with 10% fetal bovine serum (FBS), maintained in a humidified atmosphere of 5% carbon dioxide (CO2), and subcultured when cells were approximately 80% confluent. The experiments were performed with low-passage cells (<5) that were seeded at 10,000 cells/cm2, unless otherwise specified. For MSC-HSC co-culture experiments, an MSC feeder layer was established on the substrate and allowed to reach confluence over 3 to 6 days, followed by the addition of human CD34+ cells. The CD34+ cells were isolated from granulocyte-colony stimulating factor (G-CSF)-mobilized leukapheresis products obtained from healthy donors after informed consent. Hematopoietic progenitors in samples were positively selected for CD34 using magnetic affinity cell sorting (Miltenyi Biotec, Bergisch Gladbach, Germany) according to the manufacturer's instructions, as described in detail elsewhere. 24 The purity of the CD34+ population (92–99%) was verified using fluorescence-activated cell sorting (FACS). Freshly isolated CD34+ cells were cultured in long-term culture-initiating cell (LTCiC) medium (MyeloCult H5100, Stem Cell Technologies, Vancouver, Canada) that was added to established MSC feeder layers. Half the medium was replaced with fresh medium once a week.
Differentiation studies of MSCs
Studies were performed as detailed in the Supplementary Materials and Methods available online at www.liebertonline.com/ten.
Cobblestone area–forming cell assay
To quantify the frequencies of HSCs in cocultures, ECM substrates were prepared in maleic anhydride-functionalized microtiter plates. MSCs were plated in DMEM 10% FBS at a nominal density of 1.6 × 104/cm2 on the different substrates and allowed to establish themselves for 3 to 5 days to produce confluent stromal layers. In LTCiC medium, freshly isolated CD34+ cells were added to the stroma in a limited dilution format at six cell concentrations, ranging from 8 to 256 CD34+ cells, with 16 replicates per concentration. Half the medium in the plates was changed weekly, and each well was scored for the presence or absence of cobblestone area–forming cells (CAFCs) that were defined as five or more phase-dark cells underneath the feeder layer. 25 Progenitor frequencies were calculated using Poisson statistics with the software package L-Calc 1.1 (Stem Cell Technologies).
Colony-forming units in culture
ECM substrates were prepared on coverslips as detailed above, and cocultures were initiated as in the CAFC studies but using a single seeding density of 625 cells/cm2 of CD34+ cells. Half of the medium was replaced weekly with fresh medium, and colony-forming units in culture (CFU-Cs) were determined at 2 and 5 weeks. At the indicated time point, medium was aspirated; cultures were washed with PBS, digested for 25 min with collagenase (2 mg/mL in PBS) followed by trypsin–ethylenediaminetetraacetic acid (EDTA) for 5 min, and neutralized; and the cell pellet was resuspended in 1 mL of methylcellulose (MethoCult GF+, Stem Cell Technologies) and plated. Cultures were incubated at 37°C in 5% CO2, and the number of colonies was scored at day 14. For reference purposes, freshly isolated CD34+ cells were routinely subjected to the CFU-C assay and plated at 500 cells/mL in methylcellulose, cultured as described above, and scored.
Angiopoietin-1, CXCL12, and melanoma-associated antigen MUC18 quantification
MSCs were plated on the various substrates (1 × 104cells/cm2), and cytokines were quantified in medium conditioned for 3 to 14 days using an enzyme-linked immunosorbent assay (R&D Systems, Minneapolis, MN) according to the manufacturer's guidelines. Cytokine levels were normalized to correct for possible differences in cell numbers. Cell surface expression of melanoma cell adhesion molecule (MCAM, also known as CD146) was verified using FACS. In brief, cells were plated on respective substrates as detailed above and maintained in culture for 14 days. Cells were then harvested using collagenase and trypsin digestion as detailed above and subsequently incubated with MCAM–fluorescein isothiocyanate (FITC; R&D Systems) or the immunoglobulin G1-FITC isotype control for 30 min at 4°C in PBS containing 0.5% FBS. Cells were subsequently washed with PBS and immediately analyzed using FACS.
Ribonucleic acid microarrays and analysis of MSCs grown on substrates
Experiments were performed with second-passage cells from five different donors. For each donor, cells were grown on engineered ECM substrates, PTP, and two human cancellous bone chips (∼ 5 mm × 5 mm × 3 mm) (Deutsches Institut für Zell- und Gewebeersatz, Berlin, Germany). MSCs were applied to cancellous bone using a concentrated MSC suspension (∼1 × 105 cells in 50 μL), and cells were allowed to attach for approximately 30 min, followed by the addition of culture medium to cover the chip. For all other substrates, MSCs were plated at a density of 1 × 104 cells/cm2. Cells were collected on days 3, 6, and 14 by incubating them for 25 min in collagenase (2.0 mg/mL in PBS) followed by trypsin-EDTA for 5 min. Samples were subsequently washed with PBS plus 10% FBS, and the cell pellets from the five different donors cultured on the same substrates were pooled and stored at −80°C until ribonucleic acid extraction. Microarray studies were performed by examining the differential gene expression of time-matched PTP control samples with the respective ECM samples, as detailed in the Supplementary Materials and Methods section. The complete data set has been deposited in the (National Center for Biotechnology Information's Gene Expression Omnibus (GEO), Bethesda, MD) and can be accessed through the GEO series accession number GSE12635. To confirm the expression patterns observed using microarray analysis, 1 ng of complementary deoxyribonucleic acid from each sample served as a template and was subjected to quantitative real-time reverse transcriptase polymerase chain reaction (RT-PCR), as detailed elsewhere. 10
Electron microscopy
Scanning electron microscopy (SEM) was used to examine the growth of and interactions between MSCs grown on different substrates. At the indicated time points, samples were washed with PBS (pH 7.4), fixed for 1 h at room temperature with 2% glutaraldehyde (Serva, Heidelberg, Germany), rinsed with PBS, and dehydrated with a graded ethanol series. Next, samples were critical point dried (Bal-Tec CPD 030, Bal-Tec, Liechtenstein), sputtered with gold (Bal-Tec SCD 050), and examined using an SEM microscope (XL 30 ESMEM FEG, Philips, Amsterdam, The Netherlands). For the examination of CAFCs using SEM, samples were fixed as described above, stained with osmium oxide (1% in PBS) overnight at 4°C, dehydrated, and subjected to critical point drying. Samples where sputtered with a thin layer of carbon (∼20 nm) and examined using SEM at low electron acceleration (1 kV) to visualize surface structures and at 25 kV for deeper sample penetration due to the Kanaya-Okayama effect. 26
Cell viability
Cell viability was determined 48 h after plating cells at a seeding density of 1 × 104 cells/cm2 on the various culture substrates, using fluorescein diacetate (FDA) and propidium iodide (PI) to stain viable and dead cells, respectively. Solutions of 100 μg/mL of FDA and PI were prepared by dissolving the former in acetone and the latter in PBS (pH 7.4). A fresh working stock solution containing 100 ng/mL of FDA and 2 μg/mL of PI was prepared in PBS that was added directly to the cell cultures (i.e. no change of culture medium) at a 1:1 volume ratio. Samples were incubated for approximately 2 min and visualized using epi-fluorescent microscopy (Leica DM IRE2, Leica Microsystems, Wetzlar, Germany) with excitation filters at 490/20 nm and 555/28 nm and emission at 527/30 nm and 600/20 nm for fluorescein and PI fluorescence, respectively. Images were acquired using Openlab software 4.0.4 (Improvision, Coventry, UK) and subsequently processed in Image J 1.38x (National Institutes of Health, Bethesda, MD).
Statistical analyses
Data were analyzed using GraphPad Instat 3.06 (GraphPad Software, La Jolla, CA) and the unpaired Student t-test for intersample comparison. Multiple samples were evaluated using one-way analysis of variance (ANOVA) followed by Tukey's and Dunette's post hoc tests to evaluate the statistical differences (p ≤ 0.05) between all samples and between samples and controls, respectively.
Results
Cellular interactions, growth, and differentiation on engineered ECM substrates
Reproducible and defined ECM substrates were generated and characterized using amino acid analysis and SEM (Fig. 1 and Supplementary Table 1 available online at www.liebertonline.com/ten). Qualitative SEM studies demonstrated that MSCs were able to attach and proliferate on all studied ECM substrates, although remodelling of the ECM appeared to be restricted to fibrillar collagen-based and collagen–hyaluronic acid–based surface preparations (Fig. 1). Quantitative analysis demonstrated that all substrates supported cell proliferation and that, in particular, the collagen–hyaluronic acid substrate was consistently better than PTP (Fig. 2A). No major differences in cell viability were observed for MSCs grown on the different surfaces (Fig. 2B). Next, MSCs cultured on engineered ECMs were studied for their ability to differentiated into mature osteoblasts (Supplementary Fig. 1A available online at www.liebertonline.com/ten). Cells stimulated with osteogenic induction medium showed significantly greater alkaline phosphatase (ALP) activity than PTP control samples (Supplementary Fig. 1A). In addition, MSCs cultured on engineered ECM substrates displayed more mineralization than control PTP samples (Supplementary Fig. 1B). MSC cultures were also tested for their ability to support adipogenic differentiation. Stimulated cultures responded with an increase in glycerol-3-phosphate dehydrogenase activity and the formation of fat vacuoles, although lipid accumulation was most prominent in the control PTP samples (Supplementary Fig. 1C, D).
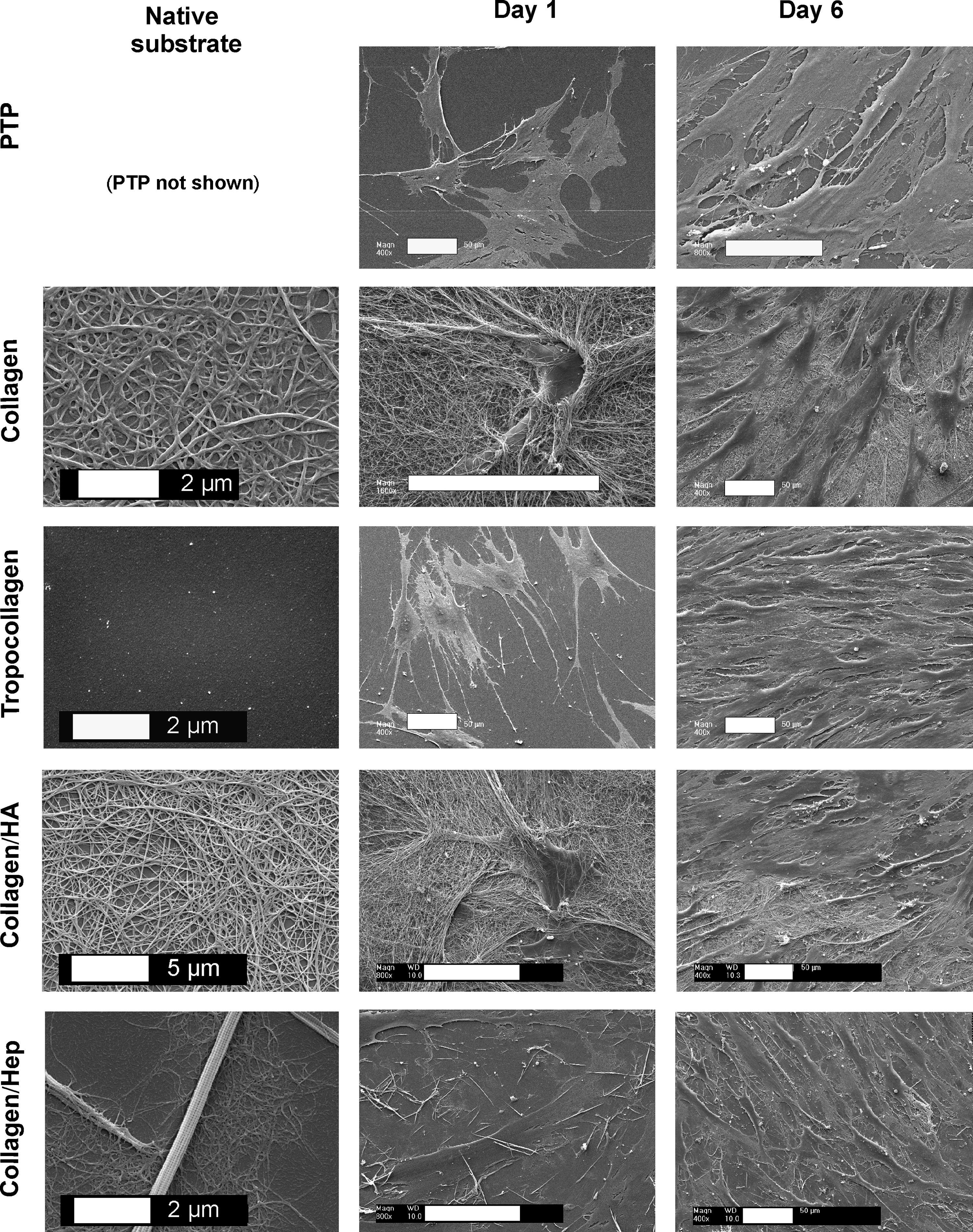
Engineered extracellular matrix (ECM) substrates support cellular interactions, remodelling, and growth. Scanning electron microscopy (SEM) images of native substrates and mesenchymal stem cells (MSCs) seeded on ECM substrates. MSCs were seeded (5 × 103 cells/cm2) on ECM preparations and examined using SEM at days 1 and 6. PTP, plasma-treated polystyrene; collagen, fibrillar collagen type I; tropocollagen, non-fibrillar collagen type I; collagen/Hep, fibrillar collagen type I supplemented with heparin; collagen/HA, fibrillar collagen type I supplemented with hyaluronic acid. Scale bars 50 μm unless specified otherwise.
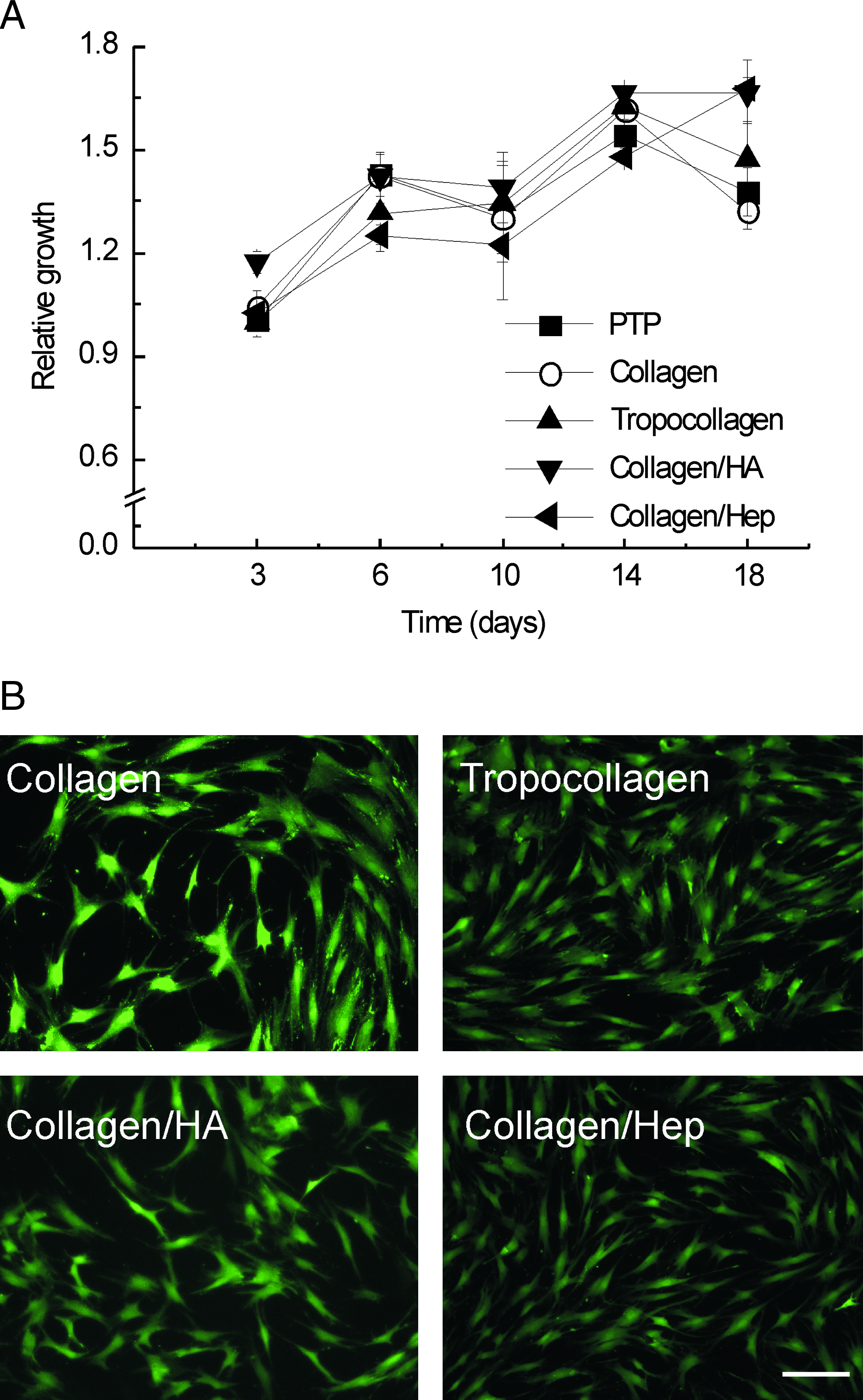
Engineered ECM substrates support the expansion of MSCs. (
Gene and protein expression studies of MSCs cultured on different substrates
MSCs cultured on engineered ECM substrates, cancellous bone, and PTP for 3, 6, and 14 days were subjected to differential gene expression analysis. This was achieved by comparing those samples with the time-matched PTP control group. First, the overall expression profiles of all data sets were compared using correlation analysis (Fig. 3A). Three main clusters were visible in the correlation matrix (Fig. 3A): one containing most samples cultured for 3 days, another with most samples cultured for 6 days, and a third containing samples cultured for 14 days and the samples cultured on cancellous bone (Fig. 3A). Gene lists were then subjected to hierarchical cluster analysis employing Euclidean distance metric and average linkage clustering. The trends observed in these heat maps were comparable with those seen using the interexperiment correlation analysis; marked differences in gene expression occurred in MSCs cultured for 3, 6, and 14 days; cancellous bone-specific cellular responses also occurred (data not shown). Using two-way ANOVA, statistical testing was performed to identify genes whose expression was changed significantly as a consequence of the time in culture or as a result of the culture substrate (identified genes, e.g., angiopoietin-1, MCAM, CXCL-12, secreted phosphoprotein 1, proteoglycan decorin) (Fig. 3B, C). The discriminatory genes identified were then assigned to functional categories based on the Gene Ontology classification (Fig. 3D). The expression of genes regulating development, receptor signalling, cell differentiation, proliferation, and cell adhesion were changed most. (A complete list of the genes in the respective categories is given in Supplementary Tables 2 and 3 available online at www.liebertonline.com/ten.) A number of differentially regulated genes were verified using real-time RT-PCR, confirming the trends observed using microarray analysis (Supplementary Fig. 2 available online at www.liebertonline.com/ten). The analysis of cell surface–associated MCAM in addition to angiopoietin-1 and CXCL-12 levels in MSC-conditioned culture medium derived from cells cultured on the various substrates demonstrated the good correlation between gene expression and protein expression (Fig. 4). Although donor-dependent differences in angiopoietin-1 levels were observed, the overall trends were comparable, with higher angiopoietin-1 levels on the control PTP substrate than on the ECM preparations at the corresponding time points (Fig. 4A). Cumulative angiopoietin-1 levels increased with time in culture on all substrates, with similar trends observed for CXCL-12 (Fig. 4A). However, for CXCL-12 there were no statistically significant differences between the control PTP substrate and the other ECM substrates at any of the corresponding time points. MCAM surface expression at day 14 indicated that MSCs cultured on the control PTP substrate expressed the highest percentage of cells that were positive for MCAM (27%), followed by tropocollagen (23%), collagen-hyaluronic acid (21%), collagen (20%), and collagen-heparin (15%) (Fig. 4B).

Engineered ECM substrates modulate the expression pattern of MSCs. Cells were plated on substrates, cultivated under cell culture conditions, and harvested at days 3 through 14. (
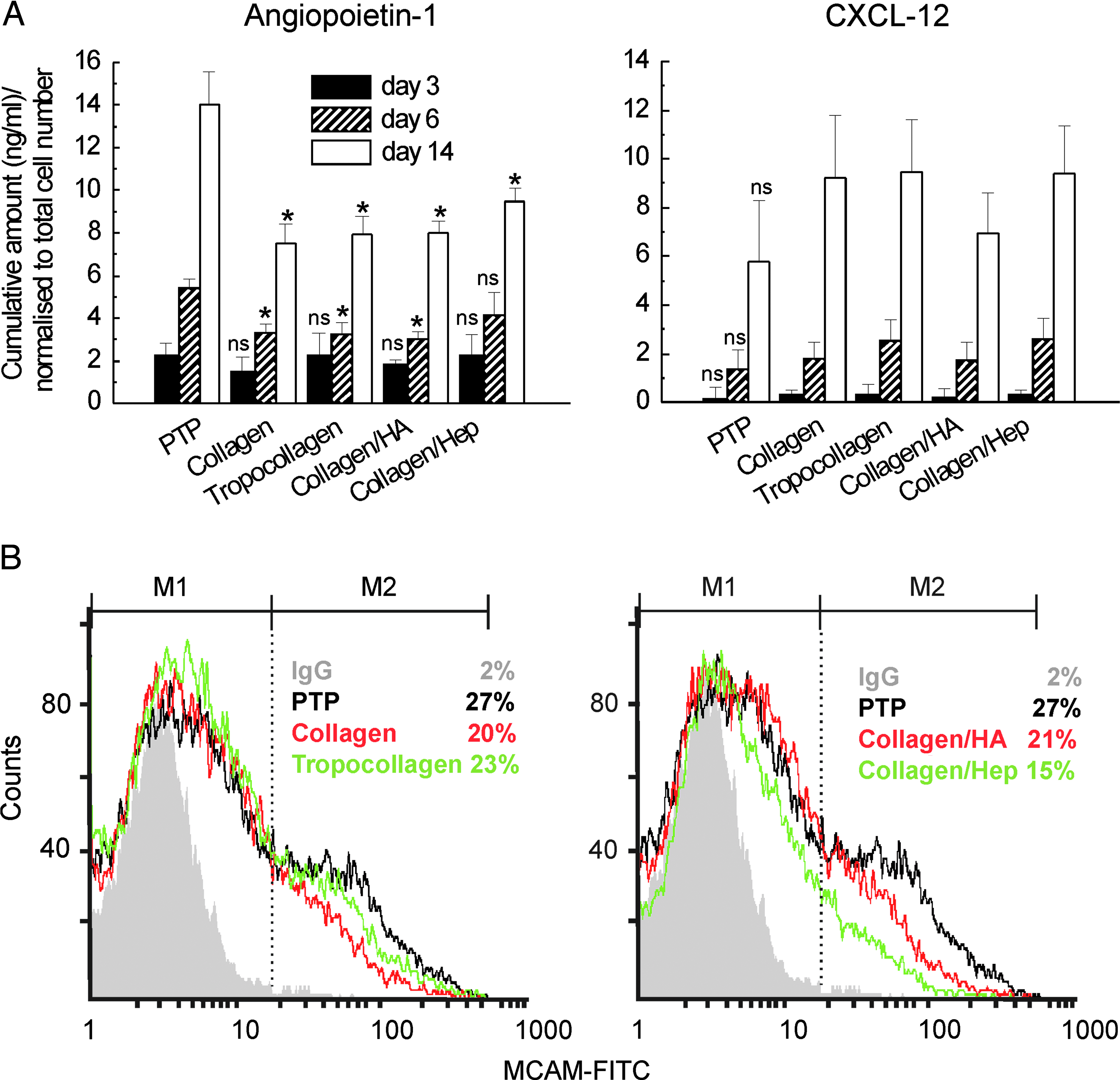
Substrate-directed expression profiles for angiopoietin-1, CXCL-12, and melanoma cell adhesion molecule (MCAM) in MSCs. (
Functional effects of substrates on MSC-CD34+ cells in cocultures
The functional effects of ECM substrates on the self-renewal and commitment of HSCs in MSC coculture experiments was examined quantitatively by measuring CAFC frequencies and CFU-Cs. The phase-contrast image of a typical cobblestone and the corresponding SEM images of the same sample demonstrated the location of CAFCs underneath the MSC feeder layer (Fig. 5A). CAFC assays over the first 2 to 3 weeks showed that all substrates yielded similar CAFC frequencies, although fibrillar collagen layers appeared to result in the highest yields of all of the ECM surface preparations (Fig. 5B). However, the PTP control substrate was marginally more effective than collagen. From week 4 onwards, PTP outperformed all ECM substrates, which supported the expansion of CAFCs for up to 6 weeks. ECM-based surface preparations supported the expansion of CAFCs for up to 4 weeks, followed by a decline and subsequent disappearance of CAFC colonies at week 6 to 7 (Fig. 5B). Comparable trends have been observed with a number of MSC donors and CD34+ cell preparations, albeit with some variation in the frequency of CAFCs. Similar trends were observed with unpurified leukapheresis products instead of CD34+ -enriched HSCs (data not shown). For studies employing unpurified leukapheresis products, the plating density of the mononuclear cells was 800 to 2.56 × 104.
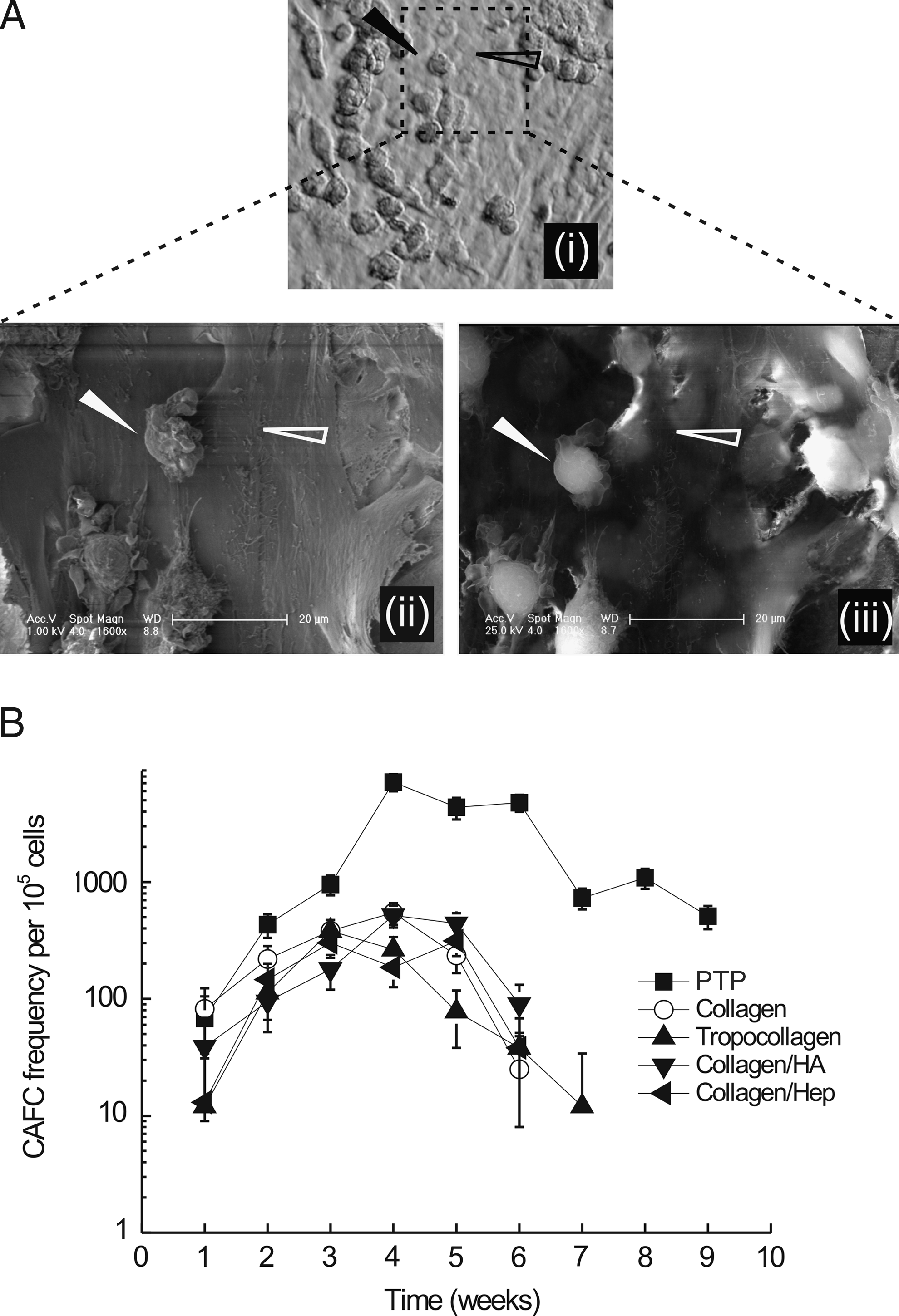
Substrate-directed maintenance of hematopoietic stem cells (HSCs) in long-term initiating cultures. (
The effect of the ECM substrates on HSC commitment was assessed by counting CFU-Cs (Fig. 6). The number of CFU-Cs from freshly isolated CD34+ cells was higher than for any other treatment group and encompassed the entire spectrum of CFU-Cs. At week 2, the number of erythroid colonies (CFU-Es) was substantially greater for cells cultured on collagen and collagen-heparin than on PTP (Fig. 6). CFU-E levels on the other ECM substrates were comparable with those on controls. Results obtained for granulocyte progenitors were similar to those observed for CFU-Es, despite greater scattering of the data. At week 5, all ECM substrates showed lower CFU-C numbers than PTP controls, despite substantial scattering in the control data sets (Fig. 6).
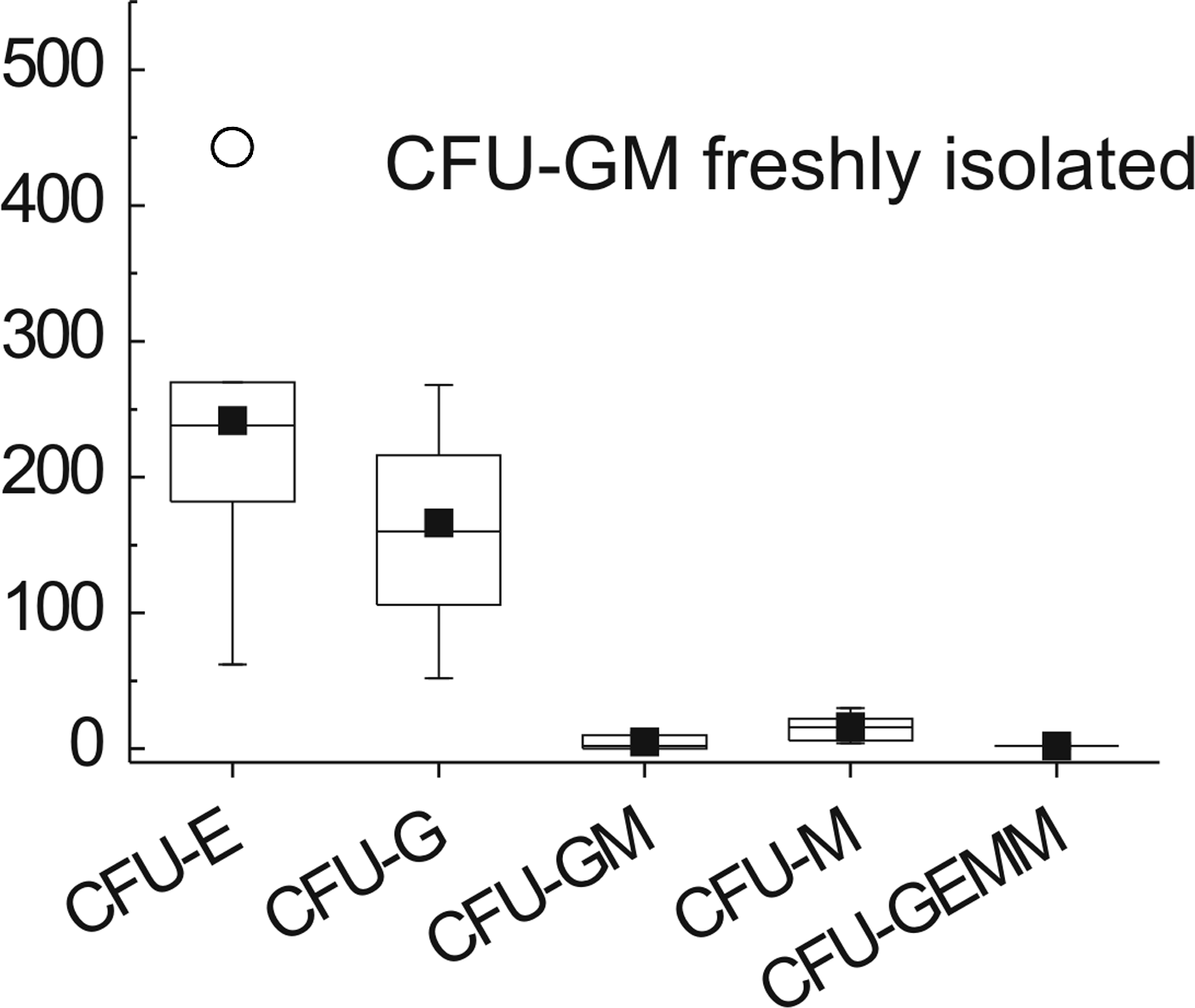
Substrate-directed colony-forming units (CFUs). Cocultures were initiated on substrates and terminated at 2 and 5 weeks, and CFUs were quantified by plating cells in methylcellulose. The edges of the rectangles, which contain 50% of the data, represent the quartiles of the box plots. Horizontal lines and squares within the boxes depict medians and means, respectively; circles represent outliers, and error bars represent the SD (n = 6 of two independent experiments). CFU: erythrocyte (E); granulocyte (G); granulocyte, macrophage (GM); granulocyte, erythrocyte, macrophage, megakaryocyte (GEMM). Statistical differences between PTP and samples were determined using one-way ANOVA and Dunnett's multiple comparison post hoc test (*p < 0.05). Substrates are denoted as in Figure 1.
Discussion
A number of studies have assessed the gene expression profiles of fetal and adult MSCs derived from bone marrow, umbilical cord blood, amniotic fluid and membranes, and adipose tissue, in addition to the expression profiles of their differentiated progeny.27–39 Although the culture microenvironment is predicted to affect cell behavior, no studies have systematically studied the effect of engineered ECMs on MSC gene expression and correlated these with their ability to support HSC clonogenicity. The ECM components examined here are found in the in vivo bone marrow microenvironment (reviewed in 40–42 ) and have also been identified by strategies aimed at characterizing the molecular signature of the HSC environment.43,44 Thus a small library of well-defined ECM substrates was generated that supported cell viability and expansion and MSC differentiation into adipocytes and osteoblasts. In particular, the enhanced osteogenic differentiation observed for cells cultured on engineered ECM substrates warranted studies to examine the ability of those systems to support MSC-HSC cocultures because osteoblast-like cells have been identified as an important regulator of HSCs in vivo. 1 Because growth and differentiation studies provide little insight into the molecular dynamics of cultured MSCs, we examined the gene expression profile of MSCs over time. Human cancellous bone was included as a reference material, because cancellous bone implanted at heterotopic sites is known to induce the formation of a hematopoietic microenvironment of recipient origin. 6
Gene expression differences between MSCs cultured on PTP and the other culture substrates were most striking for cancellous bone, because time in culture appeared to be of secondary importance. Statistical analysis of the expression data in relation to the culture substrate (i.e. cancellous bone and engineered ECM substrates versus PTP) revealed a number of differences; for example, MCAM and angiopoietin-1 were significantly downregulated during the entire study period, whereas CXCL-12 appeared to be downregulated initially and similar to control thereafter. These factors are critical components of the bone marrow microenvironment. 1 For instance, CXCL-12 produced by bone marrow stromal cells not only acts as a chemoattractant for HSCs, but also regulates the quiescence of HSCs.45,46 The angiopoietin-1/Tie-2 signalling axis between MSCs and HSCs facilitates the close association between HSCs and the bone marrow stroma, thereby contributing to stem cell quiescence in vitro and in vivo. 47 The stromal cell population that express angiopoietin-1 also appears to express high levels of MCAM. It has only recently been discovered that MCAM provides optimal support for HSCs in vitro and in vivo.48,49 Although it is not clear through which mechanism(s) those engineered culture substrates regulate those critical components of the marrow microenvironment; culture systems with low angiopoietin-1 and MCAM levels would be expected to support HSC differentiation.
Although only a limited number of the factors regulating the HSC-MSC-signalling synapse 1 have been discussed, culture substrates were able to regulate many of these factors. Because many of the genes involved in HSC self-renewal were downregulated when MSCs were cultured on engineered ECM substrates, it might be supposed that those substrates would be less able than PTP surfaces to support the long-term culture of HSCs. Quantitative analysis of the fate of HSCs was accomplished using the cobblestone assay. The assay closely resembles the in vivo situation with respect to early and late HSC engraftment, with the latter corresponding to long-term in vivo repopulating HSCs. 25 CAFC frequencies determined in this study were similar to those in previously published reports for human HSCs and displayed a typical parabolic profile.25,50,51 As predicted from the expression data, PTP outperformed engineered ECM substrates from week 3 onwards. In line with the CAFC frequencies, granulocyte-macrophage progenitors determined at week 5 indicated that the ECM-based culture system had exhausted the hematopoietic progenitor pool and yielded significantly fewer colonies than controls. However, none of the culture substrates maintained the initial clonogenic potential of HSCs, indicating that current strategies are still open to improvement.
Complex mixtures of bone marrow–derived ECMs have been reported to enhance the outgrowth of marrow stromal cells and the expansion of HSCs. 52 However, these effects appeared to depend critically on the origin of the ECM, with those derived from the Engelbreth-Holm-Swarm tumor (i.e., Matrigel) or endothelial cells not supporting long-term bone marrow cultures.52–54 The supportive function of marrow-derived ECMs could in part be attributed to the presence of hemonectin, which is found in bone marrow–derived ECM but not in Matrigel. 55 Although the engineered ECM substrates used here involved major constituents of the bone marrow matrix, the results suggest that it is necessary to identify and select ECM components and combinations carefully to enhance the feeder properties of MSCs effectively. Accordingly, it was noticed that colony size, morphology, and progenitor spectrum (CFU-E/G/granulocyte, macrophage/granulocyte, erythrocyte, macrophage, megakaryocyte) were much greater on cancellous bone than on the other substrates (data not shown), suggesting that analysis of the ECM structures contained in such bone should instruct the design of future culture substrates.
In conclusion, time-resolved expression profiles of human MSCs cultured on engineered ECM culture substrates have been generated. They provide insights important for the future design of optimized culture substrates for MSCs and MSC-HSC cocultures. The ability to regulate the expression of stromal factors using reconstituted ECM is exciting and should facilitate the engineering of an improved in vitro bone marrow microenvironment.
Footnotes
Acknowledgments
We would like to thank Prof. Katrin Salchert and Prof. Terry D. Allan for helpful discussions of ECM substrates and CAFC SEMs, respectively. Dr. Armin Springer and Tina Lenk are gratefully acknowledged for performing carbon sputtering of SEM samples and high-performance liquid chromatography analysis, respectively. This work was supported by the Deutsche Forschungsgemeinschaft, “Collaborative Research Centre: Cells into tissues— stem cell and progenitor commitment and interactions during tissue formation” (SFB 655, Dresden, FPS, CW and MB).
Disclosure Statement
The authors have no competing financial interests.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.