
Abstract
Introduction:
The successful integration of stem cells in adult brain has become a central issue in modern neuroscience. In this study we sought to test the hypothesis that survival and neurodifferentiation of mesenchymal stem cells (MSCs) may be dependent upon microenvironmental conditions according to the site of implant in the brain.
Methods:
MSCs were isolated from adult rats and labeled with enhanced-green fluorescent protein (eGFP) lentivirus. A cell suspension was implanted stereotactically into the brain of 50 young rats, into one neurogenic area (hippocampus), and into another nonneurogenic area (striatum). Animals were sacrificed 6 or 12 weeks after surgery, and brains were stained for mature neuronal markers. Cells coexpressing NeuN (neuronal specific nuclear protein) and GFP (green fluorescent protein) were counted stereologically at both targets.
Results:
The isolated cell population was able to generate neurons positive for microtubule-associated protein 2 (MAP2), neuronal-specific nuclear protein (NeuN), and neurofilament 200 (NF200) in vitro. Electrophysiology confirmed expression of voltage-gated ionic channels. Once implanted into the hippocampus, cells survived for up to 12 weeks, migrated away from the graft, and gave rise to mature neurons able to synthesize neurotransmitters. By contrast, massive cell degeneration was seen in the striatum, with no significant migration. Induction of neuronal differentiation with increased cyclic adenosine monophosphate in the culture medium before implantation favored differentiation in vivo.
Conclusions:
Our data demonstrated that survival and differentiation of MSCs is strongly dependent upon a permissive microenvironment. Identification of the pro-neurogenic factors present in the hippocampus could subsequently allow for the integration of stem cells into nonpermissive areas of the central nervous system.
Introduction
Therefore, the central aim of the present report was to clarify whether survival and differentiation of MSCs in the host brain are facilitated in a permissive microenvironment in which local conditions support neurogenesis during adult life.
To this end, we isolated MSCs from young Sprague-Dowley rats, transfected them with lentivirus carrying the gene for enhanced-green fluorescent protein, and implanted them stereotactically into the brains of adult rats from the same strain. One group of animals received implants in the hippocampus, and a second group in the striatum at a point 2.5 mm lateral from the ventricular wall, that is, a nonneurogenic brain area. The main hypothesis was that survival and differentiation differ in these two areas. Animals were sacrificed after 6 or 12 weeks, and brains were then fixed and sliced, and subjected to immunhistochemistry for different neuronal markers. Additionally, the yield of mature neuronal cells derived from the implanted cells was determined by stereological quantification under an epifluorescence microscope.
Mature neuronal cells, positive for neurofilament 200 (NF200), neuronal-specific nuclear protein (NeuN), and βIII-tubulin, were identified 6 weeks after implantation and survived for at least 3 months. Synthesis of gama amino-butyric acid (GABA) and glutamate neurotransmitters was demonstrated by immunhistochemistry. Young neuronal cells were observed distal to the implantation site. Our results indicate significantly enhanced cell survival and differentiation in the hippocampus compared to striatum. In this latter structure, a lower absolute number of mesenchymal-derived cells, together with a smaller yield of differentiated neurons, indicated more marked cell degeneration with secondary astrocytic reactions within the graft. By contrast, no significant astrocytic reaction was seen in the hippocampus where optimal integration of the graft was evident. Moreover, neuronal differentiation was also facilitated when cells were implanted at relatively late stages of in vitro maturation. To the best of our knowledge, this report constitutes the first description showing clear dependence of the MSC neuronal differentiation process on the implantation site within the CNS, confirming the importance of a permissive microenvironment for full development of stem cells in the brain.
Materials and Methods
Isolation of MSCs from bone marrow
The protocol for isolation of rodent MSCs from bone marrow was adapted from a previous report based on extraction of human cells. 9 In summary, 4-week-old Sprague-Dowley rats were anesthesized with intraperitoneal injection of cetamin and sacrificed by cardiac incision, according to a protocol approved by the Ethics Comitee of the University of Freiburg. Femures and tibias were dissected from the circumventing muscles and freed at the joints by opening the joint capsules. The epiphysis were then cut with surgical scalpel and the bones were carefully perfused with phosphate-buffered saline (PBS) (Gibco) and collected in 15 mL Falcon tubes. The solution thus obtained was filtered with 40 μm mesh Falcon filters for removal of the bone spicules. This solution was added to a Biocoll® solution (Biochrom AG) with 1.077 g/mL density and centrifuged at 1500 rpm for 25 min. At the end, a tetraphasic solution was obtained, being the first layer comprised of PBS and plasma, followed deeply by a thin layer of mononuclear cells, then Biocoll solution, and, finally, eritrocytes at the bottom. The mononuclear cell layer was collected in 15 mL Falcon tubes and centrifuged again for 5 min at 1200 rpm and finally resuspended in culture marrow stem cell basal medium (MSCBM®; Cambrex-Poietics Cambrex), complemented with 10% serum supplement, 2 mM glutamine, penicillin, streptomycin, and amphotericin, seeded after counting at 100.000 cells/cm2 in T150 culture flasks at 5% CO2, 37°C, and 80% humidity. The first medium change was performed after 24 h and each second day. Passages were performed once a week, time usually necessary to reach 80% confluency, and then seeded again at 3000 cells/cm2 in T150 flasks containing 30 mL of the medium. After 3 weeks, the stromal cell type strongly predominates in the culture, due to their adherence property to the culture flask, since hematopoiethic cells, which grow in suspension, were removed from the system during medium changes. In this way, we could generate 5–40 million stromal cells per milliliter of marrow aspirate by the second passage.
Lentiviral transfection
An eGFP construct developed in-house by one of the authors (B.S.) was used, which ensured high expression levels of GFP that remained stable for the duration of the experiment (3 months). The lentiviral transfection was conducted under an S2-security level. Specifically, mesenchymal cells were cultivated until confluence in T75 Flacon flasks. Upon reaching confluency, the medium was removed and replaced by a solution containing 2 × 106 viral particles per milliliter suspended in 2–3 mL of medium, giving a count of five viral particles per cell in 8 μg/mL of polybrene (Sigma). Cells were incubated for 8 h, after which time further medium was introduced to reach 12 mL. On the following day, the medium was completely changed. After 2–3 passages, GFP expression levels were sufficiently strong and stable to proceed with the experiments.
Neuronal differentiation in vitro
To prove the multipotency of the isolated cells and their capacity to transdifferentiate into neurons, we first tested against a differentiation protocol that had been applied satisfactorily in human mesenchymal cells. 3 Briefly, neuronal differentiation was induced in three stages. (1) Stromal MSCs were isolated as described. (2) An immature neural phenotype was induced using basic fibroblast growth factor (bFGF; 20 ng/mL; Sigma) and epidermal growth factor (EGF; 20 ng/mL; Peprotech) in a basal medium supplemented with 5 μg/mL heparin (Sigma) and 10 ng/mL leukaemia inhibitor factor (Millipore), which was added to the original protocol for long term expansions. After 1 week, cells formed spheroid aggregates. At different time points, the spheres were enzymatically and mechanically dissociated and plated at 10,000 cells/cm2 onto 24-well plates (Becton-Dickinson) that had been previously coated with fibronectin (Sigma). (3) In the third stage, a more mature neuronal phenotype was achieved by adding 25 ng/mL brain-derived neurotrophic factor (R&D Systems), 1 mM de dibutyril-cyclic adenosine monophosphate (cAMP) (Sigma), and 0.5 mM iso-butyl-metyl-xantine (Sigma) to a basal medium. MSCs can be expanded for up to 2 years using this method. Cells used in the present report, however, were expanded for 1–3 months before differentiation.
Electrophysiology
Electrophysiological recordings were performed as already described. 3 Rodent MSCs were patched in voltage-clamp technique after cultivation in bFGF-EGF for 7 days and iso-butyl-metyl-xantine-cAMP for 3 days. The standard extracellular recording saline was comprised of 125 mM NaCl, 25 mM NaHCO3, 2.5 mM KCl, 1.25 mM NaH2PO4, 25 mM glucose, 2 mM CaCl2, and 1 mM MgCl2 (equilibrated with 95% O2 and 5% CO2). The pipettes were filled with an intracellular saline consisted of 140 mM KCl, 10 mM ethylene glycol tetraacetic acid (EGTA), 2 mM MgCl2, 2 mM Na2ATP, and 10 mM HEPES (pH was adjusted to 7.3 with KOH). Patch pipette resistances ranged from 3 to 7 MΩ, and seal resistances were 2.02 ± 0.2 GΩ and were at least three times higher than the input resistances (mean 0.7 ± 0.1 GΩ). Signals were filtered at 5 or 10 kHz, series resistances with mean 20.7 ± 2.1 MΩ were compensated to 90% with 20 μs lag. The mean capacitance of the recorded cells was 41.7 ± 10.7 pF. The sampling frequency was 10 or 20 kHz. The holding potential was −80 mV. For off-line data analysis we used Igor 5.02B software (Wavemetrics). Fifteen consecutive pulses were applied to the patched cells in voltage clamp, from −70 to +70 mV at 10 mV increment for 100 ms. Current traces were averaged from at least five sweeps. The leakage and capacitive currents were subtracted on-line using a P/−4 protocol (four negative correction pulses at an amplitude 1/4 of that of the test pulse). Data plots of the I-V relationships were fitted with a nonraised Boltzmann equation multiplied with a driving force considering the reversal potential for each ion (calculated from the used solutions according to the Nernst equation), in the form I(V)ion = [(V − Eion)Gmax]/{1 + exp[−(V − V½)/k]}, where V is the membrane potential, Eion is the Nernst reversal potential for the ion, Gmax is the maximal conductance, V½ is the potential at which the value of the Boltzmann function is 0.5, and k is the slope factor.
Stereotactic implantation
To minimize the trauma related to the stereotactic implantation of the cell solution, we used a microcapillary technique previously described by the senior author,10,11 illustration in Figure 1. Accordingly, borosilicate glass capillaries (100 mm length, OD 1.09 mm, ID 0.8 mm; Drummond Scientific Co.) were elongated and hot cut using the appropriate equipment (Sutter Instruments model P97; Novato). The tips were then polished by abrasion at 40° on a Narishige EG-40 polisher (Analytical Instruments), to achieve an outside diameter of 50 μm with a thin tip 8–10 mm in length. Finally, the resulting capillary was then heated again in a Narishige MF900 polisher (Analytical Instruments) to remove imperfections on the glass surface. The glass capillary was then fitted to a 1 μL Hamilton syringe (Hamilton Company).
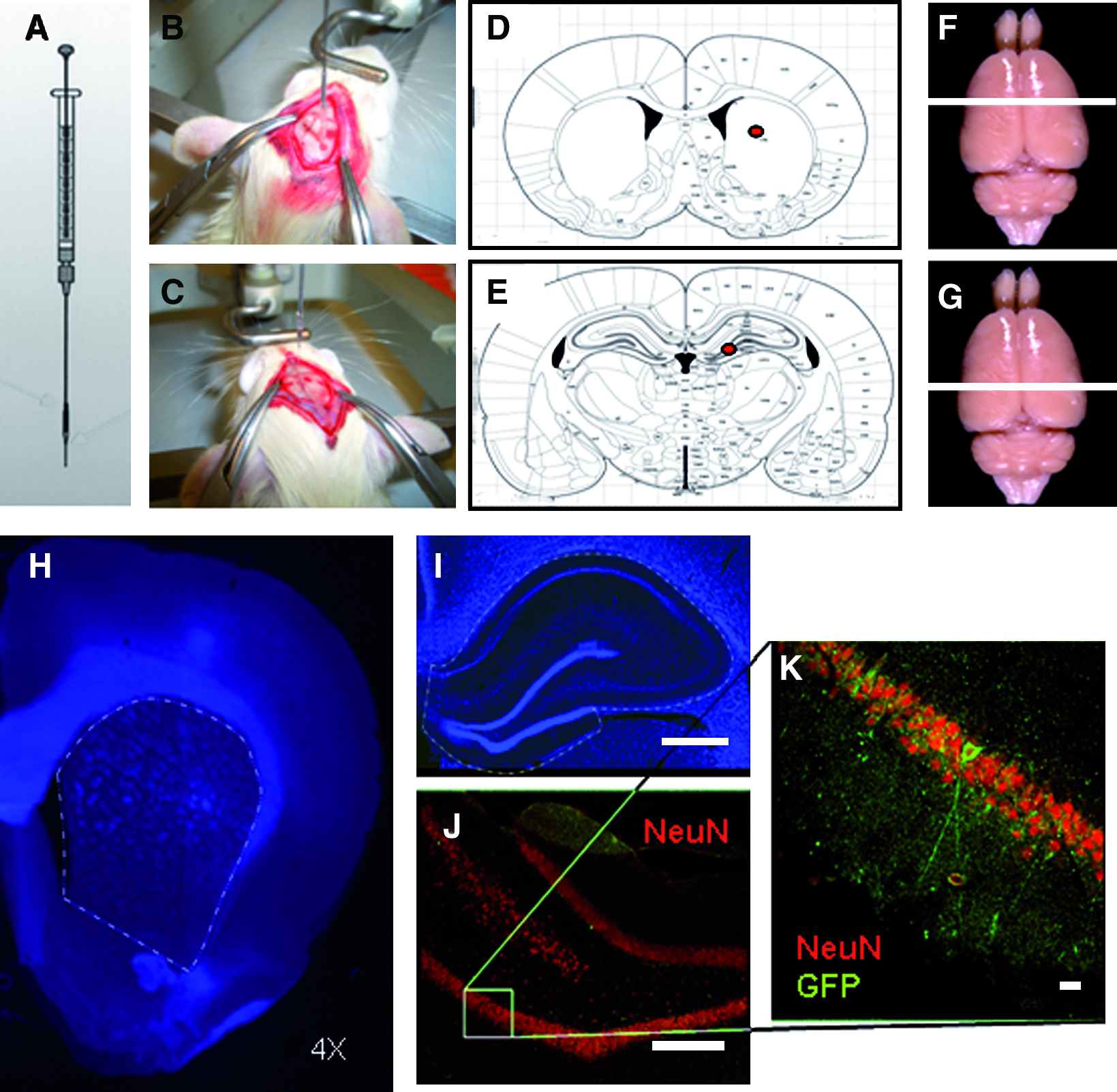
Methodology employed for stereotactic implantation of cells and stereological quantification. (
Six-week-old Sprague-Dowley rats (approximately 190 g) from the same strain used for cell isolation were submitted to general intraperitoneal anesthesia of cetamine and diazepam, and then secured to a Cunningham stereotactic frame (Stoelting Co.). All surgical and experimental procedures were approved by the Research Ethics Committee of the Albert-Ludwig University in Freiburg. A longitudinal incision was performed along the midline, and the galea was gently pared from the bone, the periost removed, and bregma identified. The coordinates used for implants into the striatum were anteroposterior (AP) +0.5 mm, lateral (L) +2.5, and horizontal (H) −4.7 (from dura). Coordinates used for the location of the hylus of the dentate gyrus were AP −3.3 mm, L +1.2, and H −3.4. Thus, 20 animals received implants in the dentate gyrus, and 20 animals in the striatum, at a point away from the ventricular wall.
The cells were detached from the culture flasks after 3-min incubation using trypsin–ethylenediaminetetraacetic acid (Gibco), and resuspended at 100,000 cells/μL diluted in Dulbecco's modified Eagle's medium containing 0.05% DNAse (Sigma) to avoid clotting. A total volume of 150,000 cells were implanted per target/animal. Cell viability was assessed immediately before implant by incorporation of trypan-blue (Merck). The stability and efficiency of the transfection was also tested under epifluorescence microscopy before implantation, and a yield of cells expressing high levels of GFP was noted.
In total, 50 animals were implanted, 30 in hippocampus and 20 in striatum. From these, 30 survived for 6 weeks and 20 for 12 weeks before sacrifice and brain slicing. A detailed description of the experimental conditions is shown in Table 1. Control animals were implanted with the culture medium devoid of cells. For analysis of the importance of the maturation stage of the cells for proper integration and differentiation in the host brain, 20 animals with hippocampal implants sacrificed 6 weeks after surgery were studied. Among these, 5 animals served as controls and were implanted with the medium alone, 5 received implant of undifferentiated stromal cells cultured in the basal medium, 5 were implanted with neurospheres (cells induced to a neuronal phenotype with bFGF and EGF), and 5 animals received differentiated cells (cultured under high cAMP). To analyze the effect of surviving time on neuronal differentiation in vivo, 40 animals implanted with either medium or stromal cells were studied, according to Table 1. One out of five slice series of all 50 animals was stained with NeuN for stereological quantification, as will be explained later, and the other series of 10 animals were stained with other neuronal markers.
Animal care
Animals were confined in large cages with an enriched environment allowing them to exercise on running wheels and seek food and pellets. This method has been previously reported to promote visuo-spatial and motor learning, due to enhanced generation of new neurons in the rostral migratory stream and hippocampus.12–15
Animal perfusion and brain slicing
At 6 or 12 weeks after implantation, the animals were anesthesized using intraperitoneal cetamine and the carotid-jugular compartment perfused with 500 mL cold PBS, followed by 500 mL fresh, cold paraformaldehyde 4%. The skull was opened and the brains removed and preserved in 30% glucose in PBS for 2–3 days. After sinking, brains were kept in antifreeze solution (30% glycerol and 30% ethylene glycol in PBS) at −20°C until sectioning. For sectioning, the brains were washed in PBS, placed onto the platform of a cryostat microtome, cooled to −40°C, and sectioned at 40 μm in the coronal plane. The slices were divided and ordered into a series of five vials and preserved in antifreeze solution until staining.
Immunohistochemistry
The staining procedure was performed on free-floating slices. After successive washings with Tween 0.05% (Merck) diluted in PBS, slices were incubated in blocking solution containing 5% goat serum (Sigma), 0.3% TritonX-100 (Sigma), and PBS for 1 h at room temperature over a shaker. The tissue was then incubated with primary antibodies diluted in blocking solution at 4°C for 24 h. After this period, the slices were rinsed again five times in PBS-Tween and incubated for a further 24 h at 4°C with secondary antibodies diluted in 0.3% TritonX-100 in PBS with the introduction of 4′,6-diamidine-2-phenylindole dihydrochloride (Sigma) 1:10,000 for nuclear staining. After 24 h the slices were rinsed again, anatomically ordered, mounted on glycerine-coated glass slides, and coated with antifade solution (DAKO).
The following primary antibodies were used: anti-glial fibrillar acidic protein (GFAP) (polyclonal, produced in rabbits, purchased from Millipore, and used at 1:600), antidoublecortin (polyclonal, guinea pig, 1:3,000; Santa Cruz), anti-βIII-tubulin (monoclonal, mouse, 1:400: Sigma), anti-microtubule-associated protein (MAP)2ab (monoclonal, mouse, 1:200; Millipore), anti-NeuN (monoclonal, mouse, 1:250; Millipore), anti-NF200 (monoclonal, mouse, 1:200; Millipore), anti-GABA-amino-transporter 1 (polyclonal, rabbit, 1:500; Millipore), and antiglutamate (monoclonal, mouse, 1:5000; Millipore). The secondary antibodies included anti-rabbit, anti-mouse, or anti-guinea pig AlexaFluor 594 (Molecular Probes) used at 1:200. The nuclei were stained using 4′,6-diamidine-2-phenylindole dihydrochloride (Sigma, 1:10,000). The original GFP signal was not enhanced with specific antibodies.
Confocal microscopy
Images were obtained with a Leica (Munique) TCS SP2 confocal system (405 nm diode, ArKr 488 nm, Ar 594 nm, and Xn 633 nm lasers) using the 20 × /0.7 or 63 × /1.2 objectives, with automatic adjustment of the pinhole to obtain an airy-unit of 1 to ensure maximal confocality. Tissue was scanned at a 2 μm thickness and pictures were digitized at 2,048 × 2,048 pixels, saved in “tif” format, and subsequently assembled using Adobe Photoshop CS2.
Stereology
The stereological quantification applied here was described previously.16–19 Briefly, coronal sections of brains from 48 animals were cut on a freezing microtome at 30 μm thickness and processed for NeuN, MAP2, βIII-tubulin, and NF200 fluorescence immunohistochemistry. Coexpression of neuronal marker and GFP was analyzed under an Olympus BX61 (Olympus Europe) epifluorescence microscope equipped with a high sensitivity digital camera DP70 and Olympus stereology system C.A.S.T.® (computed assisted stereological toolbox). The cross-sectional area of the striatum or the hippocampus was outlined in the computer screen, and cells expressing both markers were counted by scanning the slide with an automatic microcator (Heidenhain), at 40 times magnification using an optic frame of 50 × 50 μm, taking care to extend focus across the entire slice thickness (40 μm) in each frame. Finally, the entire hippocampus or striatum of each animal was scanned.
Statistical analysis
Statistical analysis and graph configuration were performed using JMP 8.0 software (SAS Institute, Inc.). The distribution of the data residues was first analyzed by histograms and normal quantile plots, and the hypothesis of Gaussian distribution was tested by the Shapiro–Wallis test. On the basis of this analysis, which revealed a non-Gaussian distribution, the Mann–Whitney U-test was selected for mean comparisons, and its equivalent Kruskal–Wallis for multiple comparisons. Quantifications of the stereology are shown in mean diamond graphs, where the width of the diamond is directly proportional to the sample size, and the height represents the variance. An intersection between the diamonds in the area between the horizontal bars implies confirmation of the null hypothesis for an α error of 5%. The nonparametrical comparisons of the means are graphically represented by intersection of circles, where the size of the circle is inversely proportional to the sample size; an intersection angle between circles of less than 90° precludes statistical difference for an α error of 5%. In the bar graphs, error bars represent SEM. The significance value p is expressed using asterisks as follows: *p < 0.05, **p < 0.01, and ***p < 0.001.
Results
Distinct neurogenesis in striatum and hippocampus
To clarify the distinct neurogenic potential in the rostral striatum and in the dentate gyrus, 6-week-old rats were sacrificed and the brains were immunohistochemicaly stained against doublecortin (Fig. 2). It was demonstrated the distribution of neuroblasts doublecortin positive in the subgranular layer of the dentate gyrus, which extended dendrites to the superficial layer. By contrast, neurogenesis in striatum is restricted to a thin layer in the lateral wall of the frontal horns in the lateral ventricles, and continues ventrally in the rostral migratory stream toward the olfactory bulb. In normal conditions and in young Sprague-Dowley rats used in our further experiments, no significant neurogenesis was demonstrated at the implantation site 2.5 mm lateral from the ventricular wall.
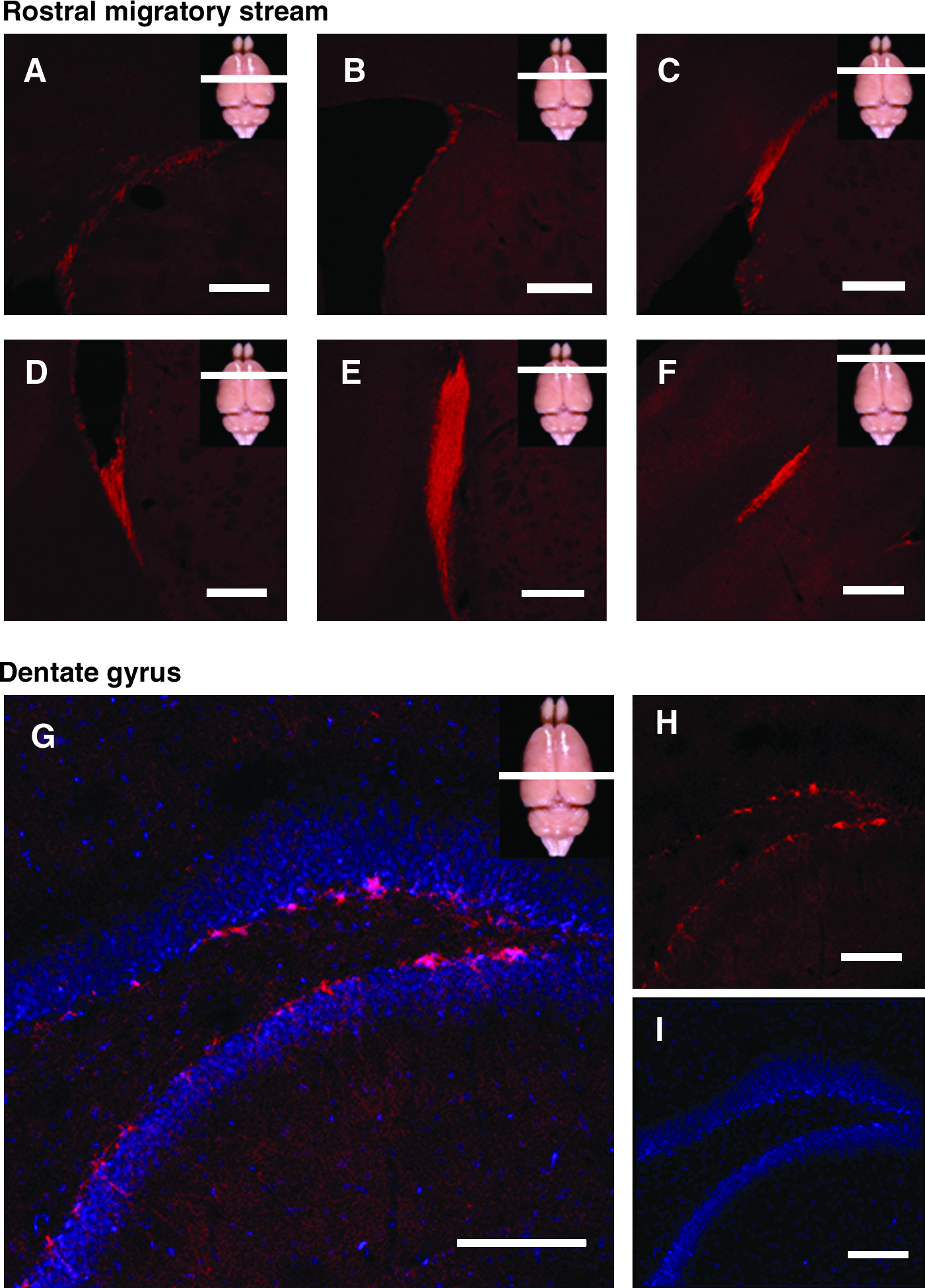
Neurogenesis in the rostral migratory stream and in the dentate gyrus. Confocal pictures of the frontal lobe (
In vitro neuronal differentiation of GFP-expressing MSCs
A population of stromal cells was successfully isolated from rodent marrow, in a similar manner to that previously described for humans. These cells have large and flat cell bodies, few elongations, and nuclei with loose cromatin. Their replication rate is high and 99.3% ± 0.5% of cells incorporate BrdU (bromodeoxyuridine) after 6 h of exposure. In this regard, very fast growth kinetics had been pointed out as important characteristic of MSCs. 20 At this stage, the cells stain strongly positive for vimentin, a mesenchymal cell marker (Fig. 3A), and weakly for the neural progenitor markers glial fibrilar acidic protein (GFAP) and nestin. The fluorescence-activated cell sorting (FACS) analysis revealed that the isolated cells expressed various clusters of differentiation from the β-integrin/fibronectin-receptor family like CD29, 44, 49d, and 49e and the stem cell marker CD90, and were negative for the hematopoietc cell marker CD45. Additionally, expression of major histocompatibility complex (MHC)-class I, but not MHC-class II, was observed (Supplemental Fig. S1, available online at www.liebertonline.com/ten). These results are in accordance with our previous report on human MSCs, 3 as well as with the previous observation of Pittenger et al. by the description of a population of stem cells on human bone marrow. 9 On the basis of the superficial cell markers, these cells cannot be considered multipotent adult progenitor cells (MAPCs, which are negative for CD44, CD45, an MHC-class I and II), considering a nomenclature proposed by some authors.
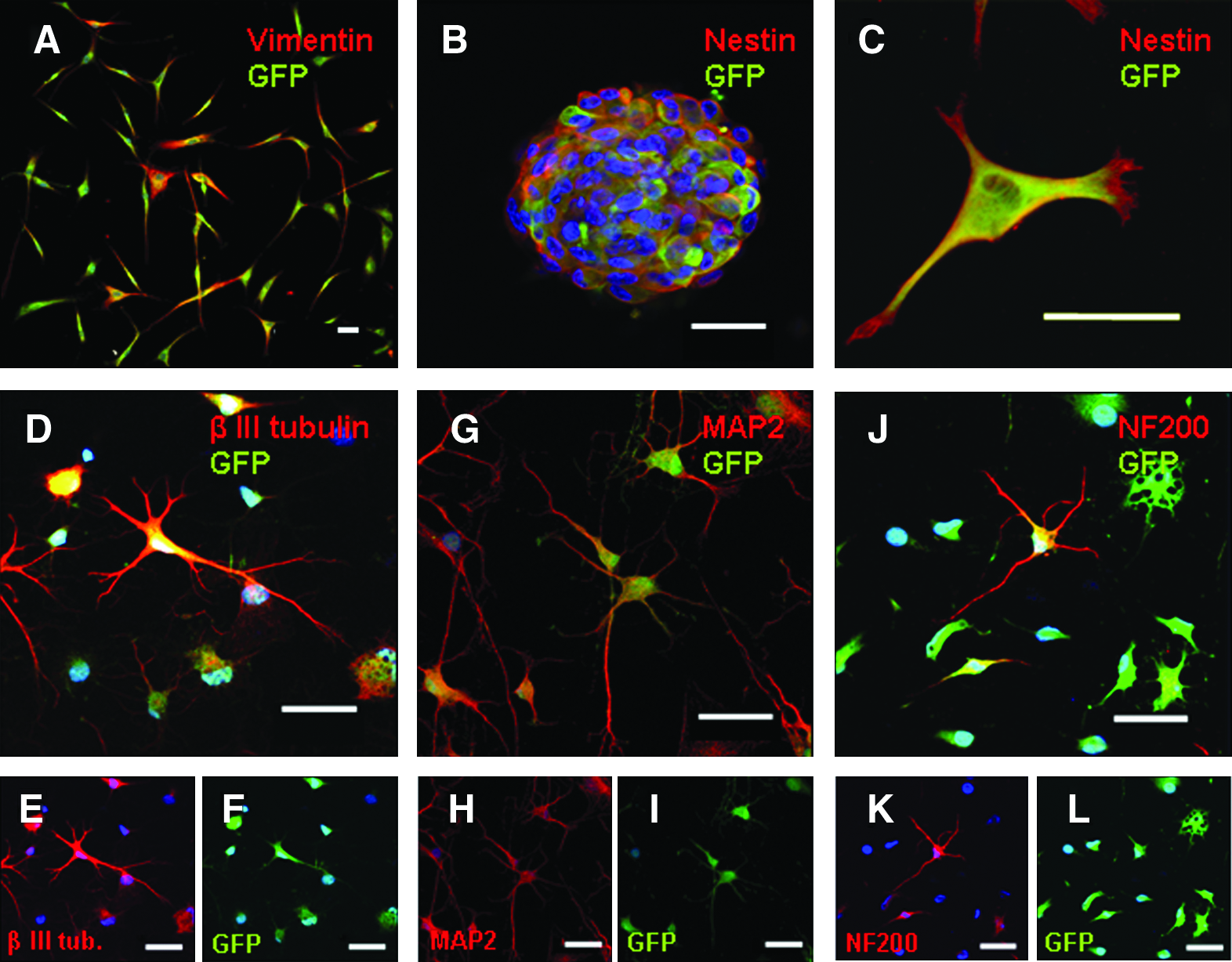
In vitro neuronal differentiation of GFP-expressing mesenchymal stem cells (MSCs). Confocal images of cell cultures at various differentiation stages. (
Cells were then transfected as described in the Materials and Methods section, and after 3 weeks, 82.5% ± 3.4% of cells expressed high levels of GFP, which remained stable throughout the experimental period (Supplemental Fig. S2, available online at www.liebertonline.com/ten). After 1 week of incubation in bFGF, EGF, heparin, and leukaemia inhibitor factor, cells build free-floating aggregates resembling neurospheres, structures formed during expansion of fetal neural tissue. These structures test strongly positive for nestin and GFAP (nestin staining is shown in Fig. 3B, C). After plating onto poly-
Functional evidences of neuronal differentiation in vitro
At the end of differentiation period, cells were transferred to a recording chamber of an electrophysiological setup and were patched in voltage-clamp mode (Fig. 4). We were able to record outward K-currents with mean amplitude of 807 ± 230 pA and inward Na-currents of 110 ± 22 pA. To prove Na+ conductances, inward currents were completely blocked by 1 μM tetrodotoxin applied to the bath solution. The current–voltage relationship showed a clear voltage dependence of the activation curve, nice superimposed to a mathematical model derived from the Boltzmann and Nernst equations, which predict current flow through semipermeable membranes in neuronal cells. 3

Electrophysiological evidence of neuronal differentiation of rodent MSCs. Electrophysiological recordings in voltage-clamp on rodent MSCs after cultivation in bFGF-EGF for 7 days and iso-butyl-metyl-xantine (IBMX)-cAMP for 3 days. Each trace represents an average from five sweeps at one voltage pulse. The leakage and capacitive currents were subtracted online using a P/−4 protocol. (
Different graft survival and integration in the host brain
Survival and integration of the grafted cells in the host brain significantly differed depending on the implant site (Fig. 5). While in the hippocampus a small graft was seen in all animals, with no surrounding astrocytic reaction or cell infiltration, and complete preservation of the hippocampal cytoarchitecture, in striatal large grafts were evident along with interstitial spread of GFP and exuberant astrocytic infiltration. These astrocytes were not derived from the implanted cells, since double-staining GFAP-GFP was rarely seen. This denotes a more marked migration of the implanted cells away from the graft in the hippocampus, yet degeneration in striatal cells.
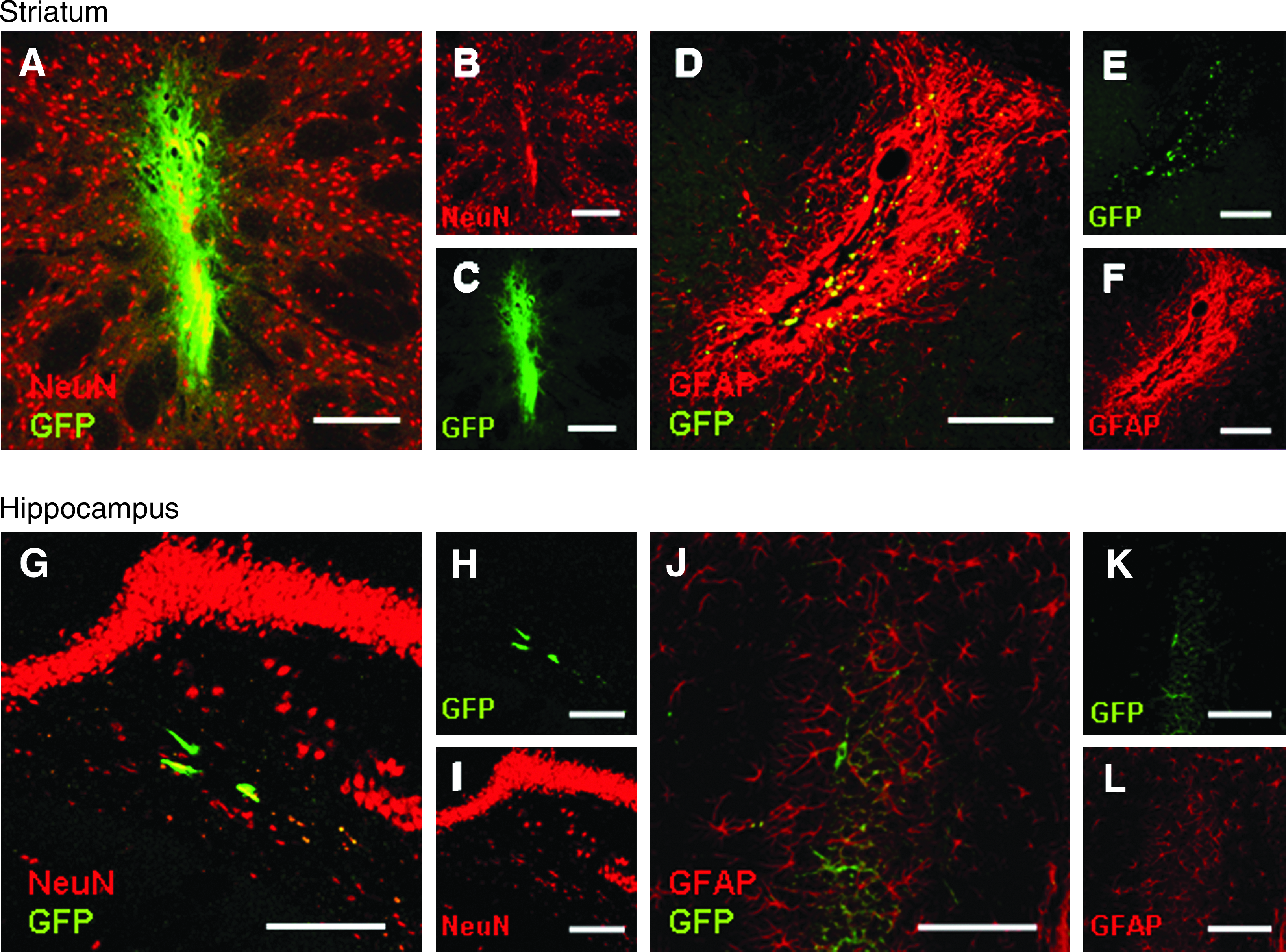
Different patterns of graft integration in the host brain. In the top row is shown the staining of the grafted area in striatum, and in the bottom row, the grafted hippocampal region. (
Mature neuronal phenotypes were observed within the hippocampus on confocal microscopy. Figure 6 depicts βIII-tubulin, NeuN, and NF200 positivity. Quantifications revealed that 90.5% ± 0.02% of all GFP-positive cells expressed NeuN, whereas 90.4% ± 0.02% were positive for MAP2, 95.0% ± 0.02% for βIII-tubulin, and 92.0% ± 0.02% for NF200 (Fig. 7G). Synthesis of neurotransmitters was demonstrated by positivity for GABA-amino-transporter 1 and glutamate. Few GFP-positive cells were seen (mostly Dcx- or βIII-tubulin-positive neuroblasts) in the accumbens, prefrontal cortex, or olfactory bulb on those animals implanted in the striatum. Among those animals implanted in the hippocampus, GFP-positive cells were detected homogeneously in all Amon horns, dentate gyrus, and temporal neocortex. Interestingly, we found no mature neurons at these distal sites (data not shown).
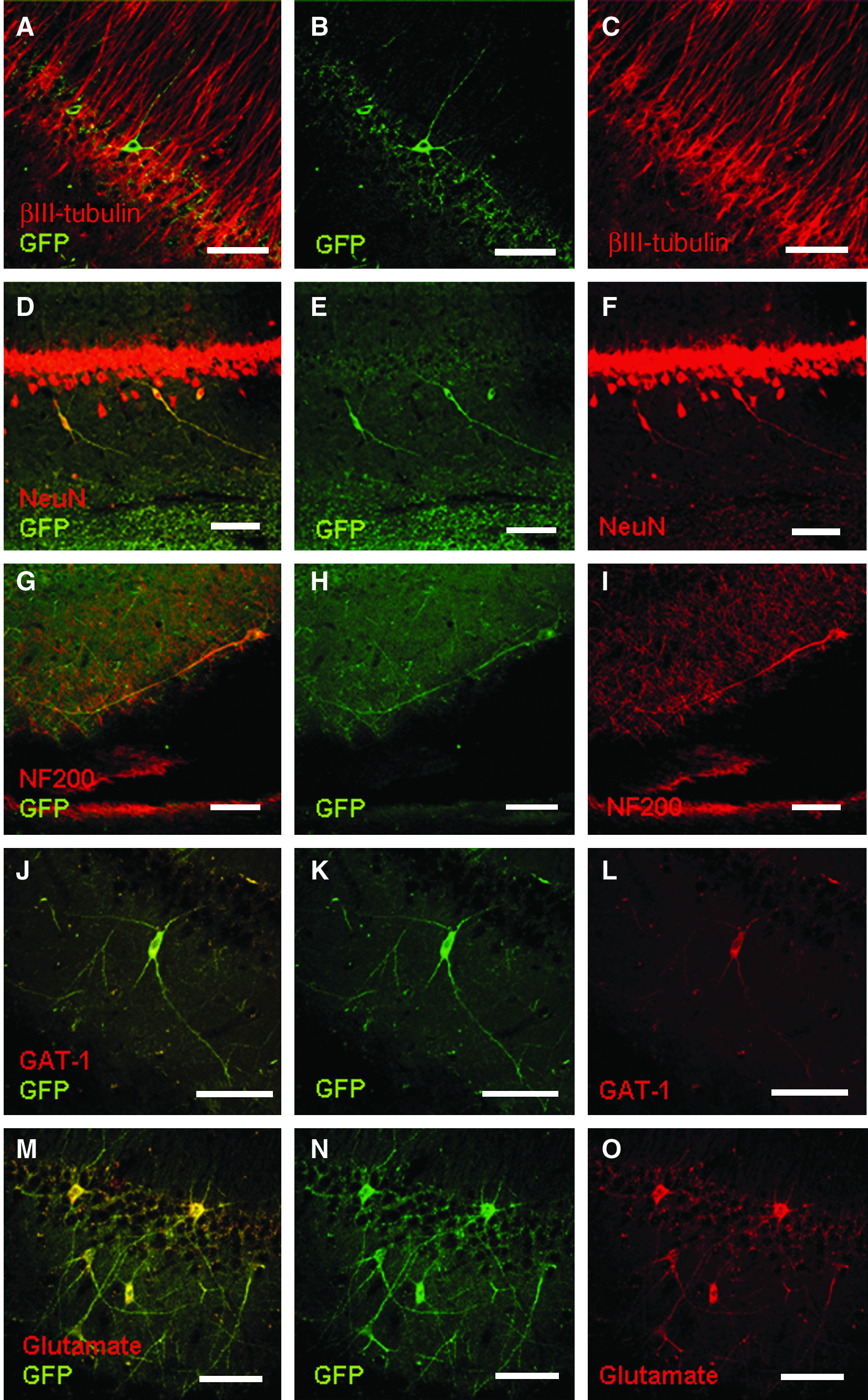
In vivo neuronal differentiation of MSCs in the hippocampal formation. Confocal images of brain slices showing neuronal differentiation of GFP-expressing cells. (
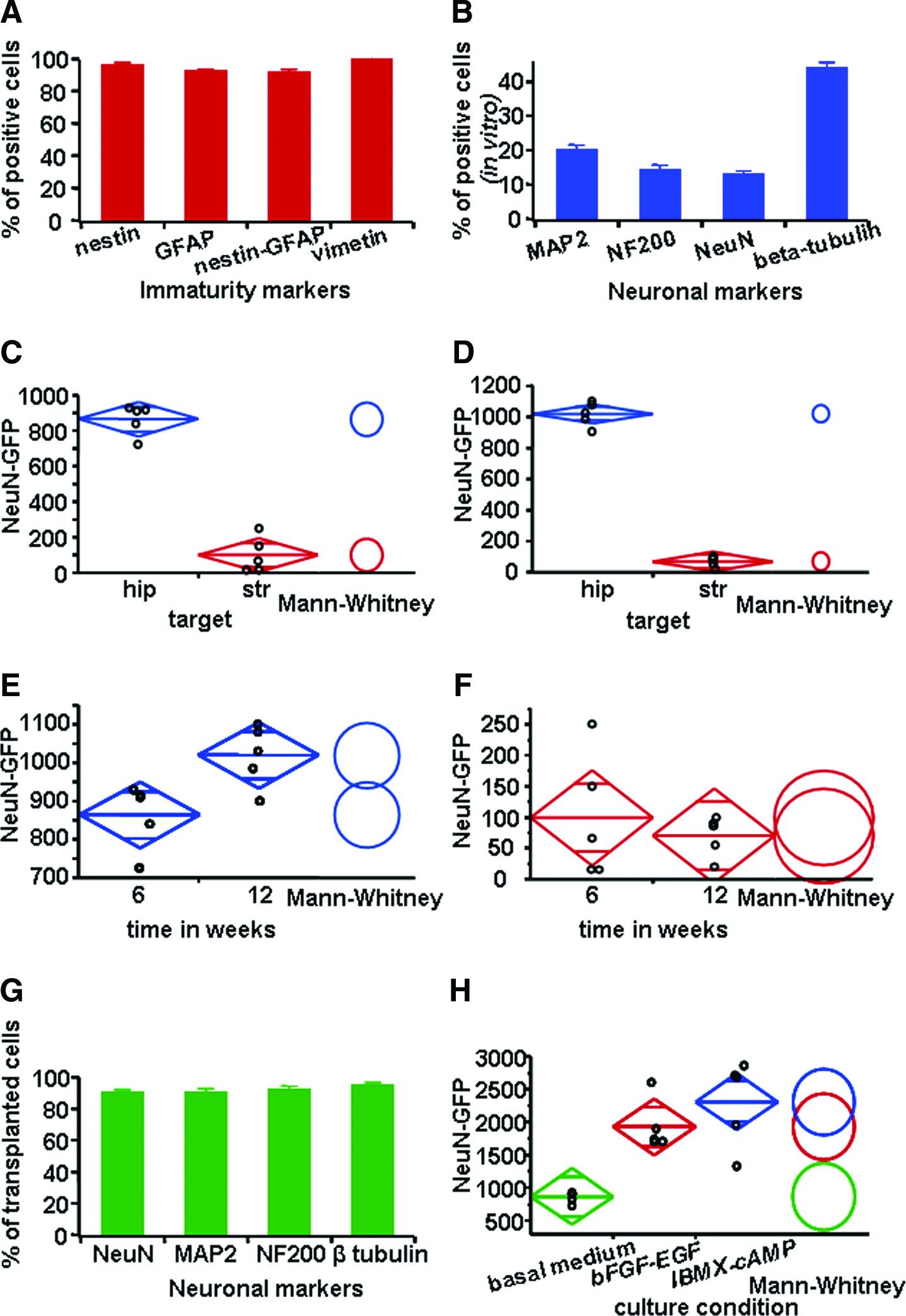
Quantification of neuronal differentiation of rodent MSCs in vitro and in vivo after transplantation. (
Neuronal differentiation in vivo differs in hippocampus and striatum
The quantification of cells double positive for NeuN and GFP revealed in the group implanted with stromal cells and sacrificed after 6 weeks 864 ± 38 cells in the hippocampus and 99 ± 45 in the striatum (Mann–Whitney U, p < 0.01, n = 10; Fig. 7C). After 12 weeks, 1020 ± 35 double-positive cells were counted in hippocampus, whereas only 70 ± 15 in striatum (p < 0.01, n = 10; Fig. 7D). Regarding the total number of living cells derived from mesenchyma (only GFP positive), we observed at 6 weeks 1075 ± 28 cells in hippocampus and 167 ± 65 cells in striatum (p < 0.01, n = 10), and at 12 weeks 1115 ± 40 and 260 ± 66, respectively (p < 0.01, n = 10). Analyzing separately the group of animals implanted in hippocampus and striatum, there was no significant difference in terms of cell survival or cell differentiation between the groups of striatal animals sacrificed at 6 and 12 weeks after implant (99 ± 45 double-positive cells in the 6-week group vs. 70 ± 15 in the 12-week group; Mann–Whitney U, p = 0.917, n = 10). By contrast, in hippocampus, neurogenesis from implanted cells continued to occur, and the absolute number of double-stained cells increased slightly from 864 ± 38 at 6 weeks to 1020 ± 35 at 12 weeks (p < 0.05, n = 10; Fig. 7E, F). No fluorescence was observed on stereology in the brains of control animals injected with the medium.
Analysis of neuronal differentiation in terms of maturation stage of implanted cells
Another important issue is whether the culture conditions, or the developmental stage of the stem cells, influence their survival and differentiation in the target structure. To address this question, we separately analyzed, 6 weeks after implantation, the animals implanted in hippocampus, where the local conditions favored a better cell survival; the four subgroups received (1) medium (control group), (2) stromal undifferentiated cells (grown in basal medium), (3) cells induced to neural differentiation under bFGF and EGF, and (4) cells differentiated under high-cAMP (Fig. 7H). We counted 864 ± 38 mature neurons derived from mesenchymal cells in the stromal cell group, 1930 ± 173 neurons derived from the induced cells, and 2313 ± 292 derived from the differentiated cells (Kruskal–Wallis p for 1 × 3 < 0.001, n = 10; for 1 × 2 <0.01, n = 10; and for 2 × 3 0.38, n = 10). Taken together, these results indicate a higher rate of neurogenesis if cells are implanted when a neuronal phenotype was already induced chemically (Fig. 7H).
Discussion
MSCs are being advocated as a promising cell source for brain repair. In general, MSCs can be easily obtained from adult tissue and isolated from other cell types in the bone marrow. They are biologically safe and have been used extensively in bone marrow transplantation in patients suffering from hematological cancer. 21 Further, the availability of MSCs allows for the transplantation of a patient's own cells, which eliminates the need for immunosuppression and its inconvenient side effects. To date, many authors have described the in vitro differentiation of MSCs into neuron-like cells.1,2 Nevertheless, complete functional maturation with electrophysiological confirmation was only demonstrated under coculture with neurons or astrocytes, and no previous work has shown the occurrence of action potentials and spontaneous synaptic activity in MSCs cultivated alone. Reports presenting evidence for transdifferentiation of MSCs into a neuronal lineage are usually based on upregulation of neuronal genes, expression of some few markers, and even synthesis of some neurotransmitters, but often weak morphological/immunocytochemical criteria and poor or no electrophysiology.22–25 Moreover, some authors claimed that the initial morphological changes undergone by MSCs during differentiation were due to the increased osmolarity of the culture media and did not imply neuronal differentiation.26,27 Another important aspect concerns the fact that undifferentiated MSCs express genes and proteins from the three embryonic layers in the early stages of maturation,28–30 and this may be misinterpreted as evidence of transdifferentiation. Recently, Fu et al. described the occurrence of trends of action potentials on one neuronal cell derived from coculture of human MSCs and human fetal astrocytes. 31 Unfortunately, there was no description on the percentage of neuronal cells, or measurements of voltage-gated ionic currents. A sporadic observation may lie in the fact that in coculture systems as described,32,33 rare neural stem cells present in the system still can give rise to mature neurons. A method of cell label would have helped to solve this question. On the basis of what was exposed, the real neurogenic potential of MSCs is still a matter of controversy.34–38 We have recently described the real but limited neurogenic potential of human MSCs in vitro in monoculture conditions, by comparing them with fetal neuronal stem cells. 3 The present work represents a further development of this previous idea, since it is demonstrated that MSCs can overcome the limitations present in the culture system in the course of neuronal differentiation, once implanted in a favorable neural microenvironment.
MSCs implanted in animal models of stroke,5,6 demyelinating diseases, 7 and spinal cord injury4,39 have been shown to promote functional recovery. The mechanisms underlying these observations, however, have remained unclear. Some evidence indicates that MSCs are able to secrete humoral factors that foster the regeneration of the CNS. 40 These factors could promote angiogenesis or recruit autologous stem cells, thus allowing tissue restoration. According to some reports, the generation of neuronal cells from MSCs proves an impossible 8 or extremely rare event. 21 In contrast, we report the generation of neuronal cells from MSCs at a significant rate of 1% of total cells implanted in hippocampus. Additionally, we could verify that 6 weeks was sufficient time for proper differentiation, where cells appeared to survive for at least 3 months with no significant cell loss.
Regarding the potential of CNS repair using MSCs, there seems to be a consensus among research groups that neuronal generation is not the central mechanism. Some authors have described motor recovery after spinal cord injury without generation of new neurons.41,42 Interestingly, generation of oligodendrocytes and axonal growth were evident, which may explain the reported observations. Similarly, other authors have failed to observe neuronal differentiation after implantation of MSCs in the retina of rats with surgical lesion to the optic nerves. 43 Given that we verified successfully neuronal differentiation in the hippocampus, it is reasonable to deduce that favorable maturation of MSCs is strongly dependent upon the implantation site. Moreover, the observation of mature neurons derived from MSCs following implant in hippocampus is supported by (1) their morphology, with long and ramified neuritic processes; (2) positivity for markers of mature neurons, such as NeuN and NF200; and (3) synthesis of neurotransmitters (GABA and glutamate).
Another cause of concern is the biological safety and low immunogenicity of MSCs. Nevertheless, these cells are currently being used for treatment of hematological cancers, with no reports of secondary malignant degeneration. Moreover, they induce very little lymphocytic activation in vitro maybe because they do not express regulatory immunogenic proteins such as CD40, 80, or 86. 44 Consequently, graft-versus-host disease is less frequently observed. In this study implantation of isogenic cells into rats did not produce a cellular infiltration within the graft or disorganization of the cytoarchitecture in the hippocampus. In the striatum, by contrast, marked astrocytic infiltration was observed along with a lower number of both surviving cells and mesenchymal-derived neurons. As it is extremely unlikely that only the animals implanted in the striatum developed graft-versus-host disease, this discrepant observation between the two brain areas may be better explained as follows. The observed astrocytic reaction may have been secondary to selective cell degeneration in this area, given that they did not differentiate in other lineages. The reason for this is most likely multifactorial, for example, the absence of trophic factors and other matrix molecules.
A striking finding in this study was the different cell survival pattern between the hippocampus and the striatum. Although neurogenesis in striatum does occur at a much lesser extent than in hippocampus in normal conditions, 45 we have demonstrated that it is restricted to a thin layer in the ventricular wall, and does not involve the core of striatum, where the cells were implanted. In a previous study, human fetal neural stem cells were grafted into both regions. Similarly, graft development and cell differentiation and migration was very different between the two target sites, 46 indicating that this phaenomenom is not dependent on the implanted cell resource but directed by local molecular cues. Among the possible factors that might support neurogenesis in specific brain areas, local astrocytes, young neurons, and blood vessels might play important roles. 47 Nevertheless, these factors remain largely unknown. In fact, neural stem cells are present in virtually the entire nervous system, but neurogenesis occurs in adult life in only two regions, namely, the subgranular zone of the hippocampal dentate gyrus, and the frontal subventricular zone/rostral migratory stream system. In other areas, cell differentiation is directed toward gliogenesis. Thus, the identification of factors that promote neurogenesis is of central relevance. Previous reports have highlighted the importance of coculture with hippocampal astrocytes to achieve functional maturation of neural stem cells in vitro.32,33 According to these authors, hippocampal astrocytes have the ability to drive neurogenesis by secreting humoral factors, and also by building GAP junctions with maturating neurons, or even by promoting synaptogenesis between mature and immature neuronal cells. Also, as previously described, electrical inputs play a central role in maturation of neuroblasts in hippocampus.48–50 Other evidence supports the notion that an astrocytic network is essential for proper maturation of neuronal precursors. 51 Accordingly, it was reported that the Wnt-catenin pathway is very active in neuronal hippocampal precursors during adult life. Specifically, Wnt3 expression was observed in these cells, where its hyper-expression promotes neurogenesis both in vitro and in vivo. Additionally, Wnt3 blockade was shown to virtually abolish neurogenesis in vivo. 52
Another secondary aspect addressed in the present report was the tendency to attain enhanced differentiation by implanting mesenchymal cells with a better defined neuronal phenotype as opposed to cells in their immature form. Maturation was achieved after induction with bFGF and EGF, and subsequent cultivation under increased cAMP conditions. Since the earlier studies on neural transplantation by Bjorklund and colleagues,53,54 it has been generally acknowledged that functional integration of grafts in the CNS is facilitated when immature/fetal tissue is used. Immature cells retain the ability to grow axons and build new synaptic contacts. With respect to MSCs, however, this dogma seems not to apply. In the case of MSCs, differentiation of the implanted MSC cells was more marked when they were already induced to acquire a neuronal phenotype. Perhaps at this stage, a “point of no return” in the maturation process has been reached. Another possibility might be that the stage reached by a mesenchymal cell after culture in bFGF/EGF and high cAMP is no different to neuroblasts formed from fetal neural stem cells, although some cells are positive for the maturity markers such as NF200 and MAP2, as demonstrated.
On the basis of our findings, we can conclude that the neurogenic potential of MSCs is strongly dependent on the implant microenvironment. The further identification of pro-neurogenic factors specifically present in the hippocampus but not in other brain areas could foster the development of restorative strategies based on MSCs and pave the way for future clinical application of autologous MSCs for CNS-repair.
Footnotes
Acknowledgments
The present study was sponsored by Deutsche Forschungsgemeinschaft, Deutscher Akademischer Austauschdienst, the German Parkinson Foundation and Bundesministerium für Bildung und Forschung, Germany. The authors would like to thank Prof. Josef Bischofberger for the assistance concerning electrophysiology; Johanna Wessolek, Brunhilde Baumer, and Donata Maciaczyk for their excellent technical support; and Dr. Jarek Maciaczyk and Dr. Anna Papazoglou for the fruitful discussions concerning the scope of this research.
Disclosure Statement
No competing financial interests exist.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.