
Abstract
Purpose:
Currently, bladder repair is performed using gastrointestinal segments; however, this technique has a high morbidity rate, and new alternatives are thus needed. The lack of native or synthetic tissue with similar properties of the bladder led us to develop autologous vesical substitutes entirely made by tissue engineering and without exogenous matrices. Watertight function and mechanical resistance are fundamental for the model. The aim of this study was to determine the structural and functional characteristics of our vesical equivalent (VE).
Materials and Methods:
Porcine VEs are produced in 55 days. The cellular types that make up the vesical wall are extracted and purified simultaneously from a small porcine bladder biopsy. Dermal fibroblasts are extracted and cultured in vitro to form cellular sheets. Endothelial cells were seeded on the fibroblast sheets before their superimposition. Urothelial cells are then seeded onto this cellular construction. VEs are characterized by histology, immunostaining, electron microscopy, and cell viability. Mechanical properties of the reconstructed substitutes are evaluated by uniaxial tensile tests, and tissue absorption is verified with 14C-urea, which quantifies the degree of impermeability.
Results:
This process allowed us to obtain a highly structured tissue with a total fusion of the fibroblast layers. As expected, histological observations showed a pseudostratification of the urothelium developing on an organized self-secreted extracellular matrix. Positive markers for cytokeratin 8/18 in immunostaining confirmed the presence of a urinary epithelium. Electron microscopy confirmed the normal aspect of urothelial cells. Our VE's permeability to 14C-urea was significantly similar to porcine bladder, and characterization of the mechanical properties indicated that our tissue could be suitable for grafting since its ultimate tensile strength compares favorably with a native porcine bladder.
Conclusion:
The construction of a VE using this method seems very promising in meeting the needs in the urological field. Our substitute has proven its efficiency as a barrier to urea and has a sufficient mechanical resistance to support suturing. Additionally, this model is completely autologous, and its possible endothelialization could promote the early vascularization process after grafting and thus significantly reducing inflammation and possible rejection.
Introduction
That is why we investigated the feasibility of constructing a tissue-engineered vesical equivalent (VE) using the self-assembly method developed at our institution. The interest of this approach was to elaborate substitutes made entirely of autologous cells with their own cell-produced extracellular matrix. It has already been used effectively in skin and blood vessel reconstruction. 6 Tissue-engineered skin is used in clinic and has a significant impact on the treatment of burn wounds and cutaneous ulcers. Therefore, we were inspired by the reconstructed skin model to develop our VE. Also, a rapid inosculation process was demonstrated between the capillary-like network of in vitro–engineered endothelialized skin substitutes and host's vasculature.7,8 On the basis of these results, we hypothesized that adding endothelial cells (ECs) to our model would accelerate the vascularization of the VE, and thus help to prevent necrosis and contraction of the graft after implantation.
Therefore, this technique allowed us to create a three-dimensional vesical graft with a minimal invasive preoperative phase. We present the characterization of the produced bladder-wall equivalents. Our results seem very promising to overcome the multiple complications associated with bladder replacement.
Materials and Methods
Extraction of fibroblasts, urothelial cells, and ECs
Fibroblasts were isolated from skin biopsies as previously described.9,10 The skin specimen was washed in phosphate-buffered saline containing 100 U/mL penicillin, 25 μg/mL gentamicin (Sigma, Oakville, Canada), and 0.5 μg/mL Fungizone (Bristol-Myers Squibb, Montreal, Canada). It was then incubated at 4°C overnight with 10 mL of a solution containing 500 μg/mL thermolysin (Sigma) in a HEPES 1 × buffer with 1 mM CaCl2, pH 7.4. The dermis was manually separated from the epidermis and put in a trypzination unit with 0.125 U/mL collagenase H (Roche Diagnostics Canada, Montréal, Canada) diluted in Dulbecco–Vogt modification of Eagle's medium (DMEM) containing 10% fetal bovine serum (Hyclone, Logan, UT), 100 U/mL penicillin, and 25 μg/mL gentamicin (fibroblast medium). Afterward, the dermis was incubated for 3 h at 37°C to isolate the fibroblasts. Fibroblasts were redistributed at 6 × 104 cells/cm2 in culture flasks with a fibroblast medium. Finally, they were placed in a 37°C humidified incubator containing 8% CO2. The medium was changed three times a week.
Urothelial cells (UCs) were extracted from a small porcine bladder biopsy (5 × 1 cm) as previously described. 11 UCs were seeded at a density of 5 × 105 cells in 75-cm2 culture flasks with 1.5 × 105 irradiated murine fibroblasts as a feeding layer. The cells were cultured in a fresh urothelial medium, DMEM-Ham containing 10% fetal bovine serum (Hyclone), 5 μg/mL insulin (Sigma), 0.4 μg/mL hydrocortisone (Calbiochem, San Diego, CA), 10−10 M cholera toxin (ICN, St-Laurent, Canada), 10 (g/mL epidermal growth factor (Austral Biologicals, San Ramon, CA), 100 U/mL penicillin, and 25 μg/mL gentamicin. They were placed in a 37°C humidified incubator containing 8% CO2. The medium was changed three times a week.
ECs were extracted from the same small bladder biopsy as previously described. 11 The ECs were seeded at a density of 5 × 105 cells in 75-cm2 culture flasks. The cells were cultured in the EGM-2 MV SingleQuots medium (Clonetics, Walkersville, MD) (EC medium). They were placed in a 37°C humidified incubator containing 8% CO2. The medium was also changed three times a week.
Production of the VE
Cultured fibroblasts at passage five were seeded at a concentration of 3 × 104 cells/cm2 on tissue culture dishes and cultured in the fibroblast medium supplemented with 50 μg/mL ascorbate (Sigma) for 20 days to form a sheet. 12 Three fibroblast sheets were superimposed and cultured for four additional days to allow sheets' adhesion. Cultured UCs at passage three were seeded on top of the fibroblast construct at a concentration of 3 × 104 cells/cm2. The seeded equivalents were cultured in a complete urothelial medium supplemented with 50 μg/mL ascorbate for 7 days under submerged conditions. VEs were then elevated at the air–liquid interface for 20 days. The urothelial medium containing ascorbate was used for the air–liquid culture phase.
VEs were compared with endothelialized VEs. Preparation was the same except that cultured ECs at passage five were seeded on the fibroblast sheets at a concentration of 3 × 104 cells/cm2, 1 day before superimposition of the sheets. Endothelialized VEs were cultured in a 1:1 mix of the EC medium and fibroblast medium with 50 μg/mL ascorbate after superimposition and in a 1:1 mix of the EC medium and urothelial medium supplemented with 50 μg/mL ascorbate for submerged and air–liquid conditions. Moreover, VEs were compared with VEs cultured with 1 mM CaCl2 for submerged and air–liquid conditions to evaluate the role of calcium in the UC stratification. All media were changed three times a week.
Histology and immunofluorescence
Sections of each sample were fixed in 1 × Histochoice tissue fixative (Amresco, Solon, OH) and embedded in paraffin. Histological sections of 5 μm were cut and stained using Masson's Trichrome. To perform immunofluorescence, parts of the samples were embedded in frozen tissue medium (OCT compound; Tissue-Tek, Bayer, Etobicoke, Canada). Sections of 5 μm of frozen VEs were cut and then fixed in 100% methanol, blocked in phosphate-buffered saline containing 1% (w/v) bovine serum albumin (Sigma), and incubated with primary antibodies. The primary antibodies used were a rabbit polyclonal IgG anti-human collagen I (dilution 1:100; Cedarlane Laboratories, ON, Canada) to stain the extracellular matrix produced by the fibroblasts, a rabbit anti-laminin (dilution 1:400; Sigma-Aldrich, ON, Canada) to delimit the basilar membrane (lamina propria), a guinea pig IgG anti-keratin 8/18 (dilution 1:1000; ARP, Belmont, MA) to identify UCs, and a mouse monoclonal IgG anti-rat pecam1 (dilution 1:600; Chemicon) to see ECs' adhesion. The secondary antibodies used were coupled with rhodamine or Alexa 488 fluorochrome. For controls, the primary antibodies were omitted. Nuclei were stained with Hoechst nuclear dye. The specimens were then examined with a Nikon Eclipse E600 epifluorescence microscope (Nikon, Tokyo, Japan), and images were processed with Adobe® Photoshop® 6.0 (Adobe Systems, San Jose, CA).
Immunoblotting
The Western blot was obtained through previously described methods. 13 Briefly, samples were mechanically dissociated in liquid nitrogen. Cell extracts were analyzed using spectrophotometry. Samples were boiled about 10 min to denature them. The electrophoresis of the samples (20 μg/lane) was done on 10% sodium dodecyl sulfate–polyacrylamide gels and transferred to polyvinylidene flouride membrane (Millipore, Tokyo, Japan). Membranes were blocked with tris-buffered saline–Tween 0.05% containing 5% skimmed milk and incubated with a 1:2000 dilution of the same IgG anti-keratin 8/18 (ARP, Belmont, MA) antibody used for the immunofluorescence. Then, membranes were treated with peroxidase-conjugated anti-mouse IgG-IgM, and signals were detected using ECL Western blotting detection reagents (GE Healthcare, Buckinghamshire, United Kingdom) and exposing the blot to an X-ray film in a darkroom. As control, membranes were blotted with an anti-alpha-tubulin antibody.
Transmission and scanning electron microscopy
Samples of VEs were fixed with 2.5% glutaraldehyde in 0.1 M cacodylate buffer (pH 7.4) at 4°C. Then, they were rinsed with cacodylate buffer and a postfixation in 1% osmium tetroxide. 14 The biopsies for transmission electron microscopy (TEM) were stained with uranyl acetate, dehydrated through a graded series of ethanol, and then embedded in Epon (Polysciences, Warrington, PA). The VEs were cut in ultra-fine sections and counterstained with lead citrate and uranyl acetate. The sections were examined with a JEM 1230 (Tokyo, Japan). The biopsies for scanning electron microscopy (SEM) were dehydrated and then critical-point dried. Samples were spattered with gold and viewed with a JEOL JSM-63060LV (Tokyo, Japan).
Permeability studies
Tissue absorption was evaluated using 0.64 cm2 standard Franz diffusion cells having donor and receiver chambers.14,15 The receiver chamber temperature was maintained at 37°C with a water jacket. Receiver chambers were filled with DMEM and stirred continuously using a magnetic stir bar. The VEs were placed between the donor and receiver chambers (urothelium facing the donor chamber), and 2.5 μCi of 14 C-urea (55 mCi/mmol; MP Biomedicals, Irvine, CA) diluted in DMEM was deposited inside the donor compartment. To evaluate the permeation rate, the bathing solution in the receiver compartment was completely removed and replaced by a fresh medium at selected time intervals (1, 2, 4, 6, 8, and 24 h). The radioactivity present in the samples was determined after digestion with NCSII (Amersham, Oakville, Canada). Scintillation fluid (Scintisafe 30%; Fischer Scientific, Fair Lawn, NJ) was added to a fraction of every sample, and the radioactivity determined with a scintillation counter (Beckman LS 6000 SC; Beckmann, Fullerton, CA). Native porcine bladders were compared to fibroblast constructions, with or without exogenous calcium, VEs at 20 days of air–liquid culture with or without calcium, and endothelialized VEs.
Cell viability
The cell viability was performed by the two same methods previously described. 13 First, the cells were extracted from reconstructed tissues with the same protocols for the fibroblast, UC, and EC extraction from the initial biopsy. In addition, the explant method was used by placing small pieces of the VE in a culture dish with DMEM. For both methods, the medium was changed three times a week, until confluency was obtained, in a 37°C humidified incubator containing 8% of CO2.
Mechanical testing
Mechanical properties of the tissue-engineered VE and endothelialized VE were measured by uniaxial tensile testing using a mechanical tester (Tytron 250; MTS Systems Corporation, Minneapolis, MN). Bone shape specimens were cut in the substitutes with an in-house-designed stainless steel punch. Both extremities were fixed with anchoring jaws, allowing for the deformation of the tissues at a constant rate of 0.2 mm/s. The stress in the samples (σ = F/S0) was obtained by dividing the force recorded by the load cell by the initial cross-sectional area of the engineered tissue. Thickness of the samples was measured on histological cuts using the ImageJ software (NIH, Bethesda, MD). Cross-sectional area was obtained from the product of the thickness and width of the specimen. The ultimate tensile strength (UTS) was defined as the highest stress recorded by the load cell before failure of the sample. Engineering strain (ɛ = [L − L0]/L0) was determined as the recorded length was divided by the initial length of the sample. The modulus (E = [σ2 − σ1]/[ɛ2 − ɛ1]) was defined as the ratio of stress over the strain and was calculated from the linear slope of the stress–strain curve for each sample. Failure strain was established as the level of deformation corresponding to the UTS. Three distinct samples were tested for both the VE and the endothelialized VE. Data were analyzed using Minitab® (Minitab, State College, PA). A two-sample t-test was used for statistical comparison of the mechanical properties of the engineered tissues. Results are presented as mean ± standard deviation, with p < 0.05 accepted as significant.
Results
Macroscopic aspect
During construction, we already could note the easiness to handle our model (Fig. 1). This preliminary observation indicated the capacity of our VE to support manipulation and sutures for grafting. The surface seemed to be homogeneous, which allowed us to believe in the uniformity of our results in any area of the substitutes.
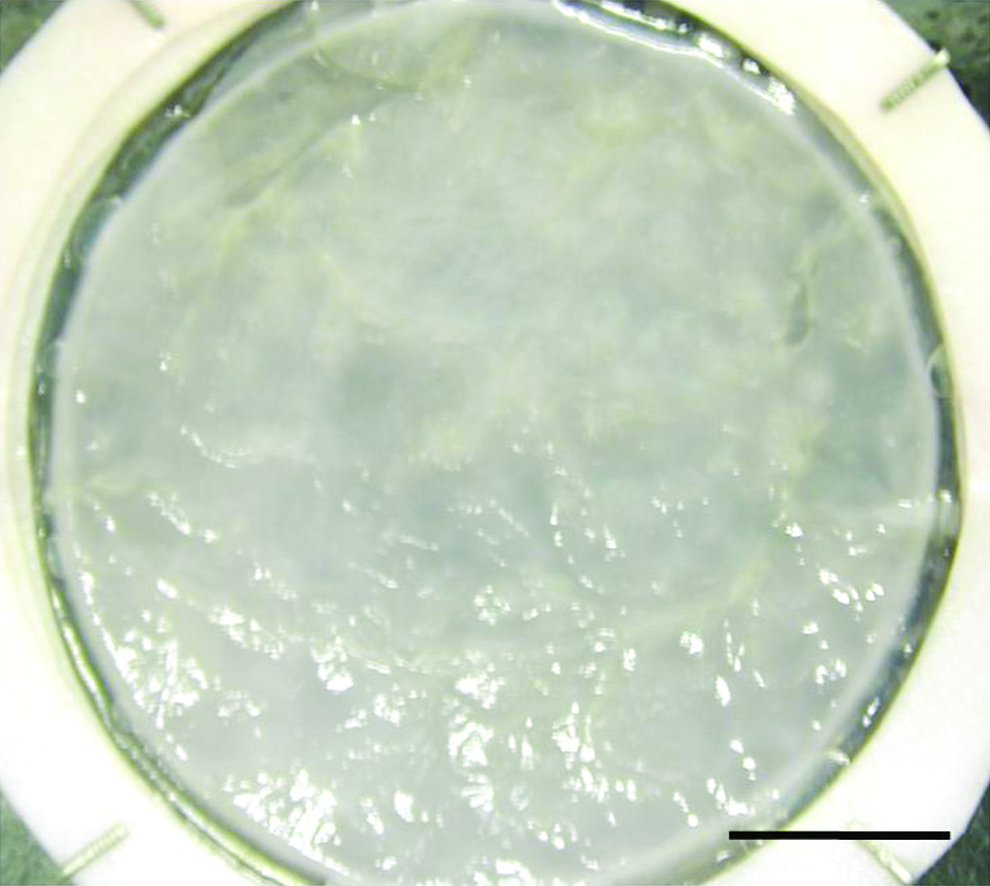
Macroscopic view. VE after 20 days of culture at the air–liquid interface (bar: 1 cm). VE, vesical equivalent. Color images available online at www.liebertonline.com/ten.
Histology
The cellular morphology appeared to be characteristic of normal bladder urothelium (Fig. 2A–D). A well-organized extracellular matrix formed by the dermal fibroblast sheets having evenly fused was observed in the VE. UCs were clearly identified and appeared as a stratified epithelium containing three to six layers of cells. It was noted that the VE cultivated with calcium had less UC layers.
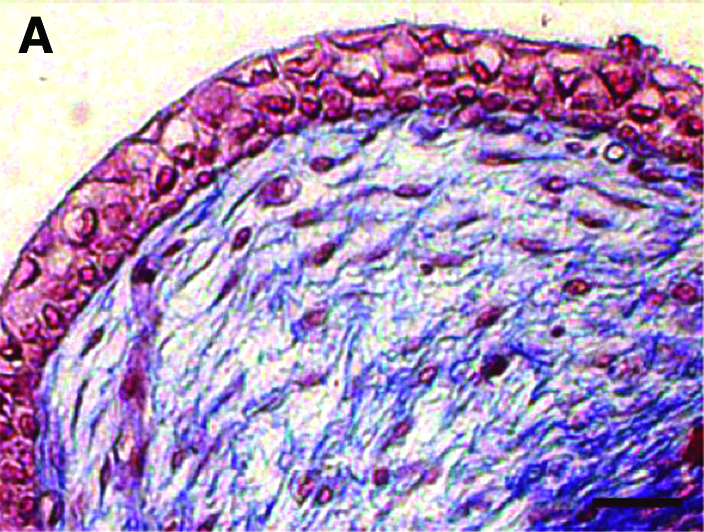
Histological analysis of VE. Masson's trichrome staining of normal porcine bladder (
Immunoblotting and immunofluorescence
The presence of UCs in our VE was confirmed by the expression of cytokeratin 8/18 in Western blot (Fig. 3). Also, the epithelium stained with anti-keratin 8/18 antibody (Fig. 4F) had the multi-layered appearance, and it was like normal porcine urothelium (Fig. 4E). The matrix stained uniformly with anti-collagen type I antibody, demonstrating a highly organized matrix of collagen type I (Fig. 4B), which is, with collagen type III, the most common type of collagen in the bladder. Moreover, the presence of laminin suggested the formation of a basal membrane under UCs (Fig. 4D). The endothelialized VE stained with anti-pecam1 antibody revealed the ECs' adhesion, and some primary organization in a capillary-like structures (Fig. 4H–J).

Immunoblotting. Western blot results of K8, K18, and alpha-tubulin. Lane 1, endothelialized VE; lane 2, VE with addition of exogenous calcium; lane 3, VE without exogenous calcium; lane 4, porcine bladder.
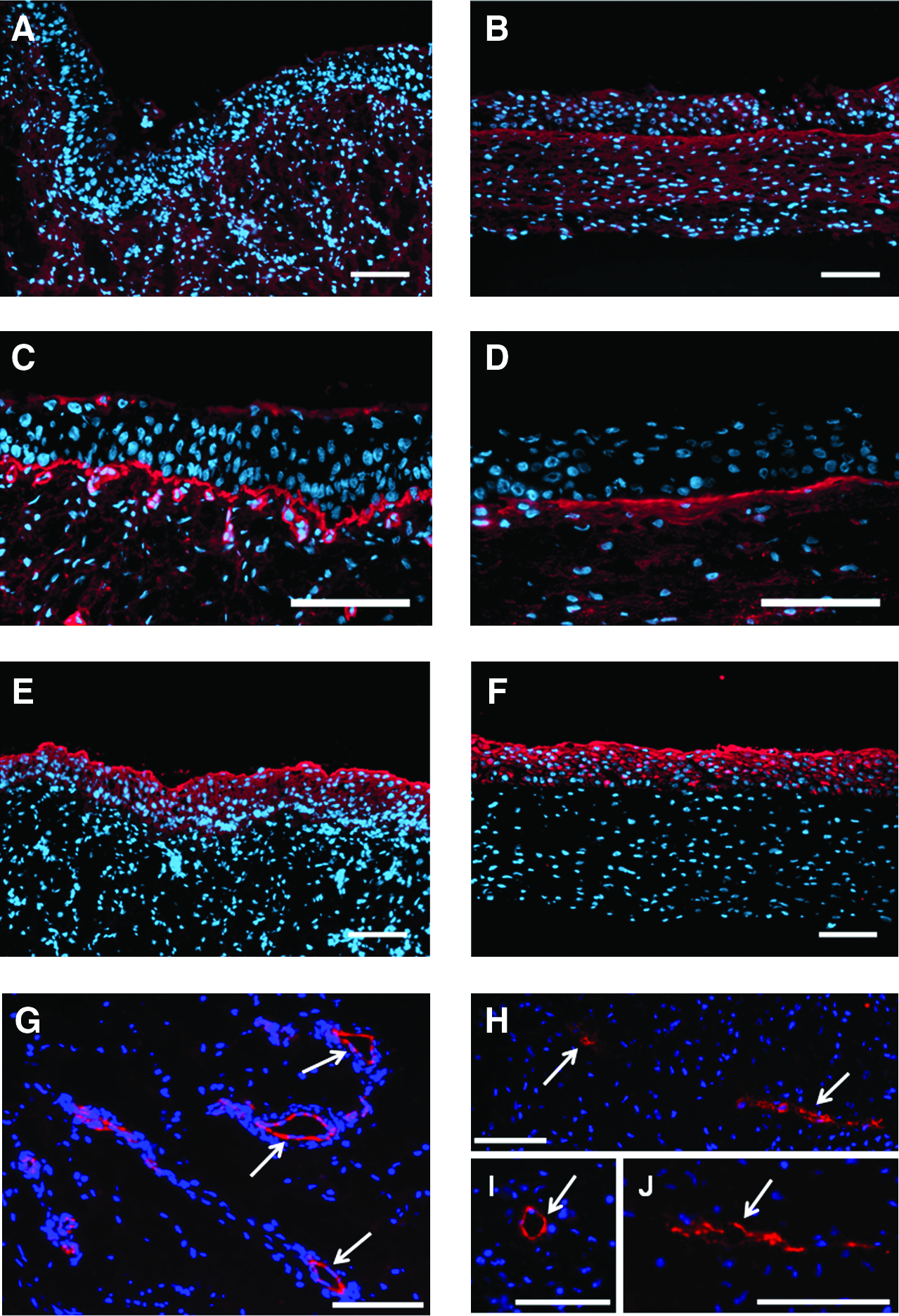
Immunofluorescence. Expression of collagen type I (
TEM and SEM
The TEM ultrastructural analysis revealed tight junctions, desmosomes, and interdigitations, for UCs' cohesion to ensure the urothelium resistance. Also, asymmetric unit membrane plaque and discoid cytoplasmic vesicles were observed in UCs. These last features are characteristic of native urothelium, and were present with and without the addition of 1 mM of calcium in the culture medium (Fig. 5). However, in the VE cultivated with calcium, the surface displayed solitary elements, a characteristic of cells covered with microvilli only, and suggested a less differentiated state (Fig. 6). Contrarily to the VE cultivated without calcium, which does not present these solitary particles but does not have either the microridged surface pattern of native bladder, these molten elements on the apical surface appear similar to the ropy ridges present at the intermediate stage in the chronology of urothelial differentiation observed by Veranic et al. This morphological pattern obtained without calcium in vitro is not the same as native bladder, which is at the terminal stage of differentiation, but the ropy ridges transition observed makes it comparable at an advanced differentiation state.
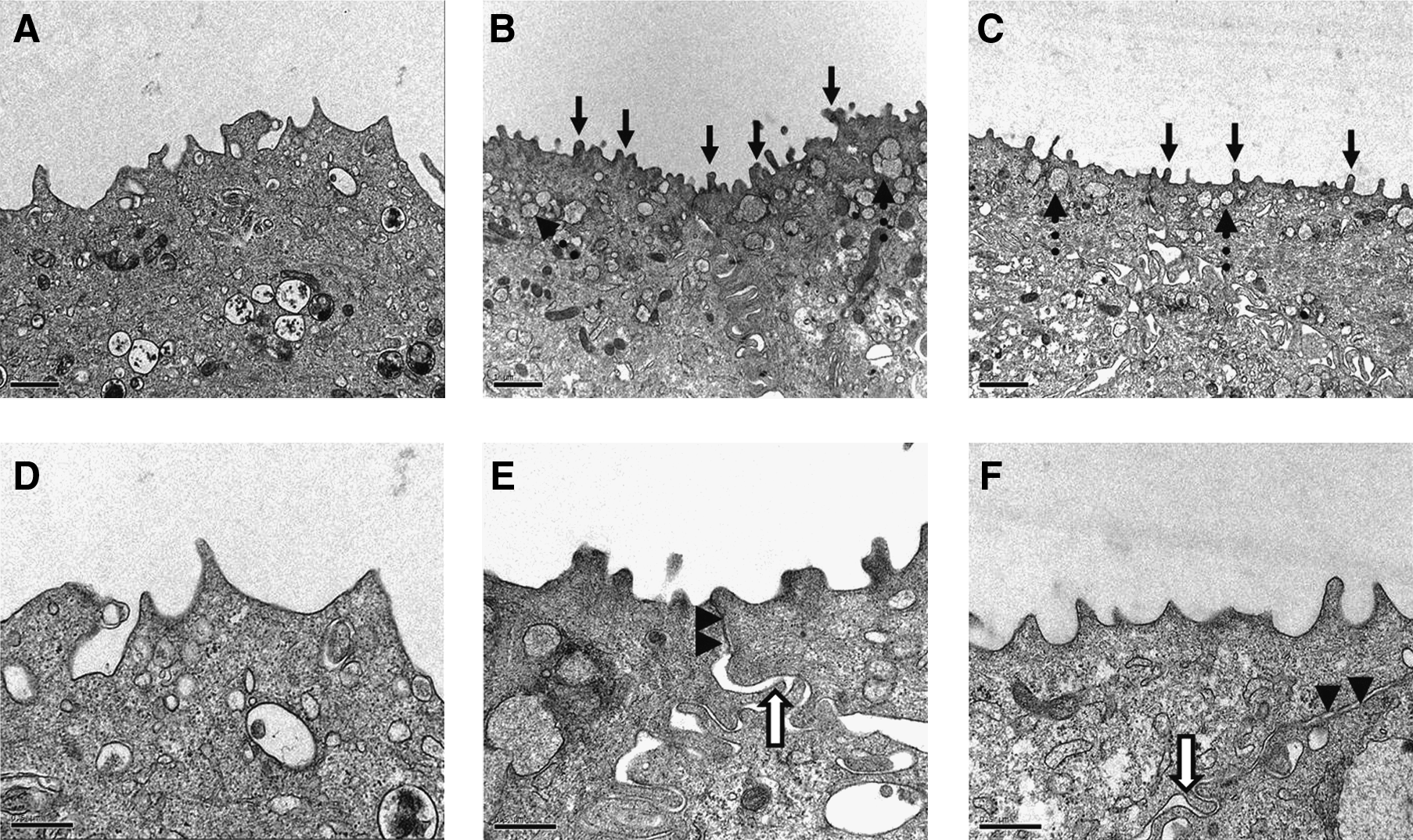
Transmission electron microscopy (TEM). TEM of normal porcine bladder (
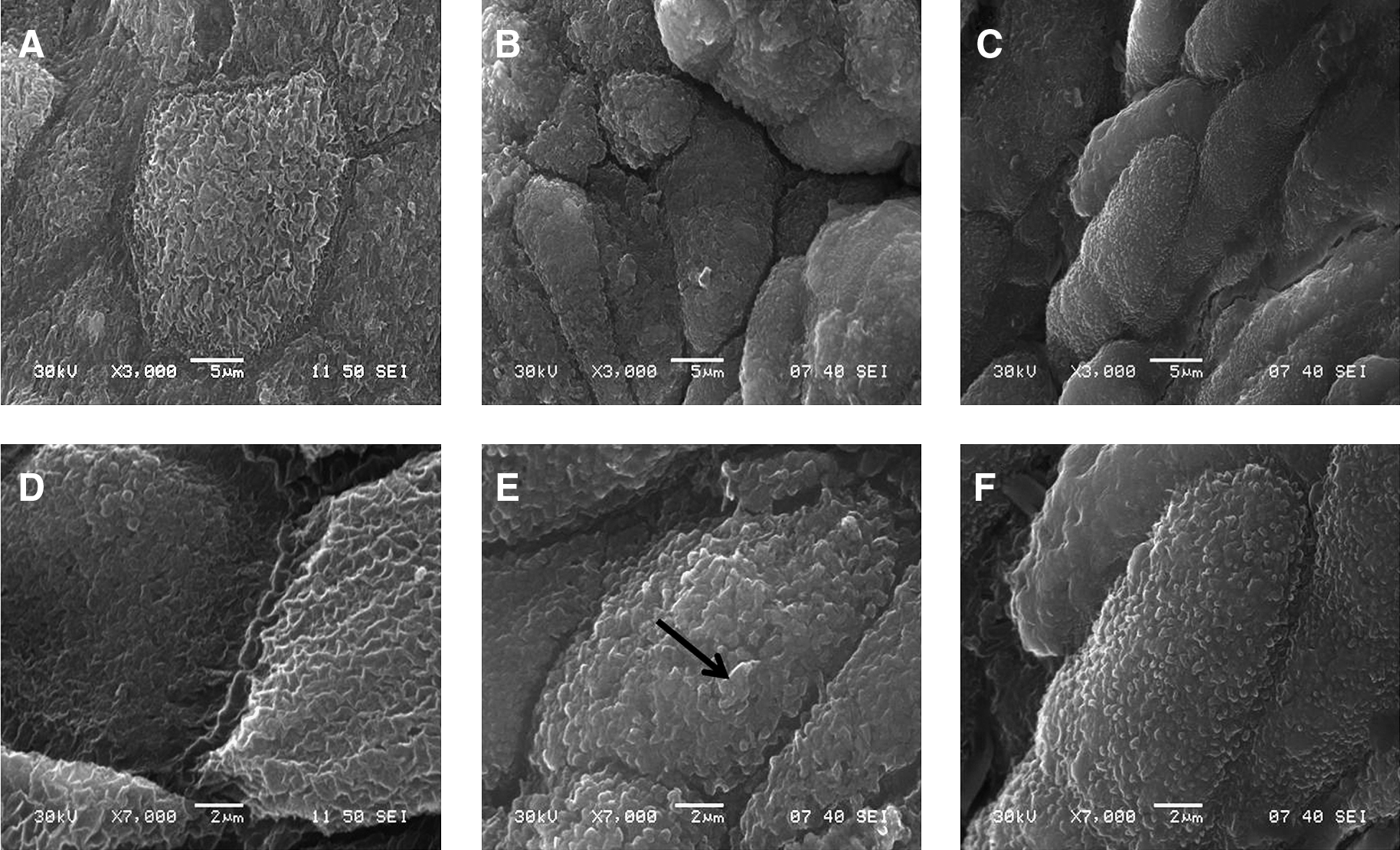
Scanning electron microscopy (SEM). SEM of normal porcine bladder (
Permeability analysis
The fibroblast constructions (Fig. 7A, B) showed a high permeability to urea, which increased quickly, contrarily to the VE (Fig. 7C, D). After only 5 h, fibroblast constructions reached the maximal diffusion, while the permeability of VE (Fig. 7D) increased slowly to reach 35% of diffusion after the same time. The presence of exogenous calcium did not modify the permeability of the fibroblast constructions, but the effect observed on the VE was different. In the absence of calcium, permeability of the VE followed significantly the same tendency as observed for native bladder (Fig. 7F), whereas in the presence of calcium (Fig. 7C), the permeability was approximately doubled. However, it remained lower than the values obtained for fibroblast constructions without UCs. It was noted that the ECs incorporation did not modify the permeability of the VE without exogenous calcium: endothelialized VE (Fig. 7E) showed the same profile as the native bladder.

Permeability analysis. Permeation tests with 14C-urea through three sheets of fibroblasts cultivated without (
Cell viability
As the engineered tissue had to sustain a long culture period, cell viability had to be performed to ensure the survival potential of the VE. After a trypan blue staining, the extraction method allowed the isolation and the reseeding of cells with a 2% mortality rate, which is standard for cell culture. Further, the beginning of migration of the three cellular types was visible after 24 h by the explants method. Colonization clearly appeared after 48 h (Fig. 8).
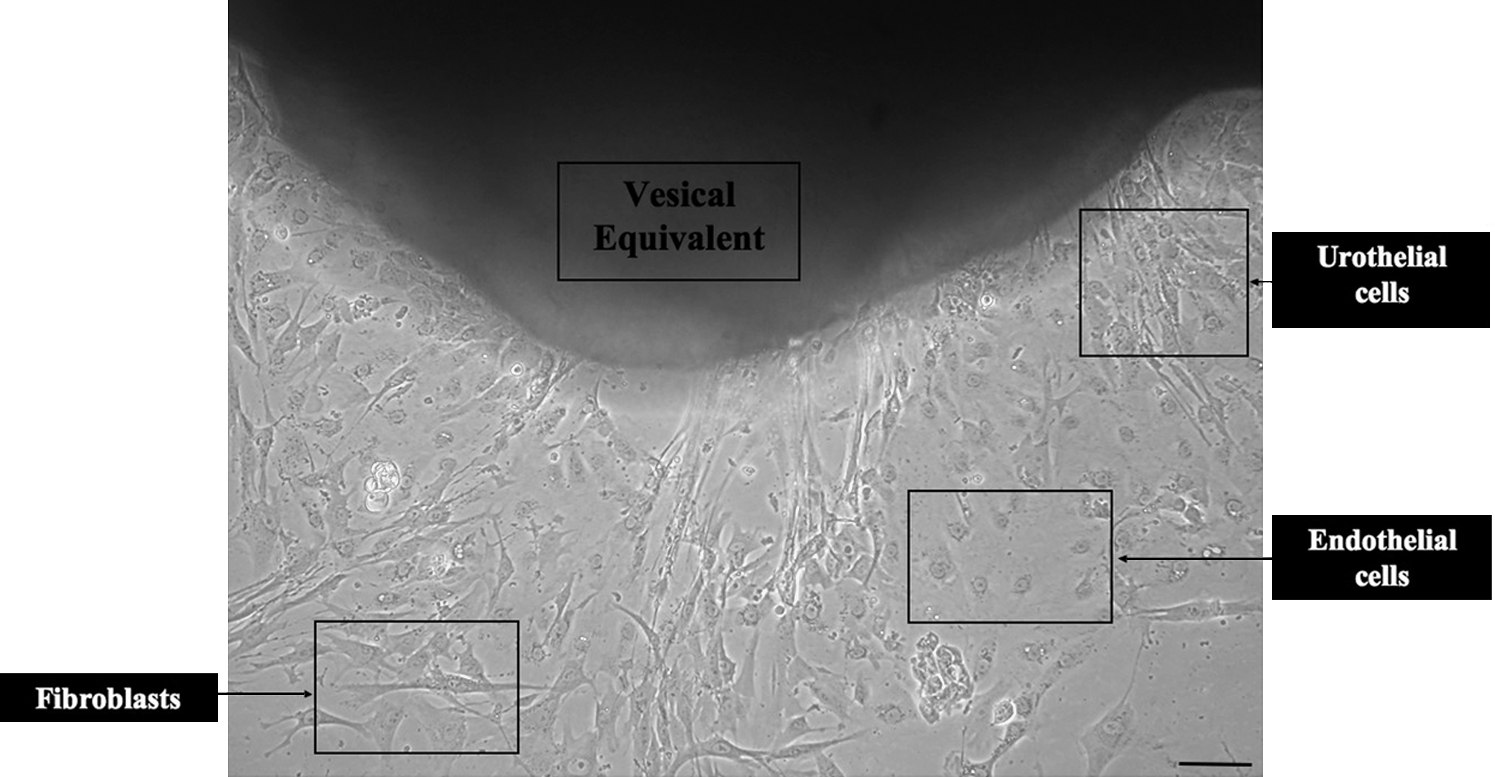
Cell viability. Colonization of fibroblasts (spindle aspect), endothelial cells (paving morphology), and urothelial cells (polygonal and tight structure), from a small piece of VE 48 h after mechanical tests (bar: 100 μm).
Mechanical testing
Mechanical properties of the engineered tissues were analyzed by uniaxial tensile testing, conducted without any slippage or failure at the grips during the testing procedures. Rupturing points were observed in the gage length for all samples tested. Stress–strain curves displayed a characteristic toe-part followed by a linear region and a failure load, indicating the viscoelastic behavior of our tissues. Results showed that ECs seeding tend to lower the mechanical properties of the VE without compromising the physiological resistance required for their tissue engineering application. UTS was shown to be higher for the VE when compared to the endothelialized VE (Fig. 9B). Results have shown that endothelialization of the VE produces a thicker tissue (Fig. 9A). This could explain the difference in UTS between the two tissues since the thickness of the samples is considered in the mechanical properties calculations. Modulus (Fig. 9C) and failure strain (Fig. 9D) were also lower for the endothelialized VE, suggesting an interaction between the cell types during the maturation period, although this hypothesis would definitely need further investigations. Cohesion between the tissue layers was confirmed since cell sheets remained firmly fixed together for all the samples during tensile testing.
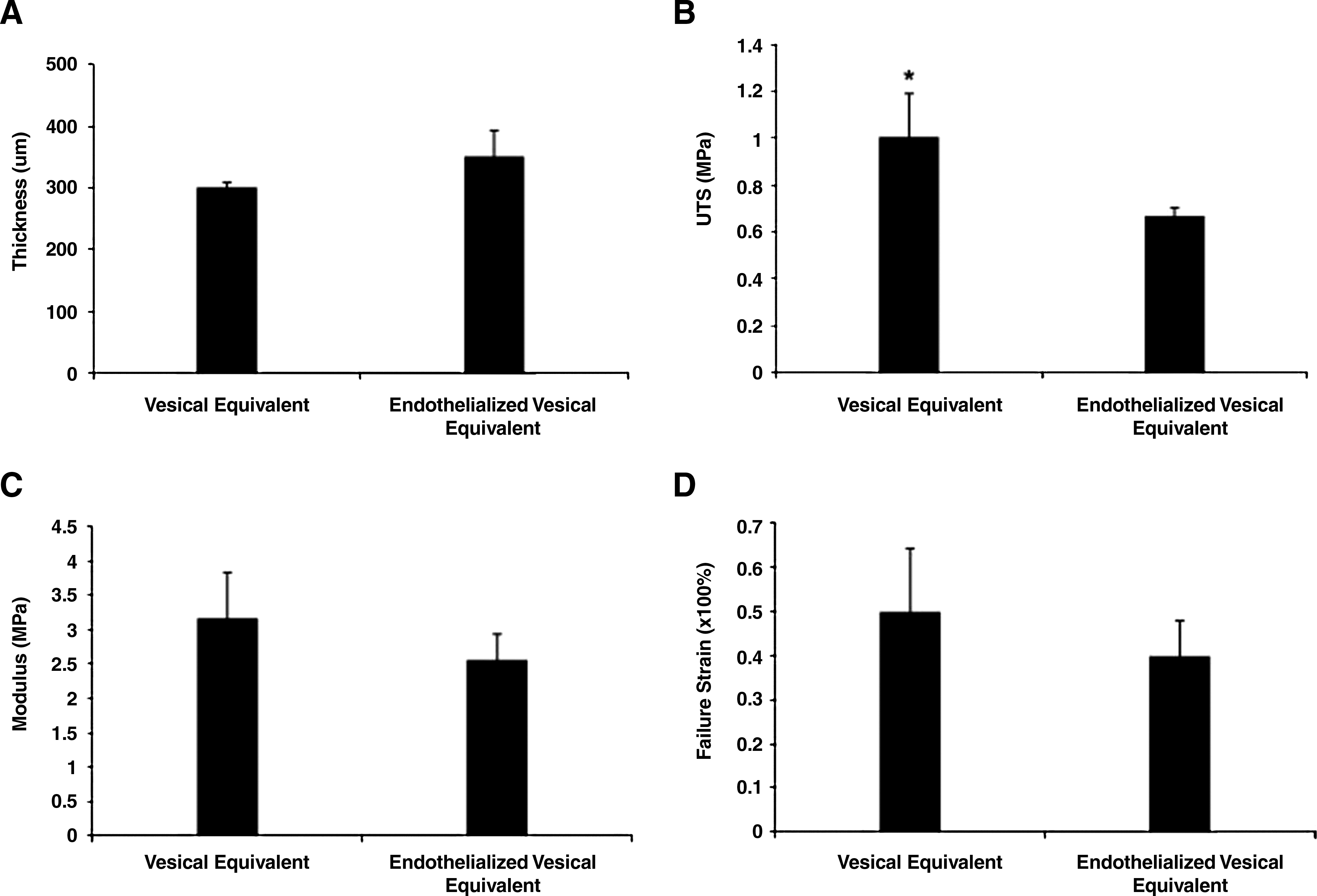
Mechanical testing. Thicknesses measured on histological cross sections on tissue (
Discussion
A new alternative for reconstructive surgery is autologous tissue substitutes using the self-assembly method, without the use of exogenous materials. The major advantages are the structural similarity compared to biodegradable scaffolds and, theoretically, the absence of immune response.16,17
As expected, our engineered VE showed a histological organization similar to a native mucosa. Further, the close cohesion of UCs observed presumes resistance to the intravesical pressure met during bladder filling. The presence of desmosomes, interdigitations, and tight junctions showed by TEM supports this hypothesis. Immunofluorescence and immunoblotting results confirmed expression of cytokeratin 8/18, a specific marker of urothelium that is found throughout the entire urothelial thickness. 18 Also, ECs showed their capacity to organize themselves into some capillary-like structures. These observations raise the need to optimize our culture conditions to favorably promote the preformation of a microvascular network, in sight of a faster inosculation with the host's vasculature. 8 Finally, a basal membrane characterized by the laminin and a lamina propria labeled by collagen I were observed, which probably promoted organization and differentiation of UCs. Thus, all these results confirm the good organization of our VE. It is important to know that these constructions contain no other material than the cells and their extracellular matrix.
To create a protective blood–urine barrier and to allow adaptation of the bladder surface during filling, the highly differentiated superficial cells had to acquire the so-called asymmetric apical membrane. The chronology of differentiation that brings basal cells to the formation of these superficial cells was described by Veranic et al. after a single dose of cyclophosphamide that destroyed UCs in the urinary bladder. The differentiation process displayed a sequence for expression and final organization of morphological markers accompanying the regeneration. The first step proposed was the coverage by microvilli on the apical surface. Then, the differentiation process continues with the progressive replacement of microvilli by ropy ridges. Finally, these apical elements organize themselves as a microridged surface, which is the typical pattern of terminal differentiation state found in native bladder. Moreover, in a recent study, Cross et al. demonstrated that calcium addition in normal human UCs supported the formation of stratified and differentiated urothelium. 19 On the basis of the morphological evolution previously described, our studies demonstrated that porcine UCs did not react as human cells to the presence of calcium. Indeed, with exogenous calcium, discoid cytoplasmic vesicles and tight junctions were observed by TEM, but the presence of microvilli only on the apical surface testifies of an early state of urothelial differentiation. On the other hand, our VE cultured without calcium presented the entire element previously analyzed by TEM, but in addition, the cellular surface observed with SEM was completely covered by molten elements, like ropy ridges. These structures are a morphological characteristic of cells covered with asymmetric unit membrane plaques and announce an advanced differentiation stage. 20 Therefore, the addition of exogenous calcium did not promote differentiation of porcine UCs. These results prove the capacity of the self-assembly method to produce a great amount of autologous tissue comparable to the morphological features of a normal urothelium and, therefore, their physiological functions of watertight membrane. Another key element of this function is uroplakins, an additional tag of UC differentiation. This protein family is more and more studied21,22 and their presence will have to be confirmed in our future experiments.
In accordance with morphological markers observed with electron microscopy, the important asset of our construction is its ability to form an effective barrier. Urine is known to contain components that are toxic for urothelium and subepithelial cells.2,23 A functional urothelial barrier might prevent inflammation once placed in vivo because less permeability would prevent urine to pass into the implanted model, thus reducing the chances of necrosis and fibrosis. The efficiency of our engineered VE is well illustrated in this experiment. The three sheets made only of fibroblasts had a higher permeability than the VE on short-time experiments. Unlike human cells, 19 the addition of calcium does not reduce the permeability to urea with porcine UCs. Our results are in accordance with those obtained for transepithelial electrical resistance measures by Turner et al. 24 This unsuitable permeability can be correlated with the SEM observations, and can be explained by the absence of transition of the microvilli profile toward the ropy ridges in the VE cultivated with calcium. However, without calcium, the VE had the same permeation profile as the native porcine bladder, and this was observed even in the presence of ECs. This means that our reconstructed urothelium, without exogenous calcium, could provide a differentiated and effective barrier to urine after bladder augmentation. Also, this model could be used to further evaluate the effect of different molecules on the permeability barrier of the urothelium.
Stress–strain curves obtained from uniaxial tensile tests were used to describe the mechanical properties of the VE. The engineered tissues demonstrated good resistance to mechanical stresses. Results showed that the UTS appeared to be higher than the gold standard of 0.32 MPa 25 measured for a porcine bladder and for all reconstructed tissues. Addition of ECs to the VE model lowered the overall mechanical properties without compromising its functionality. These results confirmed the capability of our model to withstand manipulation with forceps and sutures and thus would be qualified suitable for grafting by a surgeon. Endothelialized VE could lead to an improved graft take and survival, since a better and faster inosculation between the host vascular system and the capillary-like network of our constructions is expected.8,26
In our upcoming experiments, a full-thickness autologous bladder graft including smooth muscle cells will be develop to provide contractility of our VE. Therefore, we want to optimize the model, and for that we need to prevent the contraction of the VE while including smooth muscle cells and improve the cellular differentiation. This will be possible by a dynamic culture in a homemade bioreactor with a variable hydrostatic pressure. 27 For the clinical applications, we will graft in vivo our VE in a porcine model. In sight of results, we hypothesize that our VE will be able to constitute a temporary watertight and resistant barrier to urine that would allow time to host's cells located around the graft to supplement it. 28
Conclusion
The self-assembly method to produce VE is a significant progress and it may have important clinical impact in urology. Our construction has proven its efficiency as a barrier to urea and has sufficient mechanical resistance to support suturing. Additionally, this model is completely autologous, and its possible endothelialization could promote the early vascularization process after grafting. Consequently, it would significantly reduce necrosis, inflammation, contraction, and, therefore, possible rejection. Finally, this tissue-engineered VE without the use of scaffold can offer an ideal replacement for bladder and in vitro studies of its diverse pathologies.
Footnotes
Acknowledgments
The authors thank Martine Magnan, Alexandre Rousseau, Laure Gibot, Stéphane Chabaud, and the LOEX team for useful discussion. This study was supported by grants from the “Fonds de la Recherche en Santé du Québec” and the Canadian Institutes of Health Research. Dr. Stéphane Bolduc has received grants from the Kidney Foundation and was a recipient of the Canadian Urological Association Scholarship Fund.
Disclosure Statement
No competing financial interests exist.