
Abstract
Restoration of the vascular supply to ischemic tissues is of high clinical relevance, and proangiogenic therapies aim to reduce morbidity and mortality rates associated with the onset of cardiovascular disease. Stem cell therapy has been proposed as a potentially useful proangiogenic therapy. Mesenchymal stem cells (MSCs) have been shown to be proangiogenic and produce a number of cytokines involved in vessel development and maturation. Preclinical studies have reported increased angiogenesis after MSC delivery to the heart, and similar outcomes have been reported in recent clinical trials. Stem-cell-mediated neovascularization has been augmented by genetic modification with overexpression of angiogenic cytokines, including vascular endothelial growth factor (VEGF) and platelet-derived growth factor, showing promising results. In this study we aimed to enhance the proangiogenic capability of MSCs. MSCs were genetically modified to overexpress a versatile molecule, Ephrin-B2, involved in tissue morphogenesis and vascular development to enhance inherent neovascularization potential. Using nucleofection, Ephrin-B2 was transiently overexpressed on the cell surface of MSCs to recapitulate embryonic signaling and promote neovascularization. Ephrin-B2-expressing MSCs adopted an early endothelial phenotype under endothelial cell culture conditions increasing expression of von Willebrand factor and VEGF-Receptor 2. The cells had an increased ability to form vessel-like structures, produce VEGF, and incorporate into newly formed endothelial cell structures. These data indicate that MSCs expressing Ephrin-B2 represent a novel proangiogenic cell source to promote neovascularization in ischemic tissues.
Introduction
Stem cell therapy has been proposed as a promising proangiogenic therapy. Since initial reports demonstrated the effectiveness of stem cell delivery to the postischemic myocardium, 9 mesenchymal stem cells (MSCs) have been studied extensively as a cardiac therapy 10 and MSCs possess inherent proangiogenic capabilities producing a number of cytokines involved in vessel development, including VEGF and platelet-derived growth factor.11–13 Preclinical studies have reported increased angiogenesis after MSC delivery to the heart, 14 and similar outcomes have been reported in clinical trials. 15 Shyu et al. 16 reported that MSC delivery as a stand-alone therapy surpassed angiogenic factor gene therapy in promoting myocardial regeneration in a mouse model of acute myocardial infarction. Further, MSCs have been shown to promote angiogenesis in peripheral vascular ischemia as well as in wound healing.17,18
Stem-cell-mediated neovascularization has been augmented by the modification of these cells to overexpress angiogenic cytokines.19,20 As in proangiogenic gene therapy, initial studies focused on overexpression of a single angiogenic factor. VEGF has proved popular, along with other factors such as Ang-1 and hepatocyte-like growth factor (HGF), and initial preclinical reports point to improvements in myocardial reperfusion over cell therapy alone.21–23 Further studies have utilized a multimodal approach, where a number of factors are simultaneously overexpressed in MSCs, resulting in further improvement in global heart function.8,24,25
These studies illustrate the importance of a multimodal approach to vascular reperfusion in ischemic tissues. In this work we aimed to develop a novel combined stem cell–gene therapy approach for treating ischemic tissues with enhanced proangiogenic capability compared to nonmodified cells. We genetically modified MSCs to overexpress a versatile molecule, Ephrin-B2, involved in tissue morphogenesis and vascular development to enhance inherent neovascularization potential.
The erythropoietin-producing hepatocellular carcinoma (Eph) receptors and their cell-surface-anchored ligands, the Ephrins, comprise the largest of the receptor tyrosine kinase families with 14 receptors and 8 ligands. 26 The receptors are subdivided into Eph-A and Eph-B categories based on sequence homologies and binding properties to Ephrin ligands and have known actions in the development of the vascular and nervous system. Eph receptor tyrosine kinases and their Ephrin ligands regulate a diverse array of cellular functions, including cell migration, repulsion and adhesion, and somite formation. 27 Eph-B4 and Ephrin-B2 are co-expressed in the yolk sac, the first site of hematopoiesis and vascular development during embryogenesis. 28 Ephrin-B2 is specifically expressed in arterial angioblasts and endothelial and perivascular mesenchymal cells, whereas Eph-B4 is expressed on endothelial cells (ECs) belonging to the venous lineage. 29 A recent report has shown that vessel segregation into venous and arterial fates is also controlled by expression of Ephrin-B2 and Eph-B4 through directional control. 30 Knockout mice lacking either Eph-B4 or Ephrin-B2 resulted in early lethality in the developing embryo as a result in the arrest of angiogenesis but not vasculogenesis,29,31 illustrating the importance of both molecules in vascular development. 32
Ephrin-B2 also plays a part in vessel maturation through its involvement in smooth muscle recruitment and crosstalk between endothelial cells and pericytes. 33 Ephrin-B2 has also been shown to be expressed during postnatal neovascularization and have positive effects on wound healing.34,35 Further, a recent study has shown the delivery of Ephrin-B2 chimeric proteins to be efficacious in promoting an increase in capillary density in a mouse model of myocardial infarction. 36 Another study has shown that ex vivo modification of skin autofibroblasts with Ad-Ephrin-B2 and subsequent delivery into a rabbit model of hind-limb ischemia resulted in increased arteriogenesis. 37 Both studies highlight the potential usage of Ephrin-B2 as a proangiogenic factor and confirm our approach to creating a more efficacious stem cell therapy. Using nucleofection, Ephrin-B2 was transiently overexpressed on the cell surface of MSCs to recapitulate embryonic signaling and promote neovascularization.
Materials and Methods
MSC isolation and expansion
Bone marrow aspirates were obtained from the iliac crest of normal donors; all procedures were performed with informed consent and approved by the Clinical Research Ethics Committee at University College Hospital, Galway. MSCs were isolated by direct plating of whole marrow and expanded in culture as described previously. 38 Briefly, aspirates were washed with a medium (Dulbecco's modified Eagle's medium–low glucose [DMEM-LG] containing 1% antibiotic) and centrifuged; the precipitated cells were suspended in the medium with 10% selected fetal bovine serum (FBS) and plated at a final density of ∼3.0 × 105 cells/cm2. Cells were seeded on T-175 flasks and maintained at 37°C with 95% humidity and 5% CO2 in the same medium. After 5 days red blood cells were washed off with phosphate-buffered saline (PBS) and a fresh medium was added. Colonies of adherent cells formed within 9 days. At the end of primary culture, adherent colonies were detached by treatment with 0.25% trypsin and 0.53 mM ethylenediaminetetraacetic acid. Cells were plated in the medium [DMEM-LG; 10% FBS; 1% antibiotic (all from Sigma-Aldrich)] at 5.7 × 103 cells/cm2. Cultures were passaged at 4–6-day intervals and expanded to passage 5 for experimentation.
Osteogenic differentiation assays
To assess the osteogenic potential of MSCs, cells were plated at 30,000 cells per well in a six-well plate and cultured overnight. The following day cells were treated with the osteogenic medium [DMEM-LG; 100 nM dexamethasone; 10 mM β-glycerophosphate; 50 μM ascorbic acid 2-phosphate; 10% fetal bovine serum (FB/FBS); 1% antibiotic (Sigma-Aldrich)]. The medium was replaced two times per week. Calcium deposition was measured on day 15. Wells were fixed with 10% neutral buffered formalin for 10 min, and 1 mL of 3% silver nitrate solution was added to each well and incubated at room temperature in the dark for 10 min. Wells were rinsed and exposed to bright light for 15 min and calcium deposits imaged. Quantification of mineral deposition was performed as previously described. 39
Adipogenic differentiation assay
To assess the adipogenic potential of MSCs, cells were seeded at 2 × 105 per well in six-well plates and cultured until confluent. Cultures were then placed in an adipogenic induction medium [DMEM high glucose 10% FBS; 1 μm Dexamethasone; 200 μm Indomethacin; 10 μg/mL Insulin; and 0.5 mM Isobutylmethylxanthine; 1% antibiotic (all from Sigma-Aldrich)] for 3 days and subsequently moved into an adipogenic maintenance medium [DMEM high glucose; 10% FBS; and 10 μg/mL Insulin: 1% antibiotic] for 1 day. Cells cultured in the normal medium were used as controls. After three induction and maintenance cycles, cells were fixed with 10% neutral buffered formalin (Sigma-Aldrich) and stained using Oil Red O (Sigma-Aldrich). To quantify binding of Oil Red O to accumulated lipid it was extracted with 100% isopropanol and absorbance was read at 520 nm.
Cell surface phenotype analysis
MSCs at passage 5 were analyzed for purity and epitope expression using flow cytometry analysis. Cells were blocked in 5% goat serum/D-PBS for 30 min, and incubated with the respective phycoerythrin (PE)-conjugated antibodies (anti-CD45, anti-CD73, anti-CD105, and mouse IgG 1 isotype control [BD Biosciences]) diluted in blocking buffer at 1:100. After incubation the cells were washed in PBS and analyzed using a FACS LSR flow cytometer (Becton Dickenson). Histograms of cell number versus fluorescence intensity were recorded for 10,000 cells per sample and analyzed using FCS Express 2 (DeNovo Software).
Eph-B4 Fc-chimera protein binding assay
To assess expression of Ephrin-B2 on the cell surface of unmodified and transfected MSCs, the binding ability of Eph-B4 Fc-chimera, the receptor to Ephrin-B2, was measured. Briefly, cells were detached from the flask using cell dissociation agent (MP Bio) washed in D-PBS/5% FBS twice and incubated with 2 μg/mL Fc-chimera proteins (Ephrin-B2, Eph-B4) for 30 min on ice. After a further two washes, cells were incubated with FITC-conjugated goat anti-human IgG1 antibody for 30 min at room temperature. After a further two washes, stained cells were analyzed using the GUAVA EasyCyte or the BD LSR. Histograms of cell number versus fluorescence intensity were recorded for 10,000 cells per sample and analyzed using FCS Express 2 (DeNovo Software).
Cloning of Ephrin B2 into a mammalian expression vector
The full-length coding sequence of Ephrin B2 was amplified from human coronary artery endothelial cell mRNA using a two-step reverse transcriptase (RT)-polymerase chain reaction carried out with a proof-reading taq (PFX) that added a poly-A tail at both 5′ and 3′ ends.
Coding primer sequences: Forward, 5′ATGGCTGTGAGAAGGGACTCC Reverse, 5′ TCAGACCTTGTAGTAAATGTTC
After amplification, the cloning sequence was initially cloned into the TA- pTargeT Expression Vector (Promega) in a sterile 0.5 mL eppendorf tube. Inserts were verified with DNA sequencing (MWG Biotech—Genome Sequencing Services). Subsequently, Ephrin B2 was subcloned from the pTargeT vector to the pIRES2-eGFP bi-cistronic mammalian expression vector (Clontech). Insertion was verified by restriction endonuclease mapping.
Nucleofection and transgene analysis
MSCs at passage 5 were washed with Hanks balance salt solution (Sigma) and harvested by trypsinization. Cells were counted and 5 × 105 cells were resuspended in 100 μL of prewarmed Human MSC Nucleofection Solution (Amaxa). Two microgram of plasmid DNA in TRIS-EDTA (TE) buffer (pmaxGFP, pIRES2-eGFP, and pEphrin-B2/IRES2-eGFP) was added to the cell suspension. The sample was transferred into an Amaxa cuvette and placed into the holder of the Nucleofector Device II (Amaxa Biosystems) and subjected to the high transfection efficiency program (U-23). The cuvette was removed immediately and 500 μL of the prewarmed growth medium was added to the cell suspension and transferred to a T25 cell culture flask. The cells were incubated in a humidified 37°C/5% CO2 incubator for 24 h. After 8 h of culture, viability was assessed by the proportion of cells attached to the culture surface using a coulter counter. Cell viability was also measured by trypan blue exclusion. pmaxGFP acted as a positive nucleofection control and cells minus DNA acted as a negative control. GFP/eGFP transgene expression was analyzed 48 h after nucleofection by flow cytometry. The percent of GFP/eGFP-positive cells were quantified versus negative control over 10 days using the BD LSR and analyzed using FCS Express 2 (DeNovo Software). Western blot analysis and Eph-B4/Fc binding assays were carried out as described previously to verify increased expression of Ephrin-B2 in transfected MSCs.
Matrigel assay—vessel formation assay
Growth-factor-reduced Matrigel (120 μL; BD Biosciences) was placed in each well of an ice-cold 48-well plate. The plate was placed at 37°C for 30 min to permit Matrigel to solidify. To assess the effect of overexpression of Ephrin-B2 on MSC-vessel-like structure formation, cells were detached 48 h after nucleofection and plated at 3 × 104 cells per well. Further to assess the effect of transfected MSCs on endothelial-cell-vessel-like structure formation ECs were detached, labeled with 2 μM PK26 (Sigma-Aldrich), and plated at 3 × 104 cells per well. MSCs were added to ECs at a ratio of 1:2 24 h after EC seeding. MSC and EC/MSC tube formation was observed periodically over 72 h and at 5 days of coculture. Phase images were acquired using a Zeiss Axiovert 200 microscope and Spot v4.6 digital imaging system. Magnification bars are added to one image to indicate the magnification of all images within the panel. Confocal images and z-stacks were taking with a Zeiss LSM 510 and LSM Image Examiner. Image analysis was carried out using Olympus Cell R imaging software. Lateral sprouts per vessel-like structure length, and cross-sectional diameter were measured on 10 × images (n = 5). For analysis of smooth muscle actin (SMA) expression, ECs were labeled with 2 μM Q-tracker 645 qtracker dye to distinguish the cells from GFP-labeled MSCs. PE-labeled anti-alpha SMA (α-SMA) antibodies (Abcam) were used, and increases in mean fluorescence intensity were measured as a surrogate measure of SMA expression using flow cytometry. FITC and PE compensation was carried out using BD FACSDIVA software.
MSC/Ephrin-B2—endothelial cell differentiation and phenotype analysis
T25 cell culture flasks were coated with a thin layer of growth-factor-reduced Matrigel (150 μg/mL) for 2 h. MSCs, MSCs transfected with pIRES2-eGFP (MSCs/eGFP), and MSCs transfected with pEphrin-B2/IRES2-eGFP (MSCs/Ephrin-B2) were seeded on the Matrigel-coated flasks and cultured in the endothelial growth medium-2 growth medium for 5 days. Phase images were acquired using a Zeiss Axiovert 200 microscope and Spot v4.6 digital imaging system. Cells were harvested and fixed for 15 min in 4% paraformaldehyde. Cells were washed in PBS (−)/5% FBS and permeabilized with 0.1% triton × 100/PBS for 5 min. Cells were incubated with one of the following antibodies for 1 h at room temperature: anti-α-SMA (goat anti-human; Sigma), anti–von Willebrand factor (vWF)–PE (Dako), anti-Flk-1 (rabbit anti-human; Fitzgerald), and anti-CD73-PE (BD). Isotype controls acted as negative controls, and endothelial cells and smooth muscle cells were also used to ensure specificity of the antibodies (data not shown). Secondary-only controls were used to ensure nonspecific binding when using polyclonal antibodies. After washing, cells stained with unconjugated primary antibodies were incubated with Alexa fluor 546–conjugated anti-goat/rabbit secondary antibodies (Molecular Probes) for 1 h at room temperature. Histograms of cell number versus fluorescence intensity were recorded for 10,000 cells per sample at each time point with the BD LSR and analyzed using FCS Express 2 (DeNovo Software).
Assessment of VEGF production
After a 5-day exposure of MSCs/Ephrin-B2 to endothelial conditions, 5 × 105 MSCs were seeded in six-well plates in the serum-free medium. The cells were placed in a hypoxic chamber at 0.1% O2 for 4 h. The conditioned medium was collected and the amount of VEGF produced was measured using a Quantikine human VEGF immunoassay kit (R&D Systems). MSCs transfected with pIRES2-eGFP acted as controls.
Statistical analysis
All values are presented as the mean ± standard deviation of the mean. Datasets were tested for significance using the Student's t-test or a general linear model two-way analysis of variance in combination with a post hoc Tukey test to compare between groups. A level of p < 0.05 was considered statistically significant.
Results
MSCs were isolated from the bone marrow and expressed characteristic cell surface markers
Cell isolation, expansion, and differentiation of MSCs using direct plating were established according to other reports. 38 Isolated MSCs adhered to tissue culture plastic, had a fibroblastic morphology (Fig. 1A), and proliferated up to passage 5 with a doubling time of ∼2 days. MSCs also differentiated along the adipogenic and osteogenic pathways as evidenced by the accumulation of lipid droplets in the cytoplasm and deposition of calcium in the wells (Fig. 1B, C). The isolated cells were negative for CD45, a known hematopoietic marker, but positive for MSC characteristic markers CD73 and CD105 (Fig. 1D).

MSC characterization and differentiation potential. (
Nulceofection of MSCs with pIRES2-eGFP/Ephrin-B2
MSCs were transfected with the pEphrin-B2/eGFP plasmid using the nucleofection technique previously described.40,41 Transfection efficiency was measured by analyzing expression of eGFP in cells using both microscopy and FACS analysis. A transfection efficiency of 45.6% ± 1.48% was achieved with the pEphrin-B2/eGFP vector (Fig. 2A), whereas viability was reduced to 66.3% ± 1.48% after nucleofection. GFP-positive cells were clearly seen in micrographs 48 h after transfection (Fig. 2B). Cells remained viable in culture and expanded over 6 days (data not shown). Expression of the transgene, Ephrin-B2, was examined 48 h after transfection by the binding of its cognate receptor, Eph-B4/Fc. Cells increased expression of Ephrin-B2 as there was an increase in the binding of Ephrin-B2 receptor, Eph B4/Fc, to the cell surface of transfected MSCs by 22% ± 2.3% compared with normal cells (Fig. 2C). To assess the transient nature of transfection, we analyzed the percentage of eGFP-positive cells every 2 days over 10 days in culture. There was a marked drop of expression from 46.8% to 4.5% over the time period (Fig. 2D). The pmaxGFP vector acted as a positive transfection control, and also showed a decrease in GFP expression from 73% to 18.2% over the same 10-day period (Fig. 2D).
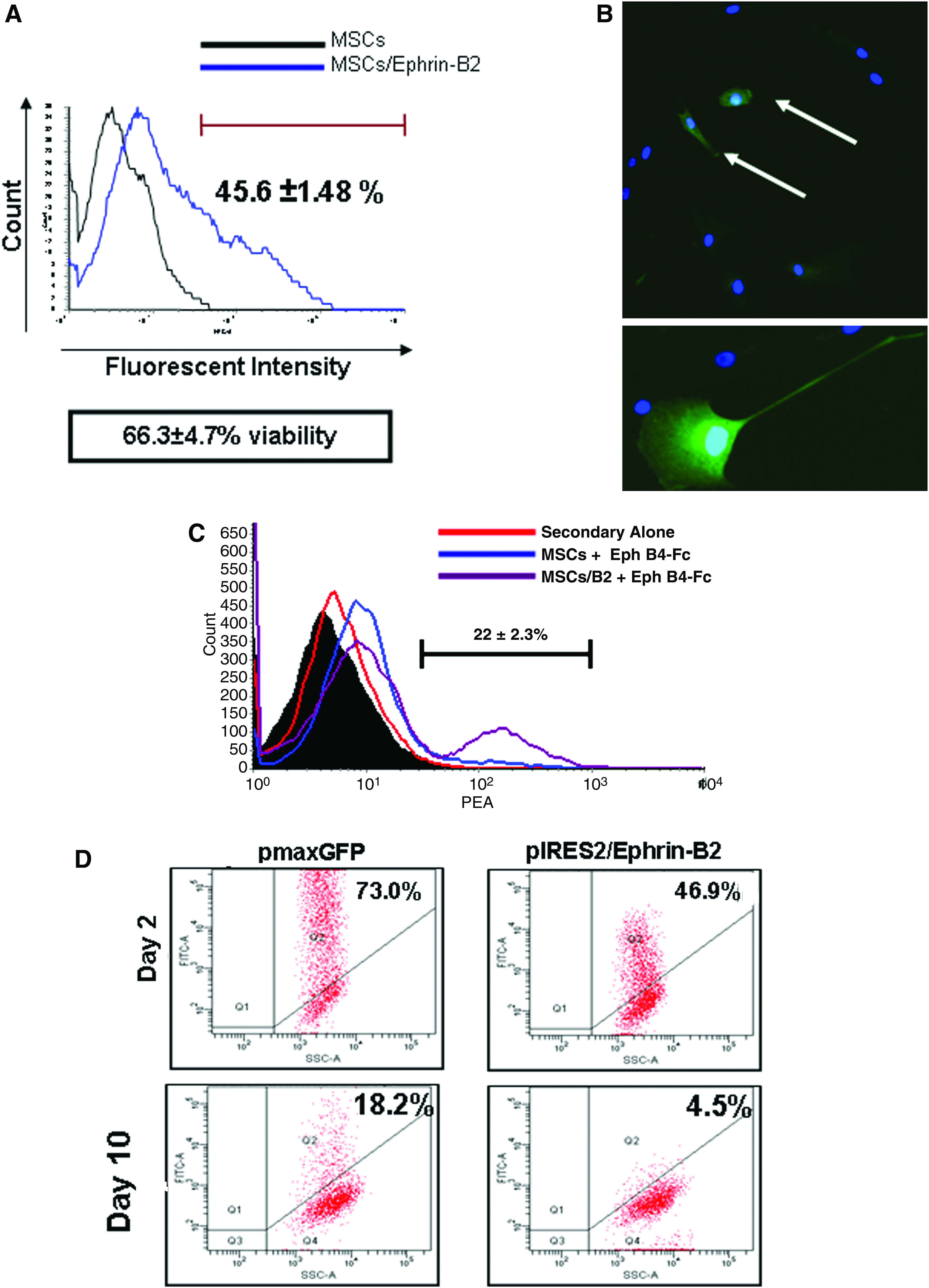
Nucleofection of MSCs with pIRES2-eGFP/Ephrin-B2. (
MSCs overexpressing Ephrin-B2 have a reduced osteogenic differentiation potential
To assess changes in the differentiation potential of MSCs/Ephrin-B2, osteogenic induction was initiated 48 h after nucleofection. It became apparent that the cells overexpressing Ephrin-B2 adopted a morphology similar to endothelial cells at days 2, 5 and 8 after osteogenic induction compared to cells alone and mature endothelial cells (Fig. 3A, inset [i]). At day 15, however, MSCs/Ephrin-B2 appeared similar in morphology to osteogenic MSCs but with a reduced cell number. Transgene expression was reverted to normal levels at this time. There was a significant reduction in the amount of calcium deposited by MSCs/Ephrin-B2 in comparison to normal MSCs at day 15 of osteogenic induction (p < 0.01; Fig. 3B). This reduction in calcium deposition is also apparent in von Kossa–stained wells of transfected cells versus normal cells (Fig. 3C). The data indicate that there was a reduction in the differentiation potential of MSCs overexpressing Ephrin-B2 toward the osteogenic lineage.

Osteogenic potential of Ephrin-B2 overexpressing MSCs. (
Overexpression of Ephrin-B2 in MSCs increases vessel-like structure formation
To further understand the changes in the cellular phenotype seen after Ephrin-B2 overexpression, the vessel-like structure formation potential of transfected MSCs was analyzed. Two days after transfection, cells were plated on growth-factor-reduced Matrigel and vessel-like structure formation was imaged 24 h later. It was immediately apparent that MSCs overexpressing Ephrin-B2 had an increased potential to form vessel-like structures on the Matrigel (Fig. 4B) compared with normal MSCs, which formed large cellular aggregates on the gel surface (Fig. 4A). This increase in vessel-like structure formation potential by MSCs/Ephrin-B2 was comparable to endothelial cells (Fig. 4 inset [i]). Using confocal microscopy, we subsequently investigated the incorporation of eGFP-positive cells into vessel-like structures formed on the Matrigel as transfected cells express both eGFP and Ephrin-B2 because of the bi-cistronic expression system (Fig. 4C, D). A large number of cells aggregated in the MSC-alone well as seen with the large number of 4′6-diamidino-2-phenylindole (DAPI)-stained nuclei in cell bundles, whereas cells in the MSC/Ephrin-B2 wells formed vessel-like structures with eGFP-positive cells incorporated into them. These vessel-like structures also remained present up to 5 days after cell seeding (Fig. 4E) in comparison to cell bunches seen in untransfected cells. (Fig. 4F).
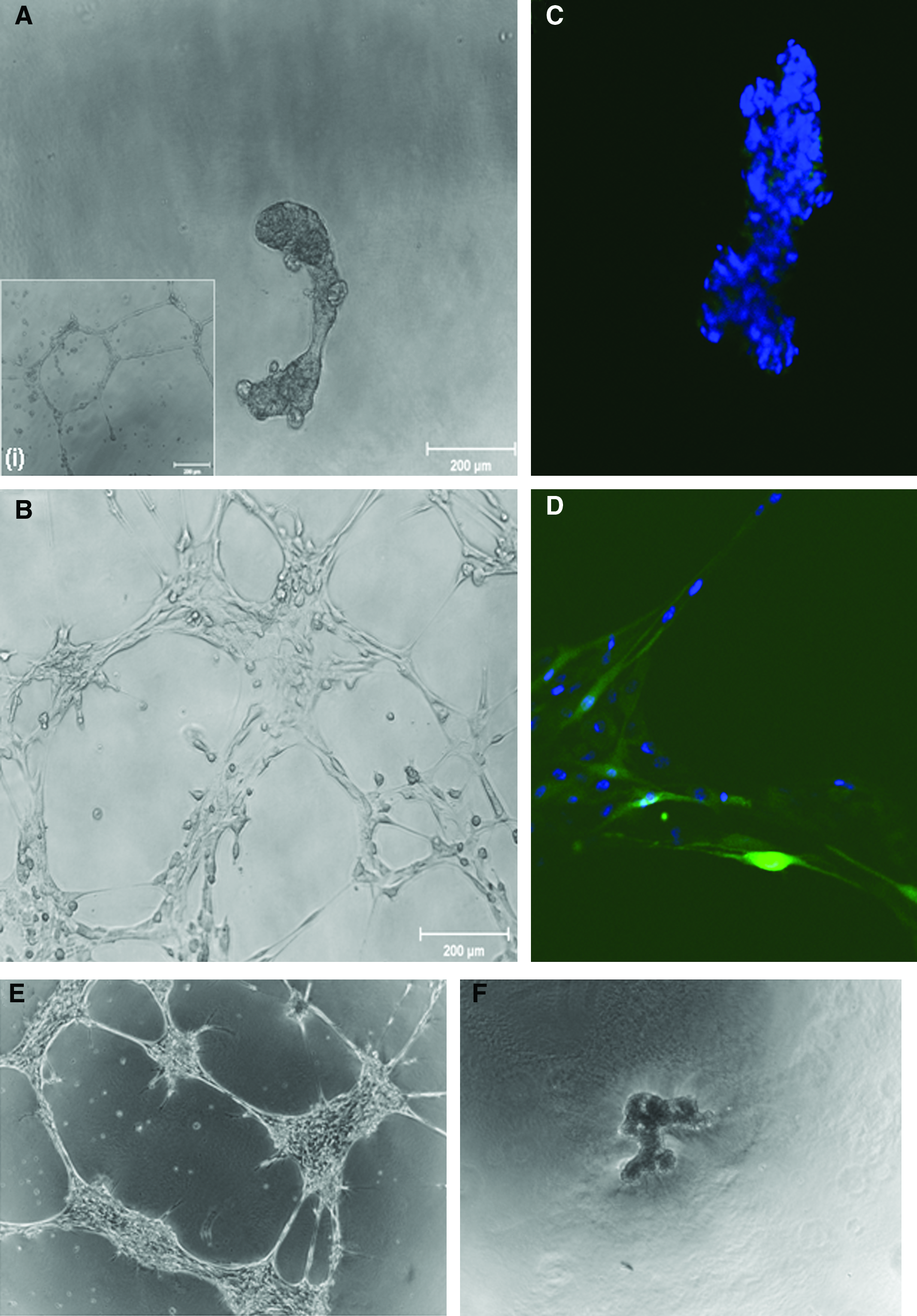
Effect of Ephrin-B2 overexpression on MSC-vessel-like structure formation. Phase micrographs of MSCs (
MSCs/Ephrin-B2 adopt an early endothelial phenotype under endothelial culture conditions
To further characterize the cellular phenotype of the MSCs/Ephrin-B2, MSCs/eGFP, and MSCs after 5 days under endothelial cell culture conditions, cells were grown on a thin layer of Matrigel in the endothelial medium for 5 days (Fig. 5A). Expression of early endothelial markers (vWF and Flk-1) was assessed by antibody staining and FACS analysis. MSCs/Ephrin B2 increased expression of both early endothelial markers, vWF and the Flk-1 (Fig. 5B), whereas there was no appreciable change in expression in MSCs/eGFP or MSCs. Cells did not initially express α-SMA and there were no changes in expression levels after culture. Interestingly, there was a decrease in the expression level of CD73 on MSCs/Ephrin-B2 compared to MSCs/eGFP and MSCs after EC culture. On further inspection this reduction corresponded to the level of CD73 normally expressed on aortic endothelial cells (Fig. 5B). These data suggest that MSCs/Ephrin-B2 adopt an early endothelial phenotype after exposure to EC cell culture conditions.
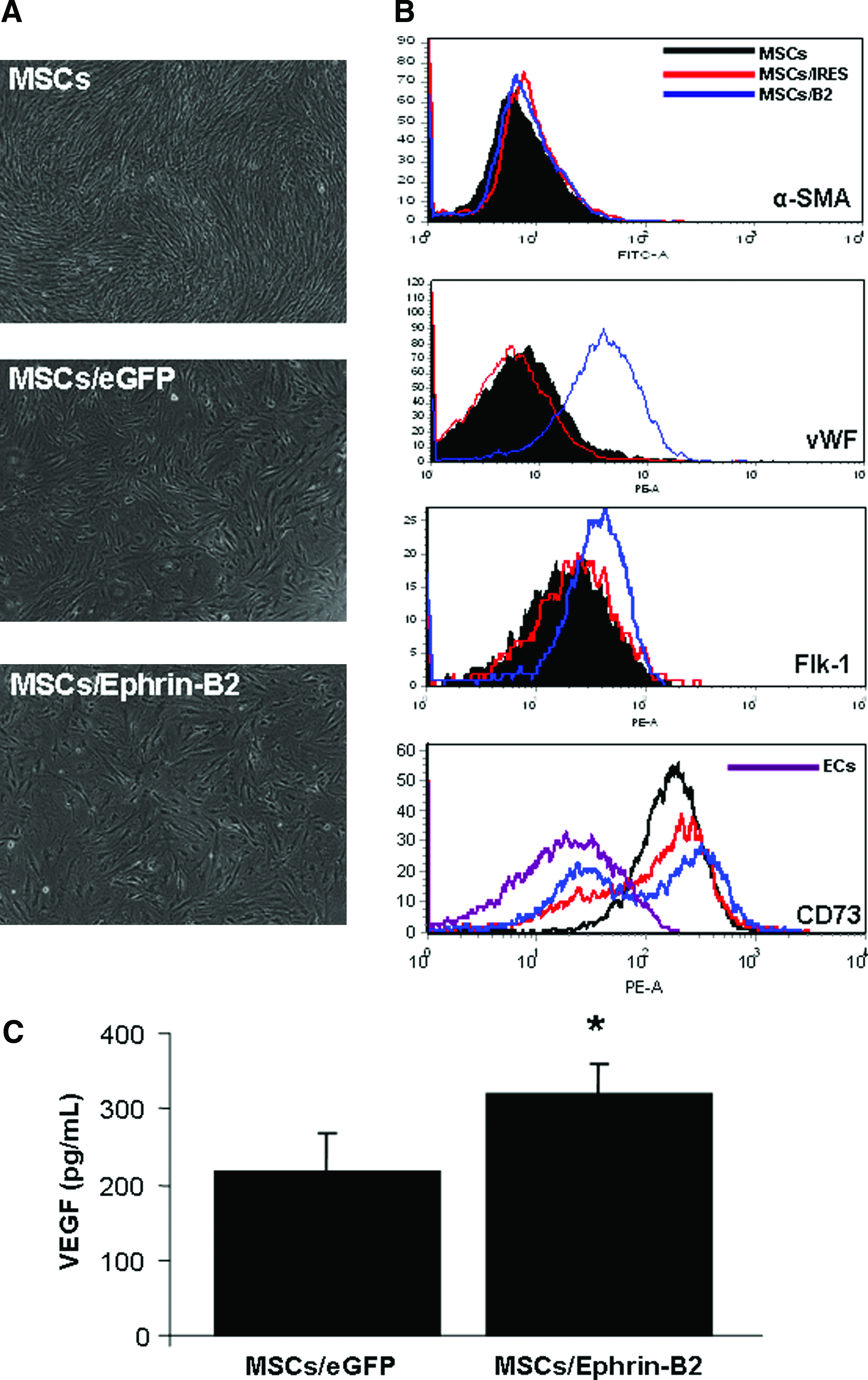
Cellular phenotype analysis of MSCs/Ephrin-B2 under endothelial culture conditions. (
To further characterize the transfected cell population, MSCs/Ephrin-B2 and MSCs/eGFP were cultured under hypoxic conditions for 4 h and VEGF production was measured. MSCs overexpressing Ephrin-B2 released significantly higher levels of the proangiogenic factor VEGF than MSCs/eGFP (Fig. 5C: p < 0.01).
MSCs/Ephrin B2 incorporate with new vessel-like structures formed by endothelial cells
To assess the ability of MSCs/Ephrin-B2 cells to incorporate into newly formed EC-vessel-like structures (Fig. 6A–D), we cocultured endothelial cells with MSCs on growth-factor-reduced Matrigel. MSCs were added 24 h after EC seeding and observed using phase and multiphoton microscopy. MSCs normally wrap around the EC-vessel-like structures and form more stable structures (Fig. 6B). Interestingly, after transfection MSCs/Ephrin-B2 incorporated into new EC-vessel-like structures rather than wrapping around the outside layer (Fig. 6C, D). There was no significant difference in lateral sprouting from vessel-like structures or cross-sectional diameter between ECs and ECs cocultured with MSCs/Ephrin-B2. However, there was a reduction in lateral sprouting and a significant increase in cross-sectional diameter in ECs coculture with nonmodified MSCs (p < 0.05; Fig. 6E, F). eGFP-positive cells can be seen directly incorporating into the vessel-like structures and elongating along the structure length (Fig. 7A). Endothelial cells alone acted as controls (Fig. 7A inset [i]). To assess whether MSCs or MSCs/Ephrin-B2 adopted a smooth muscle phenotype when in coculture with ECs, α-SMA expression was assessed using flow cytometry. There was no significant increase in the pericyte maker α-SMA on MSCs/Ephrin-B2 cocultured with ECs, whereas there was an increase in α-SMA expression on nonmodified cells when cocultured in the presence of ECs for 3 days (Fig. 7B). MSCs/Ephrin-B2 can be seen incorporating in to newly formed endothelial-vessel-like structures when viewed longitudinally (Fig. 7C)

Coculture of MSCs/Ephrin-B2 with endothelial cells and assessment of vessel-like structure formation. (

Structural organization of EC-MSC/Ephrin-B2 cocultured vessel-like structures. (
Discussion
The need for an efficacious proangiogenic therapy without adverse side effects still remains unmet. Stem cell therapies have been proposed as a potential solution to this problem, offering advantages in integration as well as the ability to supply several trophic factors involved in neovascularization. 12 Recent approaches have been undertaken to enhance the inherent angiogenic capabilities of MSCs by genetically modifying the cells to express proangiogenic factors, including VEGF, Ang-1, and fibroblast growth factor,19,22 and all have been reported to increase vascular density in ischemic tissues compared to unmodified cells. However, results from the clinical setting are still unpublished. 8
In this study we aimed to assess a novel stem cell gene therapy combination using Ephrin-B2 as the therapeutic gene. Recent reports have demonstrated increased expression of the ligand Ephrin-B2 in a number of tumor categories and to be functionally involved in tumor angiogenesis.27,42 This fact places important emphasis on considering the “The Janus Phenomenon” described by Epstein et al., 43 where an angiogenic factor aids targeted neovascularization but also promotes tumor progression by enhancing tumor angiogenesis in distal locations. It should be noted that Ephrin-B2 may be proangiogenic for tumors and could have an affect on tumor progression while enhancing tissue revascularization. To reduce this risk we chose to transiently deliver Ephrin-B2 to MSCs using a nonviral method of transfection, nucleofection.
Transfection rates of 80% have been reported in MSCs using the nucleofection technique.40,41 We achieved similar rates using a control plasmid pmaxGFP; however a lower rate of transfection of 44% was achieved with our bi-cistronic vector (pIRES2-eGFP/Ephrin-B2). This may be due to the fact that the eGFP transgene is downstream of the IRES promoter and some reports have noted significantly less expression of the second cistron transgene after transfection. 44 There was a successful increase in the expression level of Ephrin-B2 in MSCs after nucleofection by Western blotting (data not shown) and the binding of its cognate receptor, Eph-B4/Fc, to the cell surface of the transfected cells. Cell viability was reduced to ∼65% using the Nucleofection technique, but issues surrounding reduced cell viability were somewhat offset by the high transfection efficiency and increased expression of Ephrin-B2 achieved in the MSC population. This high efficiency in combination with the alleviation of safety considerations associated with viral vectors points toward Nucleofection becoming a clinically applicable transfection technique in the future if viability can be improved.
Reports have demonstrated the involvement of Ephrin-B2/Eph-B4 signaling in the crosstalk between osteoclasts and osteoblasts during bone formation and homeostasis.45,46 We examined the osteogenesis potential to ensure that MSCs modified with Ephrin-B2 did not increase bone matrix formation. After assessment MSCs/Ephrin-B2 had a reduced osteogenic differentiation potential compared with untransfected cells after nucleofection. Interestingly, the transfected cells appeared to adopt an endothelial morphology during osteogenic induction, which led to the analysis of the vessel-like structure formation capability of MSCs/Ephrin-B2 on Matrigel. We found that cells overexpressing Ephrin-B2 formed vessel-like structures in a similar manner to ECs on Matrigel, whereas untransfected MSCs formed characteristic cell bunches on the gel surface. These vessel-like structures remained present up to 5 days and cells at structure junctions adopted a more cobblestone morphology. To understand the changes in MSCs/Ephrin-B2 phenotype, expression of early endothelial markers was examined after 5 days under EC culture conditions. MSCs/Ephrin-B2 adopted an early endothelial phenotype by increasing expression of vWF and Flk-1. 47 Further, it was found that MSCs/Ephrin-B2 cells incorporated into the newly formed endothelial-cell-vessel-like structures rather than associate with the ECs by forming an outer pericyte layer as seen with untransfected MSCs in our previous study. 48 This was further demonstrated in the quantitative difference in cross-sectional diameter between the EC and MSCs/Ephrin-B2 group and the EC/MSC group, where there was only a significant increase in vessel-like cross-sectional diameter of the EC/MSC group. These findings were in line with our previous experiments, 48 where nonmodified MSCs resulted in greater vessel-like structure formation, increased stability, and increase in complexity of the structures formed. This was not the case in MSCs/Ephrin-B2 and EC cocultures where these cells coexisted with the ECs forming normal lattice structures. To understand the changes in trophic factor production between modified and unmodified MSCs, VEGF production was analyzed after exposure to a hypoxic environment. MSCs/Ephrin-B2 produced a significantly higher amount of VEGF, a known proangiogenic factor. This result further demonstrates the ability of cell genetically modified with Ephrin-B2 to assume an endothelial-like phenotype.
The differentiation of MSCs toward the endothelial lineage has been previously reported, 49 and a recent study also showed that MSCs exposed to mixed esters of hyaluronan with butyric and retinoic acid adopted an endothelial fate. 50 In this study, we show that MSCs transiently transfected with Ephrin-B2 rapidly adopt an endothelial phenotype and enhance the production of VEGF. This novel stem cell and gene therapy approach may be of relevance in promoting neovascularization. Not only do the MSCs/Ephrin-B2 have the potential to provide the endothelial hardware necessary to integrate with newly formed vessels, but the cells also produce other factors that enhance vessel formation. Future studies would have to be completed to assess the enhanced proangiogenic capability of MSCs/Ephrin-B2 in vivo on pathological ischemia and as an intervention after myocardial infarction.
Further, the use of MSCs/Ephrin-B2 to promote angiogenesis has implications in the reperfusion of engineered tissues. As biomaterial and bioreactor technologies advance, there continues to be an increase in the complexity and size of tissues/organs generated in vitro. 51 Maintaining the cells viable within these constructs will likely require vascularization. The novel cell source described here would be an ideal cellular candidate to meet these advancing requirements.
In conclusion, these data demonstrate that we have successfully created a novel proangiogenic stem cell source that may have therapeutic potential in restoring perfusion to ischemic tissues.
Footnotes
Author's Contribution Summary
Garry Duffy: conception and design, collection and assembly of data, data analysis and interpretation, and article writing.
Sinead D'Arcy: collection and assembly of data.
Tabassum Ahsan: conception and design and final article approval.
Robert Nerem: conception and design, financial support, and final article approval.
Timothy O'Brien: financial support and final article approval.
Frank Barry: conception and design, financial support, and final article approval.
Disclosure Statement
Garry Duffy, Timothy O'Brien and Frank Barry have applied for a patent.