
Abstract
The study aimed to evaluate osteogenic properties of hydroxyapatite (HA) scaffold combined with extracellular matrix (ECM) derived in vitro from rat primary calvarial osteoblasts or dermal fibroblasts. The cellular viability, and the ECM deposited onto synthetic HA microparticles were assessed by MTT, Glycosaminoglycan, and Hydroxyproline assays as well as immunohistochemistry and scanning electron microscopy after 21 days of culture. The decellularized HA-ECM constructs were implanted in critical-sized calvarial defects of Sprague-Dawley rats, followed by bone repair and local inflammatory response assessments by histomorphometry and immunohistochemistry at 12 weeks postoperatively. We demonstrated that HA supported cellular adhesion, growth, and ECM production in vitro, and the HA-ECM constructs significantly enhanced calvarial bone repair (p < 0.05, Mann–Whitney U-test), compared with HA alone, despite the significantly increased number of CD68+ macrophages, and foreign body giant cells (p < 0.05, Mann–Whitney U-test). Selective accumulation of bone sialoprotein, osteopontin, and periostin was observed at the tissue–HA interfaces. In conclusion, in vitro-derived ECM mimics the native bone matrix, enhances the osteogenic properties of the HA microparticles, and might modulate the local inflammatory response in a bone repair-favorable way. Our findings highlight the ability to produce functional HA-ECM constructs for bone tissue engineering applications.
Introduction
The recent strategies in regenerative medicine and tissue engineering assign the leading role to biomaterials that are capable of regulating and directing cellular behavior and function. Regardless of whether biodegradable or permanent, and synthetic or naturally occurring, the biomaterial scaffold has to be mechanically compatible with the native bone, integrative, porous, osteoconductive, and ideally osteoinductive. Moreover, it should not merely function as a three-dimensional framework for osteoblast growth, but also induce a specific response from progenitor (stem) cells by guiding their attachment, proliferation, differentiation, and extracellular matrix (ECM) production.3,4
Numerous attempts to develop a bone tissue engineering scaffold with both osteoconductive and osteoinductive properties failed as very few of them demonstrated in vivo efficacy. Recent experimental approaches have been conducted using mesenchymal stem cells, where the cells were induced to differentiate into osteoblasts in contact with different biomaterials.5,6 Similarities of hydroxyapatite (HA) to the bone minerals together with the HA excellent bioactivity and biocompatibility have made it a promising scaffold for bone tissue engineering. However, its effects on cellular events are not yet completely elucidated. 7
The native components of the bone ECM integrated into a scaffold may help to make it more osteogenic. 8 ECM secreted by cells on titanium scaffold in vitro effectively directed gene expression of bone marrow stromal cells.9,10 However, to our knowledge, there have been no studies aiming to generate ECM in vitro as means to increase the osteogenic properties of the HA scaffold. Thus, realizing the significance of the ECM for the progenitor cell differentiation and bone regeneration, we have aimed (1) to design a HA-ECM composite scaffold by exploiting the natural ability of the primary cells to secrete ECM proteins and growth factors in vitro, and (2) to test the osteogenic properties of the generated HA-ECM construct in vivo. To address that, we seeded rat primary calvarial osteoblasts and primary dermal fibroblasts onto synthetic HA microparticles, and cultured them for 21 days in vitro to produce HA-ECM composite constructs that were later assessed in vivo using a rat calvarial critical-sized defect model.
Materials and Methods
Isolation and culture of rat primary osteoblasts and dermal fibroblasts
Primary calvarial osteoblasts were isolated from the parietal and frontal bones, and dermal fibroblasts from the dermis of 2- to 3-day-old Sprague-Dawley rat pups by sequential digestion with collagenase as previously described.11,12 Briefly, the calvarial bones and dermal tissue fragments were aseptically dissected, cut into small fragments, and placed into Dulbecco's phosphate-buffer saline (DPBS, GIBCO, Invitrogen Inc.) containing 100 U/mL penicillin and 100 μg/mL streptomycin (Invitrogen Inc.). The calvarial bone fragments were incubated in 1 mg/mL collagenase A solution (Roche Diagnostics) for 15 min at 37°C. The supernatant was discarded, and the collagenase digestion was repeated twice for 30 min. The supernatant was collected each time, and mixed with alpha-minimum essential medium (αMEM) (GIBCO, Invitrogen Inc.) containing 10% heat-inactivated fetal bovine serum (FBS) (Invitrogen Inc.). The fractions were pooled together. The dermal biopsies were digested once with 1 mg/mL collagenase A solution for 2 h. The primary cells were dissociated from the remaining tissue fragments using a sterile 70 μm cell strainer (Falcon, GIBCO, Invitrogen Inc.), and seeded into T-75 flasks (GIBCO, Invitrogen Inc.) with αMEM containing Penicillin (50 units/mL) and Streptomycin (50 μg/mL), and 10% FBS at the density of 2 × 104 cells/cm2 at 37°C in a humidified atmosphere of 5% CO2.
In vitro-generated ECM scaffolds
In the current study, synthetic HA microparticles (CAPTAL® Plasma Biotal Ltd.) were used as a platform to generate the constructs. The HA was a kind gift from Dr. Salvador Boros (Institut Quimic de Sarrià, Universitat Ramon Llull, Spain). The sintered CAPTAL® (2 h at 1250°C) is of high purity (Ca10(PO4)6 · (OH)2, ≥97.5% by X-ray diffraction) and crystalinity with the surface area ∼6–20 m2/g. It has a similar composition with the mineral content of the human bone, and has been the precursor material in several biomaterial developments. 13 The HA microparticles were sterilized with 70% ethanol, dried out, transferred into 24-well cell culture plates (GIBCO, Invitrogen Inc.) (10 mg HA/well), and incubated overnight in αMEM supplemented with 10% FBS, penicillin (50 units/mL), and streptomycin (50 μg/mL) at 37°C in a humidified atmosphere of 5% CO2.
The primary rat calvarial osteoblasts or dermal fibroblasts were seeded onto preincubated HA microparticles at the density of 2 × 105 cells/10 mg HA/well. To increase the ECM synthesis by the rat primary cells, the culture medium was supplemented with 50 μg/mL ascorbic acid (l-AA) (Sigma-Aldrich), and 2/3 of the medium was changed every third day. The cells were cultured for 21 days (Fig. 1A).

Experimental outline
Cellular viability within HA-ECM scaffolds
The viability of the rat primary cells seeded onto HA or plastic into the 24-well plates was assessed by MTT colorimetric assay according to manufacturer's instructions (Roche Inc.). The MTT assay is based on the ability of mitochondrial dehydrogenases of the viable cells to cleave the tetrazolium ring of MTT 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide yielding purple formazan crystals that are insoluble in aqueous solutions. The crystals are then dissolved in acidified isopropanol, and the resulting purple solution is assessed spectrophotometrically.
Briefly, 30 μL of MTT reagent was added to each well and incubated for 4 h at 37°C. Then, 300 μL of solubilization solution was added to each well, and the plates were incubated overnight at 37°C. Colored formazan products were quantified by measuring absorbance at 540 nm (Labsystems Multiskan MS; Analytical Instruments, LLC). The MTT assay was performed in triplicates at days 1 and 21 of culture.
Scaffolds decellularization
HA-ECM constructs were harvested on the day 21 of static culture. Two methods were used to remove the cells from the HA-ECM constructs: (1) Triton X-100 buffer treatment, similar to previously described procedures 14 ; (2) freeze–thaw cycles in liquid nitrogen (LN2). 10 Briefly, at the end of the culture period all constructs were rinsed in DPBS, and either frozen in LN2 for 10 min, undergoing freeze–thaw cycle three times, or treated with 0.5% Triton X-100 containing 20 mM NH4OH in PBS for 3 min at 37°C. The HA-ECM constructs were washed again with DPBS and ddH2O, and stored at −80°C until the day of surgery.
DNA quantification
To assess the total DNA content within the decellularized HA-ECM constructs, the specimens were mechanically processed in RNAse-free water by using the tissue homogenizer Ultra-Turrax T25 for 10 s at 9500 rotation/min, and then sonicated for additional 30 min to completely loose the HA microparticles. The samples were centrifuged, and the supernatant optical density was assessed in NanoVue Plus Spectrophotometer (GE Healthcare) at 260 nm, being normalized to RNAse-free water.
Scanning electron microscopy of the HA-ECM scaffolds
In vitro-generated constructs (before and after decellularization) were washed three times with DPBS, and fixed in 2% glutaraldehyde in 0.1 M sodium cacodylate buffer pH 7.4 at room temperature (RT). After fixation the samples were rinsed and stored in 0.15 M cacodylate buffer supplemented with 3 mM CaCl2, pH 7.4 at +4°C until further processing. The constructs were then centrifuged (500 g), the pellet was washed in distilled water, and the suspension was transferred to poly-L-lysine–treated filters and allowed to attach for 2 min and fixed in 2% glutaraldehyde for 5 min. The filters were rinsed in distilled water and dehydrated in 70%, 95%, and absolute ethanol for 10 min each, and finally put into acetone. The filters with the pellet were then dried in a critical point dryer (Balzer, CPD 010) with carbon dioxide. After drying, the filter was mounted on an aluminum stub, and coated with carbon (Bal-Tec MED 010). The specimens were analyzed in an Ultra 55 field emission scanning electron microscope (Zeiss) at 5 kV.
Macroscopic observation and light microscopy of the HA-ECM scaffolds
Before decellularization the unfixed constructs were observed grossly at RT. Then, the constructs with and without cells were fixed in 4% buffered formalin for 24 h at +4°C. The specimens were embedded in Tissue-Tek OCT compound (Sakura Finetek Europe BV), and frozen in LN2. Sections of thickness 7 μm were cut from each frozen block. The cryo-sections were stained with hematoxylin and eosin (HE) (general morphology), Masson's trichrome (collagen fibers), and Alcian blue (glycosaminoglycans [GAGs]).
GAG and hydroxyproline assays
The samples were incubated in 1 mg/mL proteinase-K with 400 mM ethylenediamine tetraacetic acid (EDTA) solution at 56°C for 3 h to solubilize the ECM, and were processed as described previously. 15 To quantify the GAG content, 1.9-dimethylmethylene blue dye was added to diluted samples to form GAG-1.9-dimethylmethylene blue aggregate, and adapted for spectrophotometer assay in a 96-microwell plate. The light absorbance was measured at 540 nm. For the hydroxyproline (Hyp) assay, the prepared samples were hydrolyzed with 6N HCl at 110°C overnight, and later dried on a heating block at 65°C. After removal of the hydrolysis byproducts, the 4-Hyp was oxidized by n-propanol and Chloramine-T reagent, and mixed with P-dimethylaminobenzaldehyde to form chromophore. The light absorbance was measured at 540 nm. Chondroitin sulfate sodium and trans-4-Hydroxy-L-proline (Sigma-Aldrich) were used as standards to quantify GAG and Hyp content of the HA-ECM constructs. The total collagen content can be calculated by multiplying amount of the total Hyp content in each sample by a factor of 8.0, assuming that Hyp represents 12.5% of the amino acid composition of collagen in most mammalian tissues. 16 Six randomly selected constructs of the same type were used for every assay, each performed in triplicates.
Lipopolysaccharide test
The chromogenic Limulus Amebocyte Lysate (LAL) QCL-1000® test (Lonza Copenhagen, Aps) was used to assess Lipopolysaccharide (LPS) (endotoxin) level in the decellularized HA-ECM constructs. Briefly, the constructs were thoroughly vortexed and sonicated in the LAL reagent water for 30 min. Then, the samples and standard dilutions of Escherichia coli O111:B4 endotoxin were mixed with the LAL, and incubated for 10 min at 37°C. The chromogenic substrate solution was added to the LAL-sample and incubated at 37°C for additional 6 min. The reaction was stopped with 25% v/v glacial acetic acid. The optical density of the samples (in triplicates for each type) was determined spectrophotometrically at 405 nm, and the LPS concentration calculated using a standard curve.
In vivo experimental design
Twenty adult Sprague-Dawley male rats (∼350 g) were used. The rats were kept under uniform conditions for a period of least 1 week before commencement of the experiment. Free access to water and standard pelleted food was provided throughout the experiment. The experiment was approved by the Research Ethics Committee of Karolinska University Huddinge Hospital in accordance with the policy on human care and use of laboratory animals.
The rats were randomly and equally divided into four groups according to the local treatment they received: (1) decellularized constructs generated by the rat primary calvarial osteoblasts, HA-OECM (30 mg HA plus ECM); (2) decellularized constructs generated by the rat primary dermal fibroblasts, HA-FECM (30 mg HA plus ECM); (3) HA alone (30 mg HA); (4) HA mixed with TissuFleece E (TFE) a mesh-like scaffold consisting of collage type I fibrils of equine origin (Baxter AG), HA-TFE (30 mg HA plus 9 mg collagen mesh, to mimic the organic/nonorganic ratio in normal bone). Before the surgery, the constructs were washed with DPBS and uniformly minced.
The critical-sized calvarial defect model was used. 17 The rats were anesthetized by subcutaneous injection of Hypnorm (fentanyl/fluanisone, VetaPharma Ltd.) with Stesolid (Diazepam, Alpharma Aps). During the surgery the rat was maintained on a heating pad. The ViscoTears liquid gel (Novartis) was applied on eyes for the corneal protection. The rat's head was shaved, and washed with iodine solution, and an incision was made in the sagittal plane across the cranium. The skin and underlying tissues including the periosteum and temporal muscle were detached to expose the calvarial bone. An 8-mm full-thickness circular defect was created on the left parietal region using a trephine drill with a sterile saline irrigation. The defect area was evenly covered with the prepared construct or DPBS-soaked HA microparticles (HA alone) using periosteum elevator or dental plugger/spatula and forceps. The incisions were closed with single sutures in two layers.
Tissue preparation, histology, and histomorphometric analysis
All rats were euthanized by CO2 inhalation at 12 weeks after surgery. The calvarial bone was surgically retrieved, and histologically processed. The samples were fixed in 4% neutral-buffered formaldehyde overnight at +4°C, then decalcified in 12,5% ethylenediamine tetraacetic acid (EDTA), and embedded in paraffin; 5-μm serial sections were prepared parallel to the sagittal line, and stained with HE for the assessment of the general morphology and osteogenesis. Three central sections from each specimen were used for the measurement of the new bone areas using image analysis software (Adobe Photoshop CS2, Adobe Systems Incorporated; ImageJ, National Institutes of Health). The amount of the newly formed bone (NFB) was measured twice in a blinded fashion, and expressed as the percentage of the total NFB in the defect area (%).
Immunohistochemistry
The cryo-sections of the constructs before and after decellularization, and the tissue sections of the calvarial bone defects were incubated for 1 hour at RT with the primary antibodies against rat collagen type I (1:500, Cat. # AB755P, rabbit polyclonal, Millipore), bone sialoprotein (BSP) (1:500, Cat. # AB1854, rabbit polyclonal, Chemicon International, Inc.), and osteopontin (OPN) (1:200, Cat. # sc-10593, goat polyclonal, Santa Cruz Biotechnology, Inc.). Moreover, the tissue sections were additionally immunostained with the primary antibodies against periostin (1:1000, Cat. # RD181045050, rabbit polyclonal, BioVendor GmbH), and rat macrophages (1:500, CD68 [ED1], Cat. # MCA341GA, mouse monoclonal antibody, and 1:1000, CD163 [ED2], Cat. # MCA342GA, mouse monoclonal antibody, Serotec).
Briefly, after deparaffinization in xylene and rehydration, the sections were treated with 0.2 M HCl for 10 min then with 3% H2O2 to block peroxidase activity. After washing in PBS, the sections were incubated with the primary antibodies for 1 h at the RT. After additional washing in PBS, the sections were incubated with the specific secondary biotinylated antibodies for 1 h at RT. Peroxidase reactions were then observed using a commercial Vectastain ABC kit (Vector Laboratories). Finally, the sections were counterstained with Alcian blue or Methyl green, and mounted for light microscopy. Primary antibody was omitted from the sections used as negative controls.
Two tissue sections from each calvarial specimen immunostained with CD68 (ED1) antibody were used to measure the macrophage (giant cell) distribution area (in pixels) using the same image-analysis software as for the evaluation of the new bone formation.
Statistical analysis
The histomorphometric data were statistically analyzed by the nonparametric Mann–Whitney U-test using the Statistica 8.0 software package (StatSoft). Student's t-test was applied elsewhere for normally distributed data. The statistical significance was defined as p < 0.05. The corresponding graphical representation was generated using Statistica 8.0, and Microsoft Office Excel (Microsoft).
Results
Morphology of the HA-ECM constructs
The primary rat cells proliferated and actively secreted ECM that incorporated HA microparticles resulting in a dense pellet-like structure on the bottom of the wells (Fig. 1B).
Scanning electron microscopy (SEM) revealed that HA microparticles were 20–60 μm in diameter, and had a granular surface (Fig. 2e). The primary cells exhibited good attachment and spreading on the microparticles, maintaining their typical flat polygonal morphology, and displaying multiple filopodia stretched out around microparticles (Fig. 2a, c, g). The HA microparticles were abundantly covered with the continuous dense fibrillar matrix at the end of the cultivation period. SEM revealed as well that Triton-X treatment preserved the in vitro-generated ECM architecture maintaining the fibrillar network (Fig. 2b, d), and much higher porosity compared with LN2 freeze–thaw treatment that disorganized the matrix into a shrunken glue-like mass with the considerable loss of fibrillar structure (Fig. 2f).

The morphology of the in vitro-generated HA-ECM constructs
The HE staining proved the presence of the evenly distributed ECM in direct contact with the HA microparticles (Fig. 2B). The ECM fibrillar arrangement was well preserved, and the matrix was positively stained with Alcian blue and Masson's trichrome, indicating the high GAG and collagen content. No large variation between osteoblast- or fibroblast-derived constructs, before or after decellularization, was found with respect to the matrix morphology.
Immunohistochemistry (IHC) revealed the presence of the Col I, BSP, and OPN in all generated constructs (Fig. 2C). These proteins were homogenously distributed throughout the ECM, being highly expressed in the osteoblast-derived constructs, and to a lesser extent in the fibroblast-derived constructs.
Cell viability and proliferation in vitro
MTT assay was performed at 24 h after seeding onto the HA microparticles, and on day 21 before the construct harvesting and decellularization. As shown on Fig. 3A, HA microparticles supported proliferation and metabolic activity of the primary cells. The proliferation of the rat primary calvarial osteoblasts significantly increased over the cultivation period, and exceeded that of the cells grown on plastic by day 21. There was a similar increase in the proliferation of the rat primary dermal fibroblasts cultivated on HA. No significant differences between the proliferation rates of the two cell types were observed.
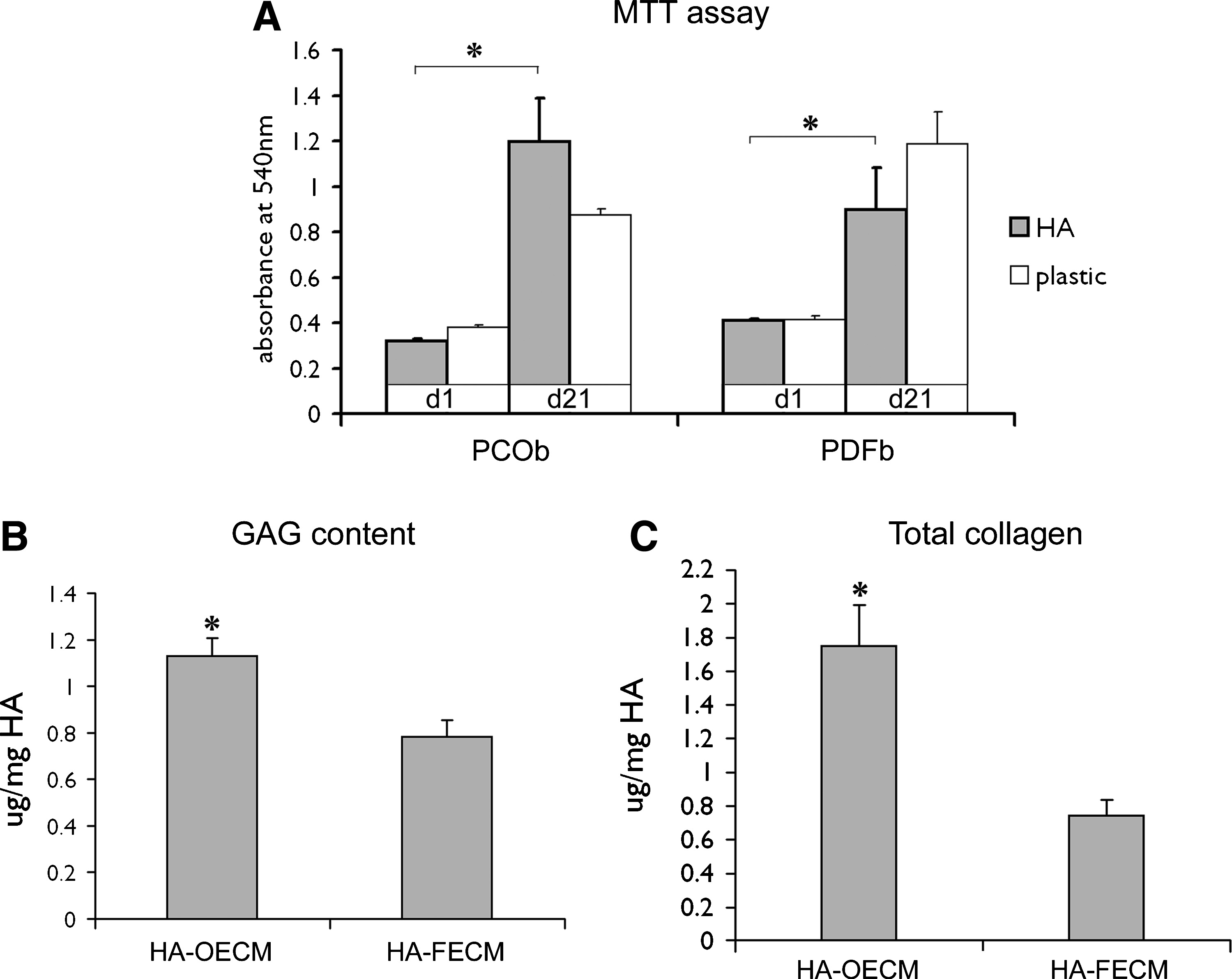
MTT activity of the rat PCOb, and PDFb after 24 h, and 21 days of culture on HA microparticles or plastic
Biochemical analysis, LPS, and DNA assays of the in vitro-generated ECM
The primary calvarial osteoblast-generated ECM had 1.5 times higher GAG content than the ECM generated by the primary dermal fibroblasts after 21 days of culture (Fig. 3B). The HA-OECM constructs also had significantly higher collagen content compare to the HA-FECM constructs (Fig. 3C). The total DNA content for both osteoblast and fibroblast-derived constructs was detected at levels lower than 0.5 ng of DNA/mg construct dry weight, as assessed by spectrophotometry (data not shown). The LPS levels were 0.008 ± 0.004 Endotoxin units (EU)/mL and 0.005 ±0.002 EU/mL for osteoblast and fibroblast-derived constructs, respectively, which corresponded to the total LPS of 2.53 mEU/animal (for HA-OECM), and 1.4 mEU/animal (for HA-FECM).
Calvarial bone repair
The HA-ECM constructs and HA-TFE complex were easy to handle (Fig. 4).
The macroscopic view of the constructs prior the implantation. HA alone
In the HA-alone-treated defects the new bone formation was mainly restricted to the areas close to the host bone margins with some samples exhibiting no NFB. The defects were filled with HA microparticles scattered among fibrous connective tissue comprising fibroblasts and blood vessels.
In all calvarial defects treated with the HA-ECM constructs (HA-OECM and HA-FECM), multiple islands of NFB were present both in the areas close to the defect margins as well as the central areas (mainly at the dural side). In most of the HA-ECM-treated defects, the HA microparticles were spread evenly within the islets of the NFB throughout the defect area being well integrated into the bone tissue. No HA-ECM-treated defects showed a complete defect restoration. However, significantly higher amount of the new bone formation was observed in the HA-ECM- and HA-TFE-treated defects, compared with HA alone treatment at 12 weeks after surgery (Fig. 5A, B). No statistically significant difference was found among defects treated with HA-ECM constructs and HA-TFE with regard to the amount of the NFB.
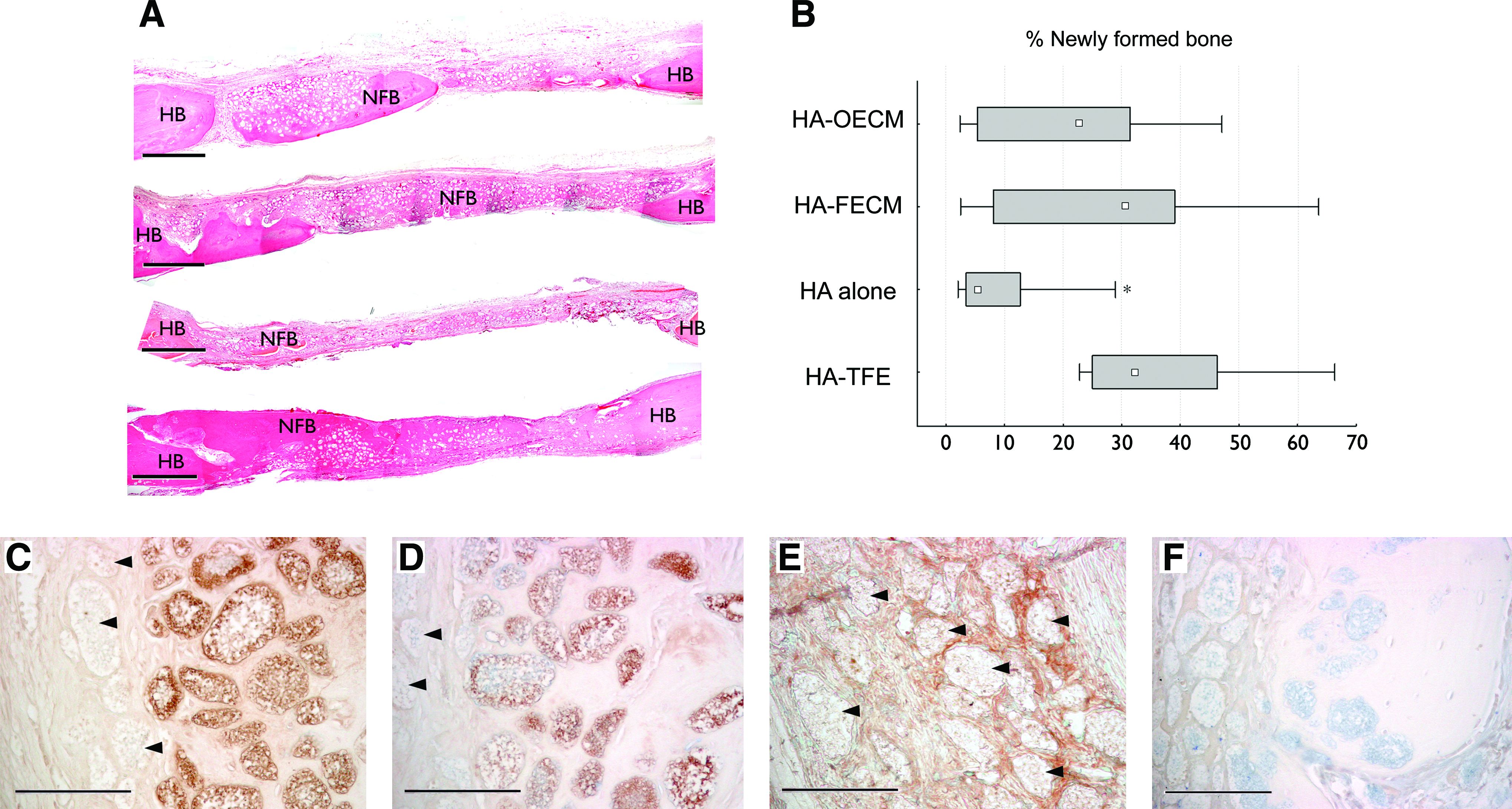
Representative histological images of the calvarial defects at 12 weeks after surgery
IHC of the biomaterial-tissue interface
The protein profile of the biomaterial–tissue interface differed between the bone-integrated and nonintegrated HA microparticles. The surface of the microparticles integrated within the NFB exhibited strong immunostaining for BSP and OPN (Fig. 5C, D). On the contrary, surfaces of the nonintegrated HA microparticles demonstrated no staining for BSP or OPN. However, the spaces between the nonintegrated HA were stained for periostin (Fig. 5E). That pattern was noted for every investigated sample, and was similar in all experimental groups.
Local inflammatory response
An active inflammatory response was still detected at the calvarial defect site at 12 weeks postoperatively proved by the presence of the large number of the macrophages and the foreign body giant cells. The HA-FECM, HA-OECM, and HA-TFE-treated bone defects revealed significantly larger amount of the CD68-positive macrophages compared with HA alone (Fig. 6). The CD68+ macrophages were mainly located onto the bone-nonintegrated HA microparticles combined with ECM or TFE (Fig. 6A–D). On the contrary, the CD163+ macrophages were scarcely present at the defect site without direct contact with the surfaces of the HA microparticles (Fig. 6E–H), nor was there any statistically significant difference in the CD163+ macrophage number among the investigated groups (data not shown).
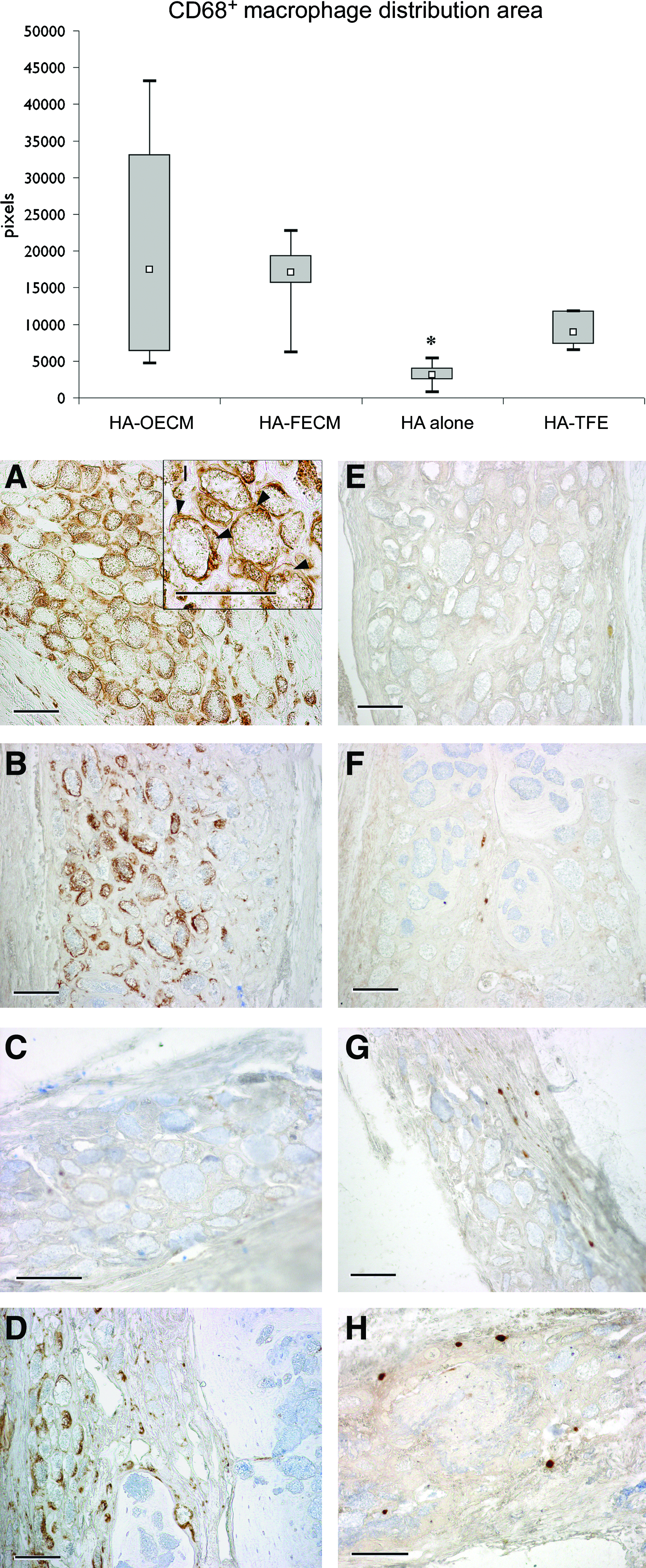
Box-plot of the CD68-positive (ED1) macrophage distribution area (pixels) (median, upper, and lower quartile; minimum and maximum values), and the representative images of the calvarial defects tissue sections immunostained with CD68 (ED1)
Discussion
Bone healing is an intricate cascade of processes involving cellular migration, adhesion, differentiation, proliferation, and synthesis of the ECM. Those processes are constantly mediated by the numerous cytokines and growth factors that instruct the phenotype of undifferentiated progenitor cells.
Alternatives to autologous bone grafts have been comprehensively investigated, including ceramics, polymers, collagen sponges, and metals. 18 One extensively investigated approach is based on the integration of the ECM proteins into a scaffold with the attempt to mimic the extracellular environment of the bone tissue. The previous study reports have indicated that in vitro-generated ECM had an impact on the osteoblastic differentiation of mesenchymal stem cells in vitro, and in vivo.9,10,19,20
In the present study it has been hypothesized that HA modified by the ECM secreted by the primary cells possesses necessary stimuli to enhance bone tissue repair, and provides a three-dimensional environment for the host progenitor cells recruitment when implanted in the bone defect area. The basic approach used in this study was to allow cells to deposit their own ECM followed by removal of the cells while preserving the native constituents and topography of the generated matrix. The primary cells required culture at high density with the media supplemented with high l-AA concentration to produce 3D ECM matrices.
The preference for the Triton-X extraction buffer method to obtain acellular constructs was based on the SEM observations. The ECM after Triton-X treatment was compared with the ECM treated with other commonly used method: freeze–thaw cycles in LN2. In the first case the ECM architecture, the fibrillar network and the porosity were better preserved, whereas after LN2 freeze–thaw treatment the ECM architecture and topography was affected, suggesting that Triton-X decellularization treatment was less harmful to the newly synthesized ECM.
Removing cellular components from the cell- or tissue-derived ECM scaffolds is considered important because of the potential adverse immune responses triggered by the cell membrane epitopes, and allogenic or xenogenic DNA. 21 To our knowledge, there are no official legal regulations regarding the DNA concentration limits in the cell- or tissue-derived scaffolds. Despite the absence of the cell nuclear structures observed with HE staining, the DNA remnants were detected by spectrophotometry in both osteoblast and fibroblast-derived constructs after decellularization. The detected DNA levels in our constructs are comparable with those in commercially available decellularized ECM scaffolds with positive clinical efficacy, suggesting that only certain higher threshold of the retained DNA may be responsible for the adverse effects. 22 Therefore, it is very unlikely that the presence of <0.5 ng DNA/mg of dry weight construct may induce a detrimental host response.
To assess the LPS levels in the implanted constructs, the LAL assay was performed according to the FDA Guidelines for the end-product endotoxin testing of human and animal parenteral drugs, biological products, and medical devices. 23 The LPS activates the innate immunity at the very low concentrations through monocytes and macrophages, and prompts the release of the pro-inflammatory mediators, therefore impairing the healing. However, the cell-derived ECM constructs in this study have had very low levels of endotoxin contamination, and thus can be considered as nonpyrogenic, according to the U.S. and European Pharmacopoeias. 23 The low pyrogenicity of the HA-ECM scaffolds may allow the reconstruction of even more extended bone defects, where several grams of the HA-ECM would be required.
In the present study we have demonstrated that HA-ECM constructs can induce a significantly better bone repair without adding the progenitor (stem) cells or growth factors, compared with the raw HA material. It is likely that the bioactive molecules, naturally resident in ECM, have stimulated the host cell–HA scaffold interactions. As demonstrated by the IHC, the major noncollagenous protein components of the bone ECM, BSP, and OPN were detected in the constructs of both osteoblast and fibroblast origin. The ability of the fibroblastic cells to express in vitro the noncollagenous proteins such as OPN was previously reported.24,25
The data obtained from the in vivo experiments have indicated that the cellular origin of the ECM is not essential for the outcomes of the bone repair induced by HA-ECM constructs. The amount of the NFB was similar in the HA-FECM, HA-OECM, and HA-TFE-treated defects. However, the dermal fibroblasts would be clinically preferred source for the ECM production in vitro as the harvesting of dermis entails much less morbidity for the patient.
Collagen matrix (TFE) in combination with HA was chosen as a positive control. The TFE is widely used as a haemostatic agent, particularly in the field of thoracic and cardiovascular surgery, able to promote granulation tissue formation, and has also been suggested as a scaffold for the tissue engineering purposes. 26 Like many other animal tissue-derived matrices, the TFE has acceptable tolerance for xenogeneic recipients since the main components of ECM are generally conserved among species. However, the in vitro-derived ECM offers many advantages, such as better biocompatibility, biodegradability, and the lack of the risk for the cross-species transmission of the infectious diseases.21,27
In the current study the levels of CD68+ macrophage infiltration were positively correlated with the presence of the ECM in the implant, and with the amount of the NFB. The lysosomal glycoprotein epitopes recognized by CD68 (ED1) antibody are expressed by most macrophage populations as well as peripheral blood monocytes, and bone marrow precursors, 28 whereas CD163 (ED2) antibody is specific for recognizing a membrane antigen of tissue-resident macrophages. Studies on functional and phenotypic diversity of the mononuclear macrophage populations in the cell-mediated immune responses have indicated that CD68+ macrophages are characterized by the production of large amounts of reactive oxygen intermediates and pro-inflammatory cytokines like IL-1, IL-12, and TNFα, 29 whereas the macrophages expressing CD163 surface marker produce high levels of IL-10 and TGF-β, inhibit the release of pro-inflammatory mediators, promote angiogenesis, and recruit cells for tissue remodeling.30–32 In our study the macrophages and the multinucleated giant cells in contact with the HA microparticles expressed CD68 but not CD163. Obviously, the presence of CD68+ cells in the intimate contact with the implanted biomaterial is critical for the fate of the implant integration; indeed, some methods have been suggested to eliminate these cells from the interface. 33 On the other hand, the CD68+ macrophages have been known to contribute to wound debridement, and may enhance the bone healing by secreting the reparative growth factors such as bone morphogenetic proteins. 34 We may speculate that the CD68+ macrophages found at 12 weeks after surgery could have shifted their phenotype from pro-inflammatory in the early healing period to pro-regenerative as healing progressed, as has been noted for bone–tendon interface repair. 35
The macrophages express multiple surface receptors, many of which interact with OPN, which is upregulated at the inflammatory sites such as the biomaterial implant area.36,37 Moreover, OPN was shown to modulate macrophage adhesion, migration, and cytokine release both in vitro and in vivo, and to play multiple roles in the cell-mediated immunity and wound healing.38,39 However, in the current study, an increase in the macrophage infiltration could not be attributed to OPN as its localization was limited merely to bone-integrated HA particles where no CD68+ macrophages were observed. Additionally, the foreign body giant cells residing on the bone-nonintegrated OPN-negative HA microparticles are most likely formed due to the lack of OPN inhibitory effect on macrophage fusion and giant cell formation, as previously shown in vitro. 40
A bone tissue–biomaterial interface layer rich in the noncollagenous proteins has been well documented. 41 In the current study we have observed a selective accumulation of the specific proteins at the tissue–HA microparticle interface, and have confirmed the importance of the BSP and OPN for the integration of HA into the mineralizing tissue. The presence of the periostin next to the nonintegrated HA microparticles reported in our study has corresponded with other observations showing the prominent periostin presence in the collagen-rich tissues opposing two mineral phases, such as periodontal ligament and cranial sutures. 42 High levels of periostin mRNA expression has been detected in preosteoblats and undifferentiated mesenchymal cells at the early stage of fracture healing, further being attenuated after 2 weeks, and resulting in the loss of periostin mRNA expression in the mature osteoblasts as mineralization progressed. 43 In the current study, however, the presence of the periostin at 12 weeks after surgery may represent the continuous fibrous connective tissue remodeling, and may impair tissue mineralization.
Taken together, the data reported herein clearly indicate that the ECM addition to the HA scaffold enhances its osteogenic properties, and leads to the increased new bone formation despite the increased inflammatory response. Apparently, the elements of the host tissue response toward the implanted biomaterial are fundamental for the successful biomaterial osteointegration, and the understanding of the principles of the precise modulation of the local inflammatory response will become the next significant advancement in the bone tissue regeneration. 44
Conclusions
The results of this study demonstrate the ability to produce functional in vitro-derived HA-ECM constructs for bone tissue engineering applications. The study reveals that in vitro-derived ECM, from the primary osteoblasts and dermal fibroblasts, mimics the native bone ECM, enhances the osteogenic properties of the HA microparticles, and might modulate the local inflammatory response in a bone repair-favorable way when implanted in vivo.
Additional studies are required to improve the osteoinductive parameters of the cell-secreted ECM as well as to fully understand the cellular and cytokine components of the host tissue response.
Footnotes
Acknowledgments
We would like to thank Kjell Hultenby for the professional assistance with SEM. This study was supported by funds from the Stockholm City Council, Karolinska Institutet, and the Swedish Institute.
Disclosure Statement
No competing financial interests exist.