
Abstract
Myoblast transplantation therapy for chronic heart failure (HF) is impaired by early donor cell death and reduced graft viability. Although epicardial implantation of cell sheets can prevent the initial loss of transplanted cells, limited vascularization subjects the sheets to apoptotic stress. We studied the efficacy of antiapoptotic bcl2 in myoblast sheet therapy for rat chronic HF. Myocardial infarction was induced by left anterior descending coronary artery ligation and HF was allowed to develop for 4 weeks. Thereafter, wild type (L6-WT; n = 16) or Bcl-2-expressing (L6-Bcl2; n = 19) myoblast sheets were transplanted epicardially. Control rats (n = 21) underwent left anterior descending coronary artery ligation and re-thoracotomy. Five rats were sham-operated in both surgeries. Four weeks after transplantation, only the L6-Bcl2 rats showed improved left ventricular ejection fraction. Their vascular density in the damaged myocardium was greater, and they had more proliferating cells. The L6-Bcl2 group had an increased amount of myocytes in the infarct area. Soluble factors from L6-Bcl2 sheets induced a 2.9-fold increase in endothelial cell proliferation, and enhanced endothelial wound healing as compared to the L6-WT sheets. These effects were inhibited by SU5416 and were thus dependent on Flt1/Flk1 signaling. In conclusion, bcl2 improves efficacy of myoblast sheet transplantation and promotes proangiogenic paracrine signaling.
Introduction
Transplantation of skeletal myoblasts has shown benefit in treatment of HF. 3 When administered as intramyocardial injections, myoblast therapy is compromised by early donor cell death and poor cell engraftment. 4 Epicardial transplantation of engineered myoblast sheets has, however, been shown to improve these shortcomings. 5 The three-dimensional myoblast sheets contain components crucial for maintaining proper cell functions: extracellular matrix, intercellular communication junctions, and adhesion molecules. 6 In transplantation therapy, the sheet structure improves cell survival as compared to conventional injections of single cell suspensions. 7 In spite of these advantages favoring myoblast sheets over cell injections, the sheets are still subject to the hostile apoptosis-promoting environment of the infarct scar. Strategies promoting donor cell survival are thus of essence in myoblast sheet transplantation therapy as well.
Recent studies have suggested that paracrine signals from transplanted cells can stimulate the dormant myocardium and induce angiogenesis. 8 Because donor cell survival and paracrine host-site stimulation are thus regarded important for effective cell therapy for chronic HF, we hypothesized that myoblast sheets genetically engineered to withstand apoptosis-promoting conditions would provide the optimal cell therapy. Moreover, in our recent study we showed that introduction of antiapoptotic bcl2 gene into myoblast sheets increased their production of potent paracrine proangiogenic meditors vascular endothelial growth factor-A (VEGF-A) and placental growth factor (PlGF). 9 Furthermore, we demonstrated that bcl2 gene therapy enhanced the efficacy of myoblast sheet therapy and promoted sheet survival in a rat model of acute MI.
In this study, our aim was to evaluate the effect of antiapoptotic bcl2 expression on myoblast sheet therapy administered on myocardial scar tissue 4 weeks after induction of MI. At this time, the acute phase of inflammation and ventricular remodeling have receded, and chronic HF has developed. This chronic model mimics the course of events and time of therapy administration in the clinical setting. We show here that the bcl2-expressing myoblast sheets significantly enhance cardiac function, neoangiogenesis via Flk1/Flt1 signaling, cell proliferation, and cardiomyocyte survival in the infarct.
Materials and Methods
Cell culture and sheets
Myoblast cell culture and sheets were constituted by our previously described method. 9 The L6 rat skeletal myoblast cell line was from the American Type Culture Collection (CRL-1458) with cells at passages 5 to 15 used for experiments. Myoblast cell sheets were formed by plating 6 × 106 myoblasts on thermoreactive cell culture dishes (CellSeed) with 15% serum and 10 mM ascorbic acid for 16 h. Intact myoblast sheets detached spontaneously from culture dishes at room temperature and were harvested for transplantation. The thickness of a single three-dimensional myoblast sheet was approximately 50 μm or 5 cell layers. 9 Human umbilical vein endothelial cells (HUVECs) were acquired from Clonetics (Lonza) and were cultured in endothelial growth medium (EGM) supplemented with 2% fetal bovine serum, bovine brain extract, epidermal growth factor, hydrocortisone, and antibiotics (all from Clonetics). Cells at passages 2 to 8 were used in the study.
Bcl-2 overexpression and transfection
The L6 cell line with constitutive human bcl2 expression was created as previously described. 9 The pBabepuro-bcl2 retroviral vector was a kind gift from Dr. Juha Klefström, University of Helsinki. 10 Cells were transfected by incubation for 48 h in the presence of retroviral vector and 8 μg/mL polybrene (Sigma-Aldrich). Transfected cells were selected by incubation in the growth medium containing 2 μg/mL puromycin for 48 h.
Animals
The study animals were 77 Wistar rats (250–400 g). Of these, 61 (79.2%) survived both operations and were divided into four groups—Group 1: control, left anterior descending coronary artery (LAD) ligation and re-thoracotomy (n = 21); Group 2: LAD ligation and L6 wild type (L6-WT) sheet transplantation (n = 16); Group 3: LAD ligation and Bcl2-expressing (L6-Bcl2) sheet transplantation (n = 19); Group 4: sham operation (repeated thoracotomy) (n = 5). Of the 16 rats that died during the study, 10 died before the second surgery, that is, before transplantation therapy was administered. Five rats (three in control and two in L6-Bcl2 groups) died during the first days of follow-up after the second surgery, and their death was associated with failure to recover from the second thoracotomy. Only one rat (L6-WT group) died of thrombus formation 2 weeks after cell sheet therapy administration. We did not detect any therapy-associated cardiac complications during the study period. All animals were euthanized at 4 weeks after the second operation. The investigation conforms with the Guide for the Care and Use of Laboratory Animals published by the U.S. National Institutes of Health (NIH Publication No. 85-23, revised 1996), and were approved by the ethics committee of the HUS/Meilahti Hospital Department of Surgery.
Surgical procedure
For all surgical procedures, rats were anesthetized subcutaneously with medetomidine (0.05 mg/kg s.c., Domitor®; Orion Pharma Inc.) and ketamine (5 mg/kg i.p., Ketalar®; Parke-Davis). Normal body temperature was maintained with a thermal plate throughout the operation. After the operation, anesthesia was partially antagonized with atipamezole hydrochloride (1.0 mg/kg s.c., Antisedan®; Orion Pharma Inc.). Buprenorphine hydrocholoride (0.05 mg/kg s.c., Temgesic®; Reckitt and Colman Ltd.) was administered for postoperative analgesia.
MI was induced by LAD ligation as described previously. 11 Animals were intubated, and respiration was maintained with a ventilator during surgery. The heart was rapidly exteriorized through a left thoracotomy and pericardiotomy. The LAD was ligated 3 mm from its origin. After ligation, the heart was returned to its normal position, and covered with pericardium to avoid adhesion to the lung and to the chest wall.
Four weeks after ligation of the LAD, all animals underwent re-thoracotomy. For animals in Groups 2 and 3, two myoblast sheets each were transplanted onto the left ventricular anterior wall. Thus, every animal in Groups 2 and 3 was grafted with a total of 1.2 × 107 cells. Group 2 received L6-WT sheets, and Group 3, L6-Bcl2 sheets. Groups 1 and 4 received no cell therapy (Supplemental Fig. S1, available online at www.liebertonline.com).
Echocardiography
All animals underwent echocardiography under anesthesia just before the second surgery (baseline) as well as 2 and 4 weeks after surgery. The echocardiographic measurements were performed with a 7.5 MHz transducer (MyLab®25; Esaote SpA). Anterior and posterior wall thickness in the diastolic phase, and left ventricular diameter in both the diastolic (LVDd) and systolic (LVDs) phase were measured in the short-axis right parasternal projection just below the mitral valves. Data were from three systolic cycles and averaged. LVDd and LVDs served in calculating left ventricular fraction shortening (LVFS) and left ventricular ejection fraction (LVEF) by the following formulas:
Histology and immunostaining
After assessment of cardiac function, the heart was excised and cut into four equal transverse parts. The two middle parts were fixed in 4% neutral-buffered formalin for 48 h, embedded in paraffin, and cut into 4-μm sections for histology and immunostaining.
Immunohistochemistry was performed with a Ventana Discovery Automate (Ventana Medical Systems Inc.). To observe vascular density, endothelial cells were stained with an antibody against von Willebrand Factor (vWF) (RB-281; Labvision Inc.). Cell proliferation was evaluated by use of anti-Ki67 antibody (RM-9106; Labvision Inc.), and cell apoptosis was detected by an antibody specifically recognizing active cleaved caspase-3 (CST #9664; Cell Signaling Technology Inc.). The sections stained for Ki67 proliferation-associated antigen were double stained for myocytes with an anti-tropomyosin antibody (MS-1256; Labvision Inc.).
Analysis of fibrosis
Amount of fibrosis was evaluated from Sirius Red–stained, paraffin-embedded sections. Fibrosis was calculated as percent fibrosis = Sirius Red–stained area/whole section area from scanned images of stained tissue sections by use of Photoshop 7.0 (Adobe Inc.).
Analysis of vascular density, cell proliferation, and apoptosis
Microscopy images ( ×100 magnification) were analyzed from six fields (two from the infracted area, two from the border area, and two from the remote area) of vWF, and Ki67/tropomyosin double-immunostained slides. Vascular density and number of proliferating cells were assessed with ImageJ (National Institutes of Health, http://rsb.info.nih.gov/ij) as described. 9 Data were collected separately for infarcted, border, and remote areas. To determine whether the proliferating cells are myocytes, we manually counted the Ki67-tropomyosin double-positive cells from six slides in each group. Apoptotic cells were calculated manually by counting the number of positive cells immunostained for cleaved caspase-3 from more than six microscopic fields per slide.
Quantification of myocytes in the infarct
The amount of myocytes in infarct area was evaluated using Photoshop 7.0 (Adobe Inc.) as the total tropomyosin-positive area within the infarct of sections stained with anti-tropomyosin antibody.
Endothelial cell proliferation, wound healing, and tubulogenesis
We collected the conditioned medium from myoblast sheets incubated in serum-free DMEM for 48 h to demonstrate the effect of myoblast cell sheet-derived paracrine mediators on endothelial cell proliferation, migration, and tubulogenesis. The conditioned medium was dialyzed with Slide-A-Lyzer 2K cassettes (Thermo Fisher Scientific) according to manufacturer's protocol to remove salts and metabolites. The dialysate was lyophilized and reconstituted in EGM for the tubulogenesis assay, and in starvation medium consisting of EGM with 50% supplement concentrations for proliferation, and wound-healing assays. For the proliferation experiments, HUVECs at 5 × 103 cells/cm2 were allowed to attach for 24 h. Cells were then washed and incubated in the starvation medium or conditioned starvation medium from WT or Bcl-2-expressing sheets for 24 h. Cells were counted from phase-contrast images (40× magnification) of triplicate wells. For endothelial cell migration analysis, HUVECs were grown in EGM at an initial density of 1 × 104 cells/cm2 for 24 h, and were growth inhibited for an additional 24 h in the starvation medium. The monolayers were wounded with a pipette tip, and before addition of the myoblast sheet-conditioned medium, the cells were washed with PBS. Cultures were photographed at 0 and 24 h.
We assessed endothelial migration by subtracting the denuded area after 24 h from the area at 0 h for each well. Analysis was by Photoshop 7.0 (Adobe Inc.). For 3-dimensional cultures in collagen lattice, we used HUVECs expressing green fluorescent protein (GFP). The lentiviral vector for GFP expression was a kind gift from Prof. Seppo Ylä-Herttuala, AIV institute, Kuopio, Finland. HUVEC-GFP collagen lattices were made from collagen I solution (Millipore), 2× DMEM, and PBS at a 1:1:3 ratio. The final concentration of collagen was 1 mg/mL. The mixture containing 1 × 104 HUVEC-GFP/60 μL was let to gel at 37°C for 15 min. EGM or conditioned EGM was then added to the wells, and was renewed after 48 h. Fluorescent images were acquired at 10× magnification after 72 h with an Olympus IX81 microscope and DP30BW camera (Olympus). Images were analyzed using Cell F software version 2.7 (Olympus). Endothelial cell proliferation, migration, and tubulogenesis assays were carried out with or without SU5416 (2 μM; Sigma-Aldrich Co.).
SU5416 is a small-molecule inhibitor of the Flt1 and Flk1 receptor tyrosine kinases. 12 For proliferation and migration assays, we pre-treated cells with SU5416 for 30 min before and during experiments with the conditioned or control medium. For the tubulogenesis assay, we included SU5416 in the collagen lattice and culture medium. No inhibitor toxicity was seen at the concentrations used in this study as evaluated by lactate dehydrogenase (Cytotoxicity Detection Kit Plus; Roche) release into the medium from HUVECs treated with the inhibitor.
Statistical analysis
All data are presented as mean ± standard error of the mean. Differences between groups were compared by analysis of variance following the Bonferroni correction. Statistical analysis was performed by Graph Pad Prism 4.0 (GraphPad Software Inc.).
Results
Cardiac performance
Cardiac performance data, as assessed by echocardiography at baseline, and 2 and 4 weeks after the second surgery, are presented in Table 1. Cardiac performance was severely reduced in LAD-ligated groups as compared to the sham-operated group at baseline, demonstrating the effectiveness of the procedure.
Values represent mean ± standard error of the mean.
p < 0.001 as compared to sham-operated group.
p < 0.05 as compared to sham-operated group.
p < 0.05 as compared to control group.
p < 0.01 as compared to sham-operated group.
p < 0.01 as compared to control group.
p < 0.05 as compared to L6-WT group.
AWTd, anterior wall thickness; PWTd, posterior wall thickness; LVDd, left ventricular diameter (all in diastolic phase); LVDs, left ventricular diameter in systolic phase (all units in mm); FS, fraction shortening (%); EF, ejection fraction (%).
At baseline, all LAD-ligated groups presented with a similar decrease in LVEF. At 2 weeks, however, the LVEF of the control group declined further, but the LVEF of the sheet-treated groups remained comparable to baseline (p = 0.011 for L6-Bcl2 and p = 0.048 for L6-WT as compared to control). At 4 weeks, the LVEF of the L6-Bcl2 sheet-treated group exhibited continuous improvement (p = 0.004 versus the control) (Fig. 1a). Moreover, at study end, animals in the L6-WT and L6-Bcl2 groups had significantly higher anterior wall thickness than did the control group (0.56 ± 0.016 for control; 0.64 ± 0.023 for L6-WT, p = 0.015; and 0.62 ± 0.020 for L6-Bcl2, p = 0.034).
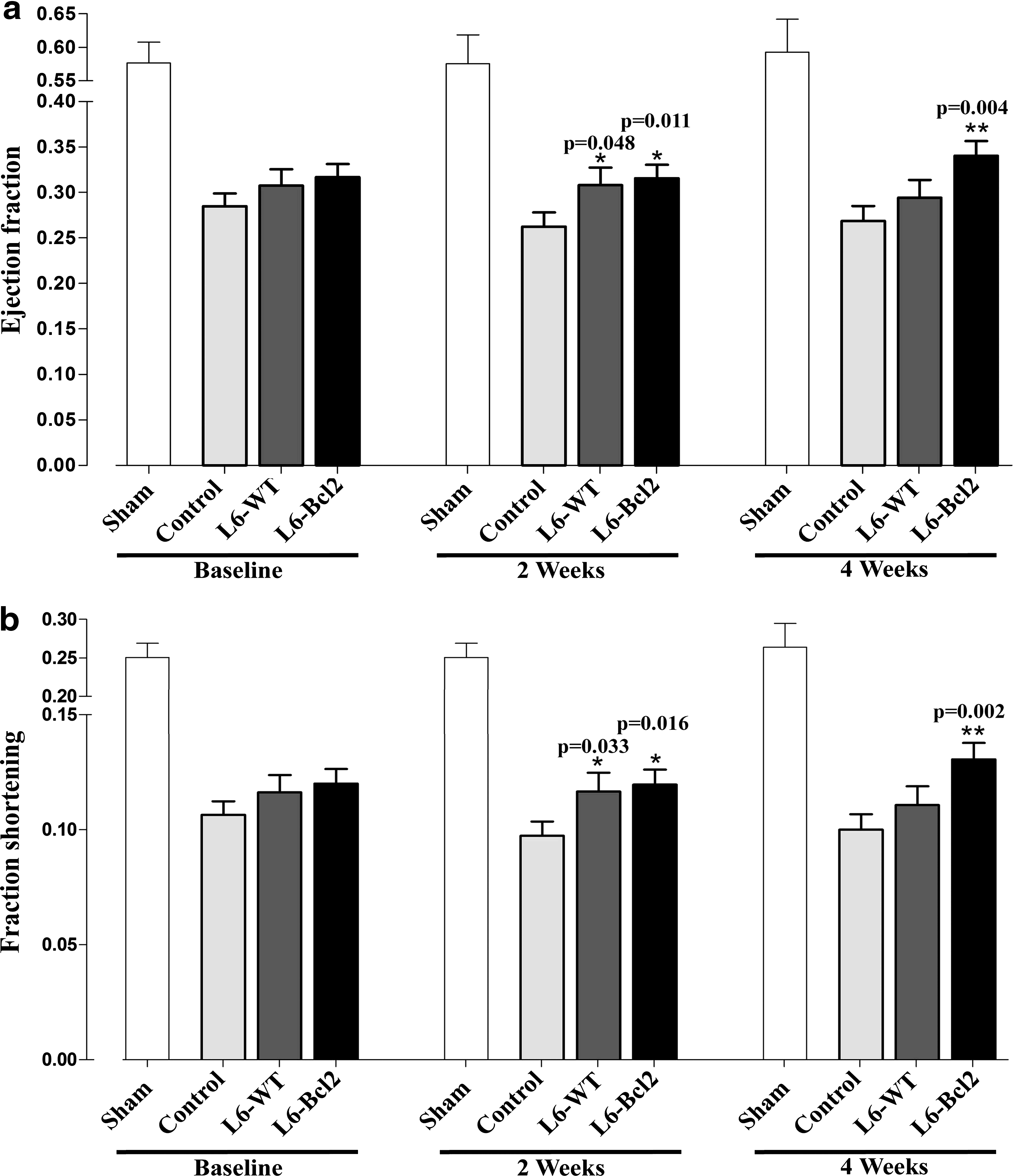
Analysis of cardiac function. Myocardial infarction was induced by ligation of the left anterior descending coronary artery (LAD) in all rats except for a sham group receiving thoracotomy only. All rats underwent re-thoracotomy 4 weeks after ligation, and wild type (n = 16, L6-WT) or Bcl-2-overexpressing (n = 19, L6-Bcl2) myoblast sheets were transplanted. Control rats (n = 21) and sham-operated rats (n = 5) received no therapy. Echocardiography data for
Fibrosis
To study whether myoblast sheet therapy can counteract or reduce the amount of fibrotic tissue already developed in chronic HF after MI, we assessed the percentages of fibrosis in histology sections stained with Sirius Red. The amount of fibrosis was 3.4% ± 1.2% in sham, 24.8% ± 1.2% in control, 24.2% ± 1.3% in L6-WT, and 24.5% ± 1.0% in L6-Bcl2-sheet treated groups. No significant differences or reduction in pre-developed fibrosis by sheet therapy thus occurred between the LAD-ligated groups with or without sheet therapy.
Angiogenesis
Paraffin-embedded myocardial transverse sections were stained for expression of the endothelial antigen vWF as a marker of vascular density. The infarcted area of the L6-Bcl2 group stained significantly more intensely than did the control group (p = 0.049). No difference emerged between vascular densities of the L6-WT and the control group in the infarcted area. In the border zone, more intense vWF staining was evident in both of the sheet therapy groups than in the control group (p = 0.033 for L6-WT and p = 0.003 for L6-Bcl2) (Fig. 2a, b). We then evaluated vascular density in the combined infarct and border area and found that the L6-Bcl2 group displayed a significantly higher amount of vasculature than did the control (p < 0.001) and L6-WT groups (p = 0.011). Moreover, the difference between the L6-WT and control groups was significant (p = 0.042) (Fig. 2a).
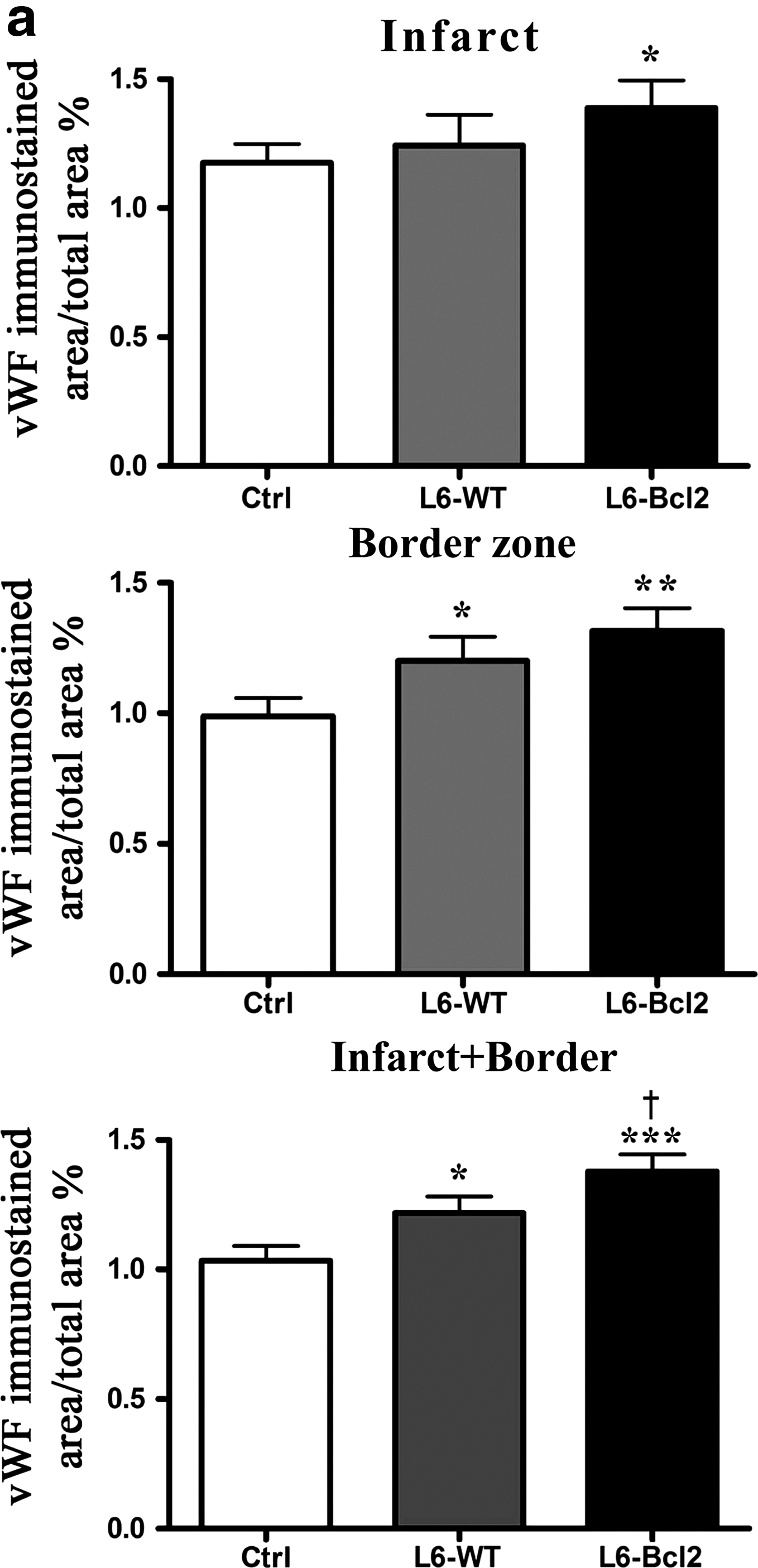
Quantitative evaluation of vascular density. Samples were collected 8 weeks after LAD ligation or sham operation (sham group) and 4 weeks after re-thoracotomy and therapy with or without (control and sham groups) myoblast sheets.

Proliferation and apoptosis in the myocardium
To investigate the effect of Bcl-2-modified myoblast sheet therapy on the balance between cell death and cell proliferation in the postinfarct myocardium, we evaluated expression of active cleaved caspase-3 and proliferation-associated Ki-67 antigen. A higher number of proliferating cells were evident in the infarct and in remote areas in the L6-Bcl2 group than in control or L6-WT groups (p = 0.028 and p = 0.007 for infarct, and p = 0.046 and p = 0.009 for remote) (Fig. 3a, b). In animals receiving L6-Bcl2 sheet therapy, the border zone displayed a significantly higher number of proliferating cells than did the L6-WT group (p = 0.044), whereas the difference from the control group was nonsignificant (Fig. 3a). Double-staining of Ki67 and tropomyosin revealed that <1% of the proliferating cells were myocytes (Control group 0.72% ± 0.31%, L6-WT 0.52% ± 0.28%, and L6-Bcl2 0.98% ± 0.34%) and that the differences were nonsignificant. We did not detect differences in number of cells positive for cleaved caspase-3 in the myocardium (Control group 40 ± 9, L6-WT 31 ± 11, and L6-Bcl2 38 ± 15).
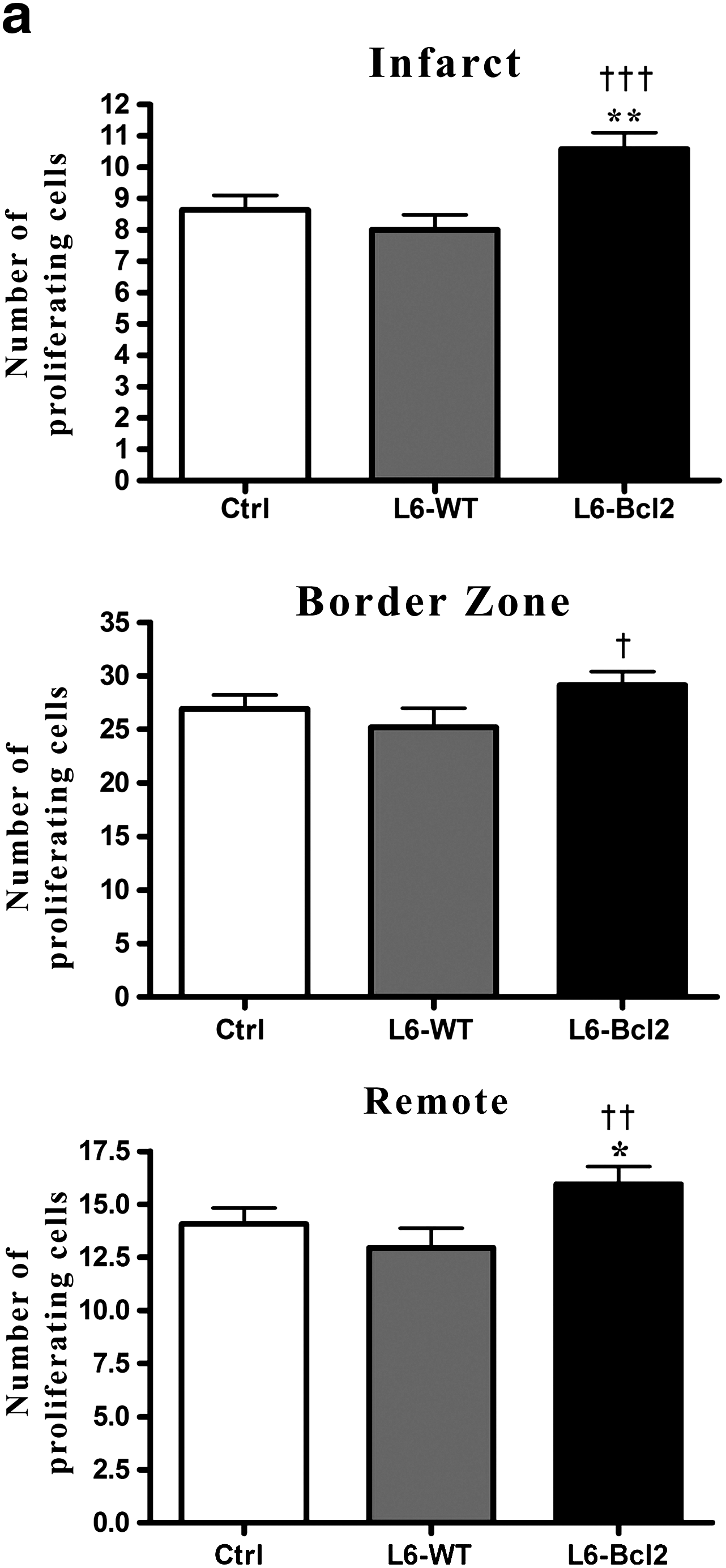
Quantification of proliferating cells in the myocardium. Samples were collected 8 weeks after LAD ligation or sham operation (sham group) and 4 weeks after re-thoracotomy and therapy with or without (control and sham groups) myoblast sheets.
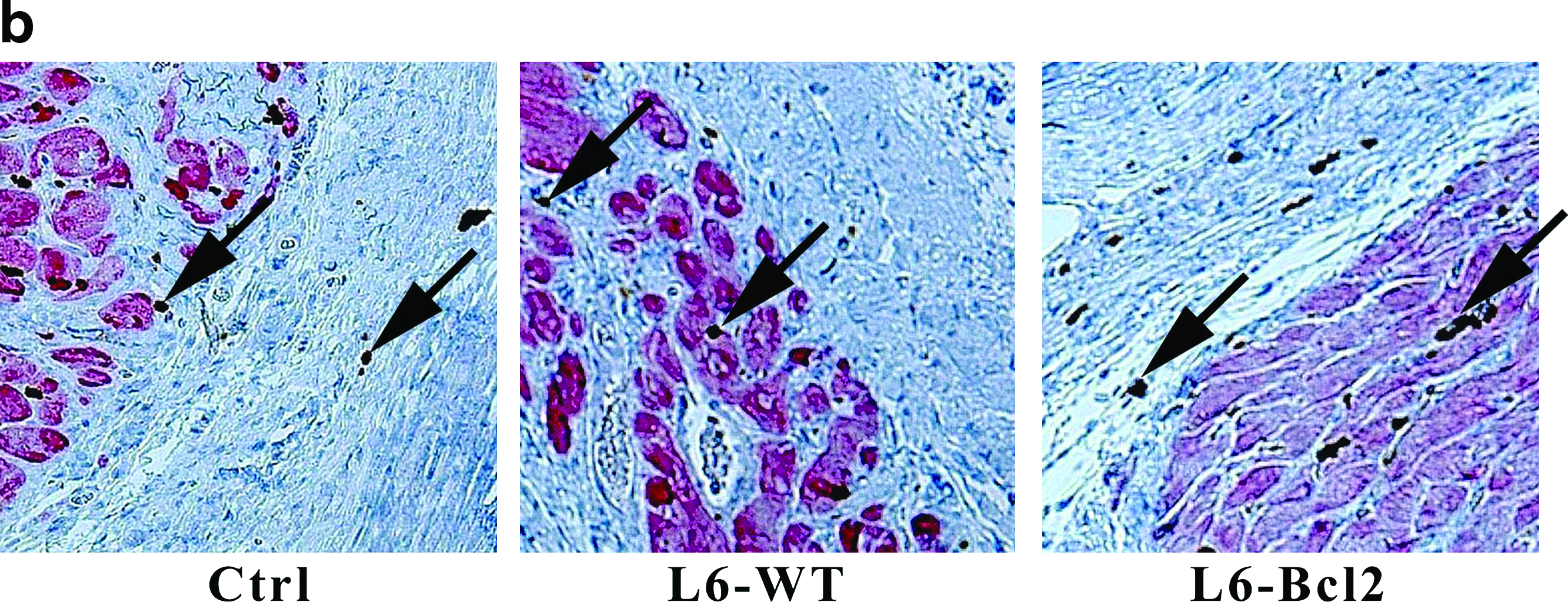
Myocyte survival in infarcts
After elucidating the enhanced angiogenic response in the infarct and border regions, we studied whether the increased number of vessels would promote myocyte survival in the infarct. Analysis of tropomyosin-positive cells from the infarcted ventricular wall revealed that animals receiving L6-Bcl2 sheet therapy had a significantly higher number of myocytes in the infarcted region (pixel count: 17594 ± 2485, p = 0.039) than did the control group (11038 ± 1275). The L6-WT group had 26% more myocytes in the infarct region than did the control group, but this difference was nonsignificant (13954 ± 1698, p = 0.122) (Fig. 4a, b).
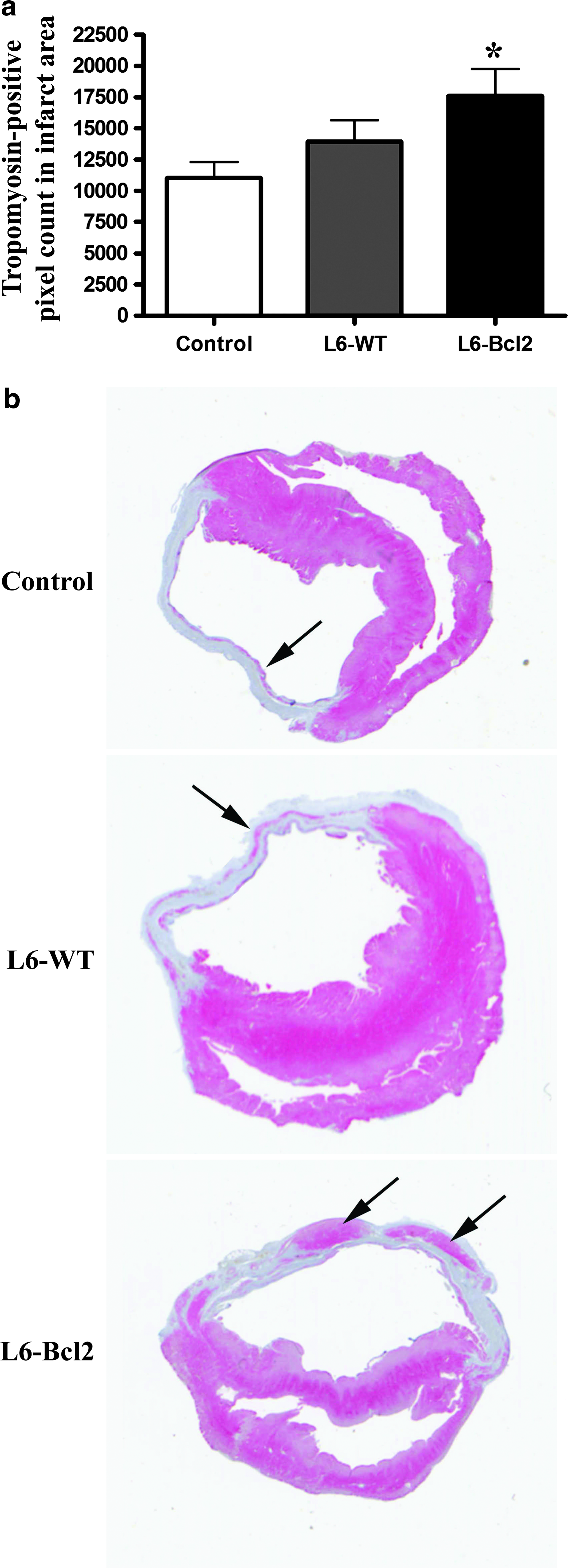
Quantification of surviving myocytes in infarct. Samples were collected 8 weeks after LAD ligation or sham operation (sham group) and 4 weeks after re-thoracotomy and therapy with or without (control and sham groups) myoblast sheets.
Endothelial cell proliferation, migration, and tubulogenesis
We incubated monolayers of HUVECs with the conditioned medium from WT or Bcl-2-modified myoblast sheets, and after 24-h incubation, evaluated the number of endothelial cells per visual field. Endothelial cell proliferation was significantly higher in L6-Bcl2 myoblast sheet-conditioned medium-stimulated cultures (961 ± 50 cells) than in the L6-WT (743 ± 26 cells, p = 0.018) or the vehicle-treated cells (628 ± 19, p = 0.004) (Fig. 5a). A lesser effect of the L6-WT myoblast sheet-conditioned medium in comparison to vehicle controls was also apparent (p = 0.023). Inhibition of signaling through Flt1/Flk1 by SU5416 (2 μM) attenuated the proliferative response in HUVECs treated with the L6-Bcl2-conditioned medium (686 ± 42, p = 0.008) and L6-WT-conditioned medium (639 ± 40, p = 0.047).
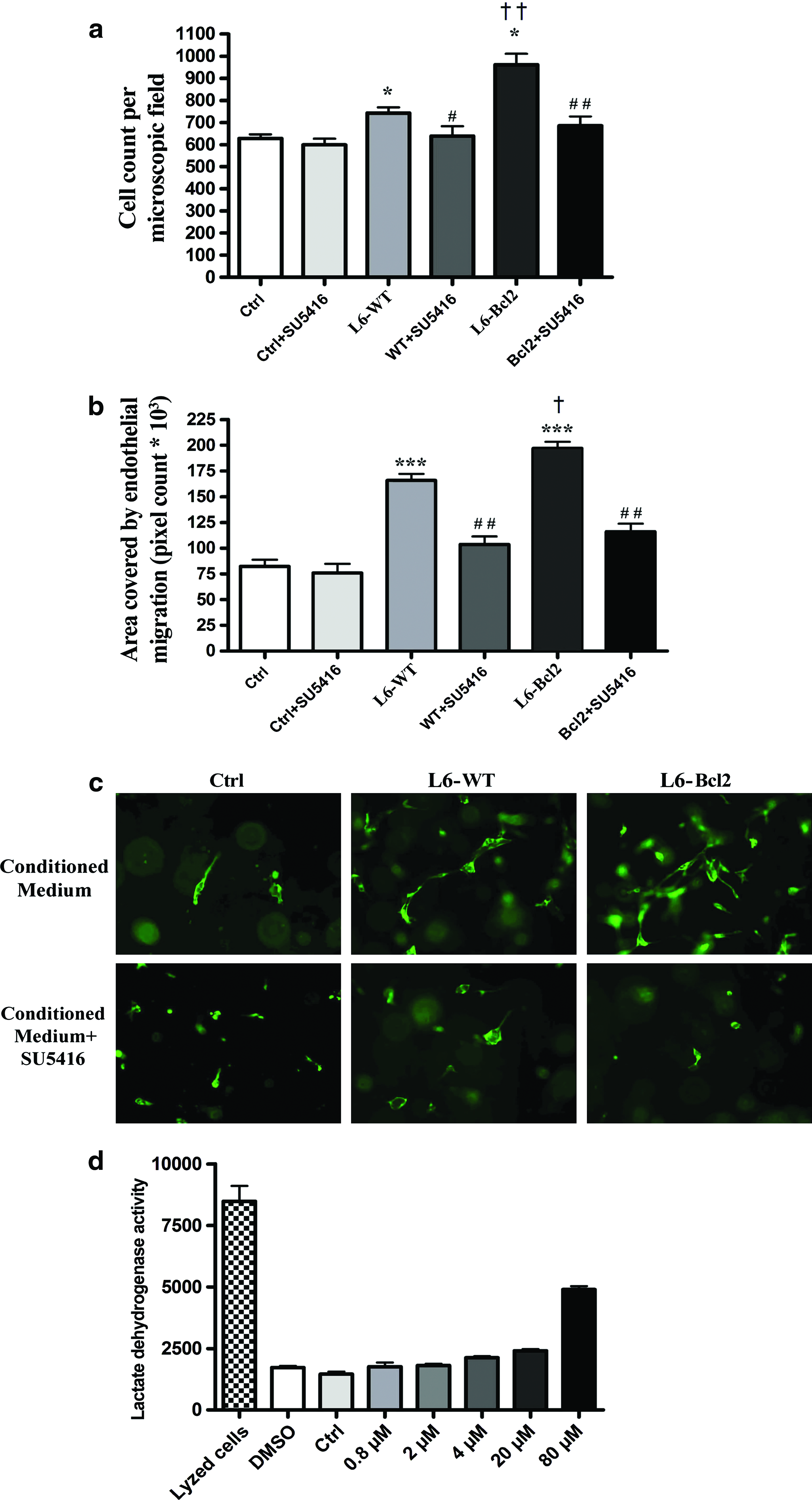
Human umbilical endothelial cell mobilization by myoblast sheet-derived paracrine factors.
We also studied the capacity of myoblast sheet-derived paracrine factors to stimulate endothelial cell motility in a wound healing assay. At the 24-h time point, HUVECs treated with the L6-WT (165900 ± 6030, p = 0.0001) and L6-Bcl2 (196900 ± 6502, p < 0.0001) myoblast sheet-conditioned medium exhibited significantly more migration than did the control group (82250 ± 6506) (Fig. 5b). Consistent with the proliferative response, the effect of the L6-Bcl2 sheet-conditioned medium was significantly higher than that of the L6-WT sheets (p = 0.025) in stimulating endothelial cell migration. Treatment with SU5416 (2 μM) resulted in a significant decrease in migration in both L6-WT (103400 ± 7108, p = 0.003) and L6-Bcl2 (115900 ± 7699, p = 0.001) groups.
We assessed the effect of myoblast sheet-derived soluble mediators on endothelial cell morphology in a collagen lattice. After 72 h, cells treated with the L6-Bcl2 sheet-conditioned medium acquired an elongated phenotype and demonstrated formation of more tube-like structures than did cells treated with the L6-WT sheet-conditioned medium. Addition of SU5416 counteracted these effects (Fig. 5c). In contrast, endothelial cells cultured in the presence of EGM displayed spherical morphology and failed to spread in the lattice.
We also determined the toxicity of the small-molecule inhibitor SU5416 and carrier dimethyl sulfoxide that we employed in the studies, finding 24-h treatments with 1% dimethyl sulfoxide and 2 μM SU5416 showing no differences from baseline or vehicle. Marked toxicity occurred at concentrations >10-fold higher as those used in this study (Fig. 5d).
Discussion
Cell transplantation therapy for chronic HF is compromised by the inability of the host-site fibrotic scar tissue, such as that developed after an MI, to promote viability of grafted cells. 13 To achieve long-term benefits of cell transplantation, the graft must resist apoptosis and restore blood flow at the host environment chronically deprived of nutrients and oxygen. Although a three-dimensional sheet structure provides the cells with intercellular and ECM contacts that in contrast to single cell injections initially protect the cells, 14 the sheets greatly suffer from the inactivity of the underlying morbid myocardium of chronic HF. In this study we investigated the ability of bcl2 gene therapy to enhance the therapeutic effect of myoblast cell sheet transplantation in a rat model of chronic HF. Here we show that this therapy improves cardiac function, enhances paracrine angiogenic signaling, and promotes cell proliferation and myocyte survival in the failing myocardium.
Bcl-2 is an antiapoptic member of the Bcl-2 family of apoptosis-related proteins. It is primarily localized to the mitochondrial membrane where it inhibits the mitochondrial pathway of apoptosis by counteracting the proapoptotic members of the family and prevents cytochome c release and caspase activation. 15 Evidence exists on the usefulness of Bcl-2 as a regulator of early donor cardiomyoblast or mesenchymal stem cell death in models of acute MI.16,17 Moreover, in a rat model of acute MI we have previously shown that Bcl-2 gene therapy of myoblast sheets enhanced their production of paracrine mediators, prolonged graft survival, and improved cardiac function. 9
In this study, we mimicked the clinical situation of chronic HF by allowing the myocardial scarring and functional decline to develop for 4 weeks after LAD ligation. Myoblast sheet therapy was thus administered on quiescent scar tissue after the acute inflammatory phase had receded. At 4 weeks after administration of therapy, only the group receiving bcl2 gene-expressing myoblast sheets showed a persistent increase in cardiac function, although some improvement had been evident in both L6-WT and L6-Bcl2 groups at 2-week time-point.
Our previous in vivo work has shown that that bcl2 is functionally antiapoptotic in the myoblast sheets. 9 However, this modification does not immortalize the cell sheets, and no sheet structures were morphologically evident at the end of the study. This in turn suggests that the increased LV wall thickness in the L6-Bcl2 group could best be attributed to paracrine stimulation of the host myocardium rather than a direct contribution by the sheet structure itself. Moreover, the infarct and border areas of hearts receiving L6-Bcl2 sheets displayed a significantly higher number of vWF-positive cells than did L6-WT or control hearts, suggesting that the sheets with Bcl-2 were more effective to induce formation of neovasculature in the diseased myocardium. These therapeutic benefits may be attributed to the prolonged survival of Bcl-2-expressing sheets, their enhanced capability to secrete therapeutic paracrine mediators that stimulate the dysfunctional myocardial tissue, or a combined action of both. It is plausible that the increased myocardical angiogenesis also provides oxygen and nutrients for the graft and thus further promotes sheet survival.
Production of paracrine effectors is currently regarded as the major mechanism by which transplanted cells such as myoblasts 18 and mesenchymal stem cells 19 exert their therapeutic actions and stimulate the damaged host tissue. 20 Although the conditioned medium from both L6-WT and L6-Bcl2 sheets induced endothelial cell proliferation, migration, and tubulogenesis, we show here that the paracrine action of L6-Bcl2 sheets is superior for induction of endothelial cell proliferation and migration than that of the WT sheets. Because positive mutual regulation between Bcl-2 and VEGF has been described,17,21 it is plausible that to some degree bcl2 expression in myoblasts could also upregulate expression of VEGF. As we have previously described, 9 introduction of bcl2 retains the normal gene expression prolife in myoblasts. In the sheets, however, it induces expression of the proangiogenic genes VEGF and PlGF.
VEGF is known to bind to the Flt1 and Flk1 receptors, whereas PlGF binds only to Flt1. In the current study, we investigated whether the small-molecule Flt1/Flk1 inhibitor SU5416 can block the stimulatory effects of myoblast sheet-derived paracrine factors on endothelial cells. Indeed, all of the endothelial cell responses described were inhibited by SU5416, suggesting that Flt1/Flk1-mediated signaling is crucial for the therapeutic angiogenesis induced by myoblast sheet transplantation. Further, it has been shown that heterodimerization of VEGF/PlGF can enhance angiogenesis by intermolecular crosstalk of these receptors, and that this heterodimer can drive therapeutic angiogenesis in myocardial ischemia even in conditions refractory to VEGF or PlGF alone. 22 Proangiogenic effects have also been described in cell sheets engineered from cardiac cells. 23 The effects of such sheets were shown to be mediated by VEGF, Tie-2 receptor, and cyclooxygenase-2, suggesting that cell sheets in general may harbor angiogenic potential, and that there may be cell type-dependent differences in the angiogenic paracrine patways utilized by sheets of different cellular origins.
It is plausible that the increased availability of oxygen and nutrients by myoblast sheet-induced angiogenesis in the ischemic area could rescue some of the cardiac myocytes struggling for survival after the acute phase of MI. 24 We found, however, no differences between study groups after analysis of cleaved caspase-3 or Sirius Red staining but found that increased vasculature in the infarct wall was associated with a higher number of cardiac myocytes in the L6-Bcl2 group. This implies that the enhancement in cardiac function and the increased LV thickness may be attributable to higher cellularity and subsequent contractility of the infarct wall. Further, we detected a significantly increased number of proliferating cells in the myocardium of L6-Bcl2 animals. Our analysis, however, revealed that only a minor portion of these cells are myocytes. The nature of these cells, stimulated by myoblast sheet therapy, requires further studies.
The induction of arrhythmias as a complication of myocardial injections of skeletal myoblasts has been raised as a safety concern of this therapy. 25 In addition to the possible inherent pro-arrhythmogenic potential of skeletal myoblasts, inflammation caused by needle injections to the already-irritated myocardium with high baseline of arrhythmias is suggested as a possible mechanism for the reported clinical cases. 26 In the current study, we did not detect any therapy-associated cardiac complication or mortality. It is possible that by avoiding needle injections, the use of epicardially implanted myoblast sheets may resolve the issue of arrhythmogenicity. Moreover, transplantation of myoblast sheets has a dose-dependent effect on cardiac function and growth factor production. 27 In the current study, we used two layers of myoblast sheets each approximately five cell layers or 50 μm in thickness. The optimal number of myoblast sheets for most efficient cell therapy remains to be solved.
In conclusion, we are, to our knowledge, the first to report that in a chronic model of HF after MI, counteracting donor cell apoptosis is a vital strategy to promote the paracrine effect of myoblast sheet transplantation. The enhanced production of paracrine effectors stimulates host-site angiogenesis via Flt1/Flk1-dependent cell signaling. Our results suggest that strategies improving tolerance to apoptosis should be applied in clinical myoblast sheet transplantation to maximize the therapeutic paracrine effects.
Footnotes
Acknowledgments
We thank Lahja Eurajoki for her help with the cell cultures and measurements, Irina Suomalainen for the immunohistochemical staining, and Anne Reijula for the tissue processing. We also thank Veikko Huusko, Virpi Norppo, Kari Savelius, and Olli Valtanen for all their help and for the excellent animal care. This work was supported by Core-to-Core funding between Academy of Finland and the Japanese Society for the Promotion of Science; Finnish Government EVO grants [TYH7201 and TYH2009/301]; RAM collaboration between Japan Science and Technology Agency (Y.S.) and Finnish Funding Agency for Technology and Innovation (E.K.), and Finnish Heart Association.
Disclosure Statement
No competing financial interests exist.
This work was carried out at Institute of Biomedicine, Pharmacology, Biomedicum, University of Helsinki, Finland.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.