
Abstract
Bone represents a highly dynamic tissue whose development is strongly dependent on vasculogenic and angiogenic processes. Neovascularization also plays an important role in fracture healing and in tissue engineering applications aiming at restoring bone function. We have previously shown in a heterotopic subcutaneous implantation model of severe combined immunodeficiency (SCID) mice that implanted human umbilical vein endothelial cells (HUVECs) gave rise to the formation of a complex functional human neovasculature. In this study, we investigated the effect of HUVEC coimplantation on mesenchymal stem cell (MSC)-mediated bone regeneration in an orthotopic calvarial bone defect model in immunocompromised mice. For this purpose, human fibrin/Matrigel-immobilized HUVECs and MSCs were seeded alone or in combination into scaffolds consisting of decalcified processed bovine cancellous bone (Tutobone) and implanted into calvarial critical-sized defects. Our results show that implanted HUVECs formed complex three-dimensional networks of perfused human neovessels that were stabilized by recruiting perivascular cells. Neovessel formation was considerably higher in the coimplantation group, suggesting that implanted MSCs supported HUVEC-triggered neovascularization. In addition, implanted MSCs effectively supported bone formation in calvarial defects. However, the HUVEC-derived neovasculature did not improve MSC-triggered bone regeneration in this orthotopic critical-sized defect model.
Introduction
In the present study, we investigated whether human umbilical vein endothelial cells (HUVECs) coseeded with human MSCs may support neovascularization and hence bone regeneration. To test this, HUVECs and MSCs were cultured alone or in combination in fibrin/Matrigel matrices and seeded into scaffolds consisting of decalcified processed bovine cancellous bone (PBCB; Tutobone). Neovascularization and osteogenesis was evaluated by implanting the scaffolds into 4.3-mm calvarial bone defects in severe combined immunodeficiency (SCID) mice. Our results showed that (co)-implanted MSCs effectively supported bone formation and that (co)-seeded HUVECs gave rise to a functional three-dimensional human neovasculature.
Materials and Methods
Cell culture
HUVECs were purchased from Promocell (Heidelberg, Germany) and cultured in endothelial cell growth medium (ECGM) supplemented with 10% fetal calf serum (FCS) at 37°C, 5% CO2 in a humidified atmosphere. Only HUVECs from passage 2 to 5 were used for the experiments.
Human MSCs were isolated and expanded as described before. 17 In brief, bone marrow aspirates were obtained from healthy adult donors by an iliac crest biopsy. Mononuclear cells were purified by density gradient centrifugation with Biocoll Separating Solution (Biochrom AG, Berlin, Germany). Subsequently, cells were filtered through 100-μm cell strainers (BD Labware, Franklin Lakes, NJ). Mononuclear cells were seeded in culture flasks at a density of 5 × 105 cells/cm2 in expansion medium (alpha-minimal essential medium [MEM], 10% FCS, 50 μg/mL gentamicin, and 5 ng/mL bFGF) at 37°C, 5% CO2. The medium was changed twice weekly, removing all nonadherent cells. Once adherent cells had grown to confluence, they were detached and reseeded at a density of ∼2000 cells/cm2 and cultivated for two further passages. Before implantation, MSCs were oesteogenically predifferentiated in vitro for 3 days in osteogenic differentiation medium (Dulbecco's Modified Eagle Medium [DMEM] supplemented with 10% FCS, 50 μg/mL gentamicin, 10 mM β-glycerophosphate, 0.1 μM dexamethasone, and 50 μM ascorbic acid).
Generation of endothelial spheroids
HUVEC spheroids (100 cells/spheroid) were generated as previously described. 18 Cells were suspended in ECGM containing 0.25% (w/v) methylcellulose and seeded on plastic dishes in a hanging drop to allow overnight spheroid aggregation. Under these conditions all suspended cells contribute to the formation of a single spheroid per drop of defined size and cell number.
Seeding of decalcified PBCB scaffolds
Discs of decalcified PBCB (Tutobone; diameter 4.3 mm and thickness 1.5 mm) were provided by Tutogen Medical (Neunkirchen, Germany). To achieve consistent seeding results, discs were incubated in ECGM supplemented with 10% FCS at 4°C for 24 h before seeding. One thousand HUVEC spheroids along with 2 × 105 HUVECs in single cell suspension and/or 1 × 106 osteogenically predifferentiated MSCs were dispersed in 37 μL fibrinogen stock solution (fibrinogen from Calbiochem, Darmstadt, Germany; dissolved in endothelial cell basal medium (ECBM) without FCS; final fibrinogen concentration in the constructs 1.5 mg/mL) containing the recombinant angiogenic growth factors VEGF-A and bFGF (R&D Systems, Wiesbaden, Germany; final concentration 1 ng/μL each). This fibrinogen-cell suspension was combined with 20 μL Matrigel (growth factor reduced; BD Biosciences, Heidelberg, Germany). Before seeding, 3 μL thrombin (0.5 units/μL; Baxter, Unterschleißheim, Germany) was added, and discs were immediately seeded with this cell suspension. As a negative control, decalcified PBCB discs were seeded with the fibrin/Matrigel matrix without cells. The constructs were incubated at 37°C for 30 min to induce polymerization of the fibrin/Matrigel matrix. After polymerization, discs were incubated at 37°C and 5% CO2 in a humidified atmosphere in ECGM supplemented with 10% FCS for 1 h before implantation.
Implantation of PBCB scaffolds into SCID mice
Six- to 8-week-old SCID mice (C.B.-17-SCID; Charles River, Kisslegg, Germany) served as recipients of the scaffolds. German regulations for care and use of laboratory animals were met at all times. All experiments were approved by the animal care committee of the University of Freiburg. The animals were housed in the veterinary care facility of the University of Freiburg Medical Center. Mice were anesthetized and a 4.3-mm-diameter calvarial critical-sized defect was created on one side of the calvarial bone using a dental drill. During the surgery every care was taken to ensure no damage to the dura mater. Mice were divided into four groups: group 1, without cells (n = 7); group 2, HUVECs alone (n = 6); group 3, MSCs alone (n = 5); group 4, HUVECs + MSCs (n = 6) and implantation of the scaffolds was performed. In addition, some animals (n = 3) did not receive any scaffold or cells and served as empty control. Six weeks after surgery, the mice were killed and the specimens were retrieved for immunohistological examinations.
Immunohistological staining
For examination of blood vessel formation, samples were fixed in Schaffer's solution (37% formaldehyde and 80% ethanol) for 2 days and subsequently decalcified in Tris-EDTA solution (260 mM Tris; 270 mM EDTA) for 14 days at 37°C. Samples were then paraffin embedded and sectioned at 5 μm before staining. Immunohistochemical analyses were then done with deparaffinized and rehydrated paraffin sections. Sections for immunoperoxidase stainings were treated with 3% H2O2 to inhibit endogenous peroxidase. After washing in PBS, the sections were incubated for 30 min with blocking solution (goat serum, ready to use; Zymed, San Francisco, CA) followed by incubation with the corresponding monoclonal mouse primary antibodies: anti-human CD34 (hCD34, 1:75; Dako, Glostrup, Denmark), anti-hCD31 (1:100; Dako), or anti-human vimentin (1:75; Dako) in a humid chamber at room temperature for 1 h. For immunofluorescence, the sections were incubated with the secondary antibody goat anti-mouse/Alexa 488 (1:200; Molecular Probes, Leiden, The Netherlands). For immunofluorescence double stainings, the sections were additionally incubated for 30 min with anti-smooth-muscle alpha actin (α-SMA)-Cy3 (1:200; Sigma, Steinheim, Germany). For immunoperoxidase stainings the sections were incubated with a biotinylated goat anti-mouse IgG antibody (1:200; Zymed), exposed to streptavidin peroxidase (ready to use; Zymed), developed with diaminobenzidine as substrate, and weakly counterstained with hematoxylin. For vimentin stainings, sections were incubated with a secondary HRP-labeled goat anti-mouse antibody (Dako) and developed with Histogreen (Linaris, Wertheim, Germany).
For Safranin O stainings, deparaffinized and rehydrated paraffin sections were hematoxylin stained followed by stainings with 0.02% aqueous fast green (Merck, Darmstadt, Germany) and 0.1% (w/v) Safranin O (Merck).
Tartrate-resistant acid phosphatase (TRAP) staining was performed on deparaffinized and rehydrated paraffin sections according to the manufacturer's instructions (Sigma, Deisenhofen, Germany). In brief, sections were stained using naphthol-AS-BI phosphate as a substrate and fast garnet GBC base solution as a detection agent for the reaction product. TRAP activity appears as purplish to dark red staining within the cytoplasm of osteoclasts.
For examination of bone formation, samples fixed in Schaffer's solution were dehydrated in graded acetone. After embedding in methylmethacrylate (Technovit 9100; Heraeus, Wertheim, Germany), 5 μm cross sections were obtained using a Leica hard tissue microtome (Leica Microsystems, Bensheim, Germany). For von Kossa staining, sections were incubated for 20 min in 5% silver nitrate in the dark and rinsed with H2O bidest. Thereafter, sections were incubated in 5% sodium carbonate, 25% formol for 2 min, rinsed with distilled H2O, incubated in 5% sodium thiosulfate, and rinsed again. Thereafter, sections were dehydrated and inspected for mineralization.
Histomorphometric analysis
Histomorphometric measurements were made using Image-J software (NIH, Bethesda, MD; http://rsb.info.com). For determination of bone formation, three von Kossa–stained cross sections per animal were captured at 25 × magnification and analyzed for new bone formation. Total defect area and total area of new bone formation was measured and the ratio between total new bone area and total defect area was calculated for each individual cross section. Values are given as mean value ± standard deviation.
For quantification of human blood vessels, three sections per animal stained with a human-specific anti-CD31 antibody and counterstained with hematoxylin were analyzed. Microscopic pictures were taken at 100-fold magnification of three randomized areas per section. To determine the microvascular density of human vessels (mean number of capillaries per mm2) the number of structures with lumen surrounded by hCD31-positive cells was counted manually. To determine the microvascular density of mouse blood vessels, the number of lumenized structures with surrounding hematoxylin-stained but hCD31-negative cells was counted. Values are given as mean value ± standard deviation.
Statistical analysis
Statistically significant differences between groups were determined by using an unpaired Student's t-test. Statistical significance was defined when p < 0.05.
Results
Scaffolds consisting of decalcified PBCB were seeded with fibrin/Matrigel-immobilized HUVECs (group 2), human MSCs (group 3), or with both cell types (group 4). As a negative control, decalcified PBCB loaded with fibrin/Matrigel alone was used (group 1). These scaffolds were implanted into 4.3-mm calvarial defects of SCID mice and retrieved for histological examination after 6 weeks. The explants were cut into halves, wherein one half was decalcified and embedded in paraffin for immunohistological examination of blood vessel formation and the other half was embedded in methylmethacrylate for evaluation of bone formation.
Analysis of neovascularization
To assess whether implanted HUVECs were able to form functional blood vessels in vivo, paraffin sections were immunohistochemically analyzed using human-specific antibodies against the panendothelial markers CD31 (Fig. 1A) and CD34 (Fig. 1B). In both stainings, human-specific vascular structures could be detected in groups 2 and 4, with considerably more human vessels in the coimplantation group (group 4). As expected, formation of human blood vessels was not detectable in groups 1 and 3. At higher magnification, perfused hCD31-positive blood vessels were detectable in groups 2 and 4, as evidenced by the presence of intraluminal hematoxylin-stained mouse erythrocytes (Fig. 1C). Double stainings for hCD34 and α-SMA on paraffin sections of implants seeded with HUVECs alone (group 2) and implants seeded with HUVECs and MSCs (group 4), revealed that the newly formed human blood vessels were covered with α-SMA-positive mural cells, suggesting that the vessels were stabilized by murine pericytes or smooth-muscle cells recruited from the surrounding mouse tissue (Fig. 2).
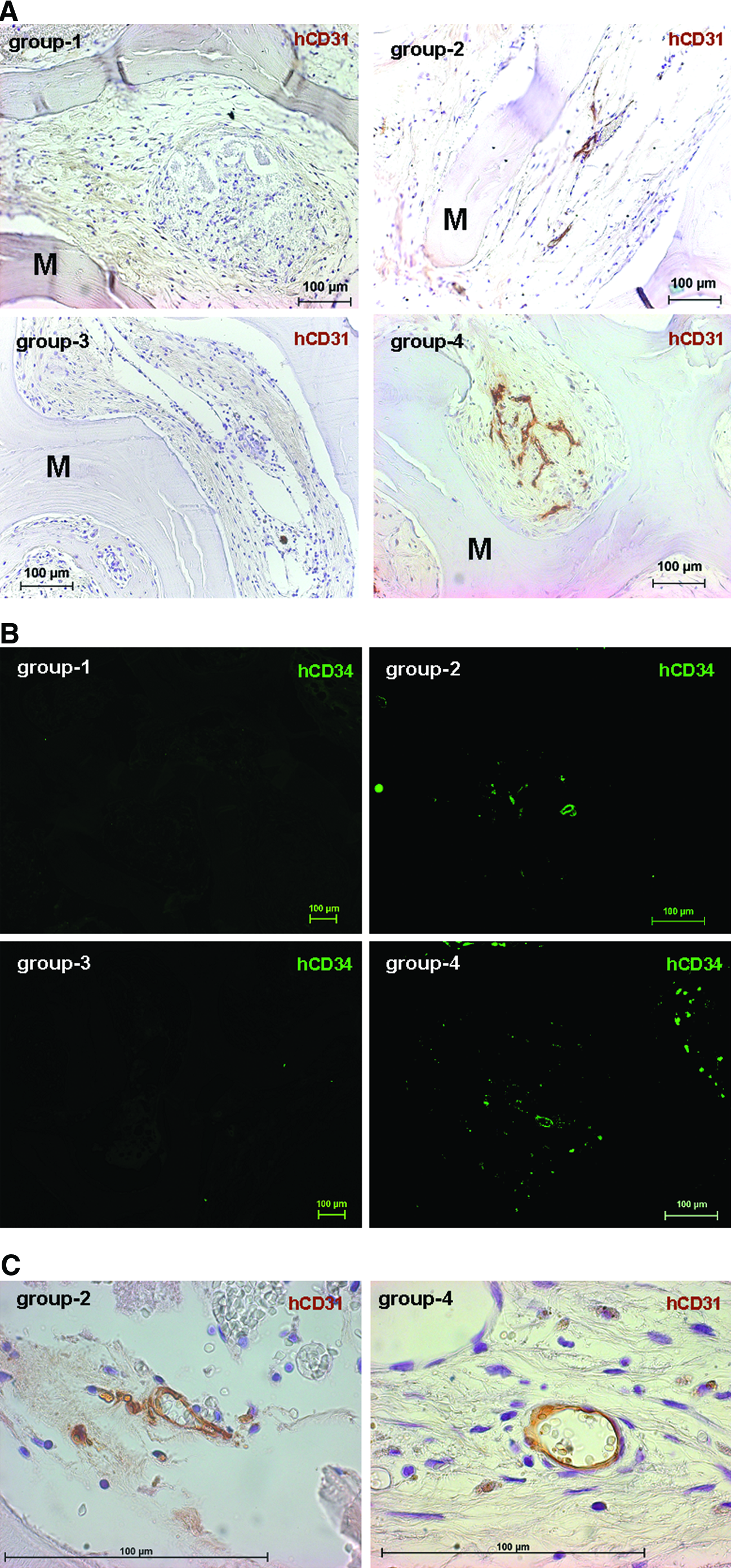
In vivo analysis of blood vessel formation. PBCB scaffolds were implanted into 4.3-mm calvarial defects and retrieved after 6 weeks. Group 1, without cells; group 2, HUVECs alone; group 3, MSCs alone; group 4, HUVECs + MSCs.

Double immunofluorescence staining for hCD34 (grafted HUVECs, green) and α-SMA (recruited murine mural cells, red). Group 2, HUVECs alone; group 4, HUVECs + MSCs. Magnification: 400 × . Scale bar: 100 μm. α-SMA, smooth-muscle alpha actin. Color images available online at www.liebertonline.com/ten.
As evidenced by the hematoxylin counterstainings of the CD31 stainings, all explants contain hCD31-negative mouse blood vessels (exemplarily shown for groups 3 and 4 in Fig. 3A). In group 4, the coexistence of mouse vessels and hCD31-positive human vessels is shown. Quantification of hCD31-negative vessels in the explanted scaffolds revealed a relatively homogeneous microvascular density of murine vessels ranging between 12.98 ± 3.625 vessels per mm2 for group 1 and 15.06 ± 6.249 vessels per mm2 for group 4 (Fig. 3B). In contrast, as already shown in Figures 1A and B, hCD31-positive human blood vessels were only detectable in groups 2 and 4 with microvascular densities of 6.79 ± 2.122 vessels per mm2 for group 2 and 28.25 ± 17.806 vessels per mm2 for group 4 (Fig. 3C). As expected, empty implants (group 1) or implants seeded with MSCs alone (group 3) failed to form any detectable hCD31-positive microvessels (Fig. 3C).
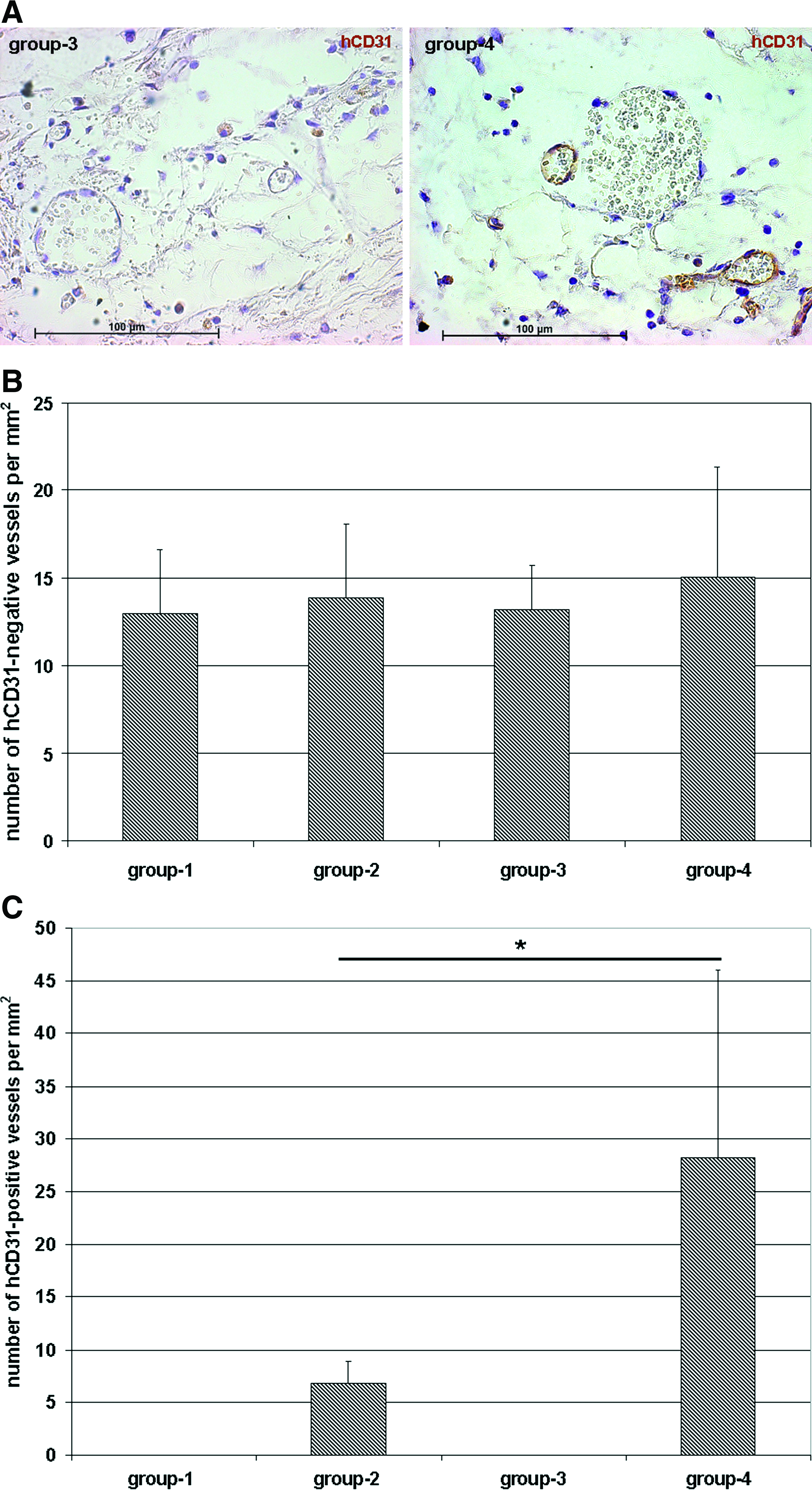
Quantification of mouse and human blood vessels.
Analysis of bone formation
The distribution of the MSCs within the explanted scaffolds was investigated on histological cross sections by using a human-specific antibody against vimentin, revealing a homogeneous distribution of MSCs within the pores of the constructs from groups 3 and 4, whereas no human-specific cells could be detected in the negative control (group 1) (Fig. 4A). Double stainings for human vimentin (green) and hCD31 (brown) showed at higher magnification the coexistence of human microvessels and MSCs in group 4, whereas as expected, only MSCs but no human vessels could be detected in group 3 (Fig. 4B).
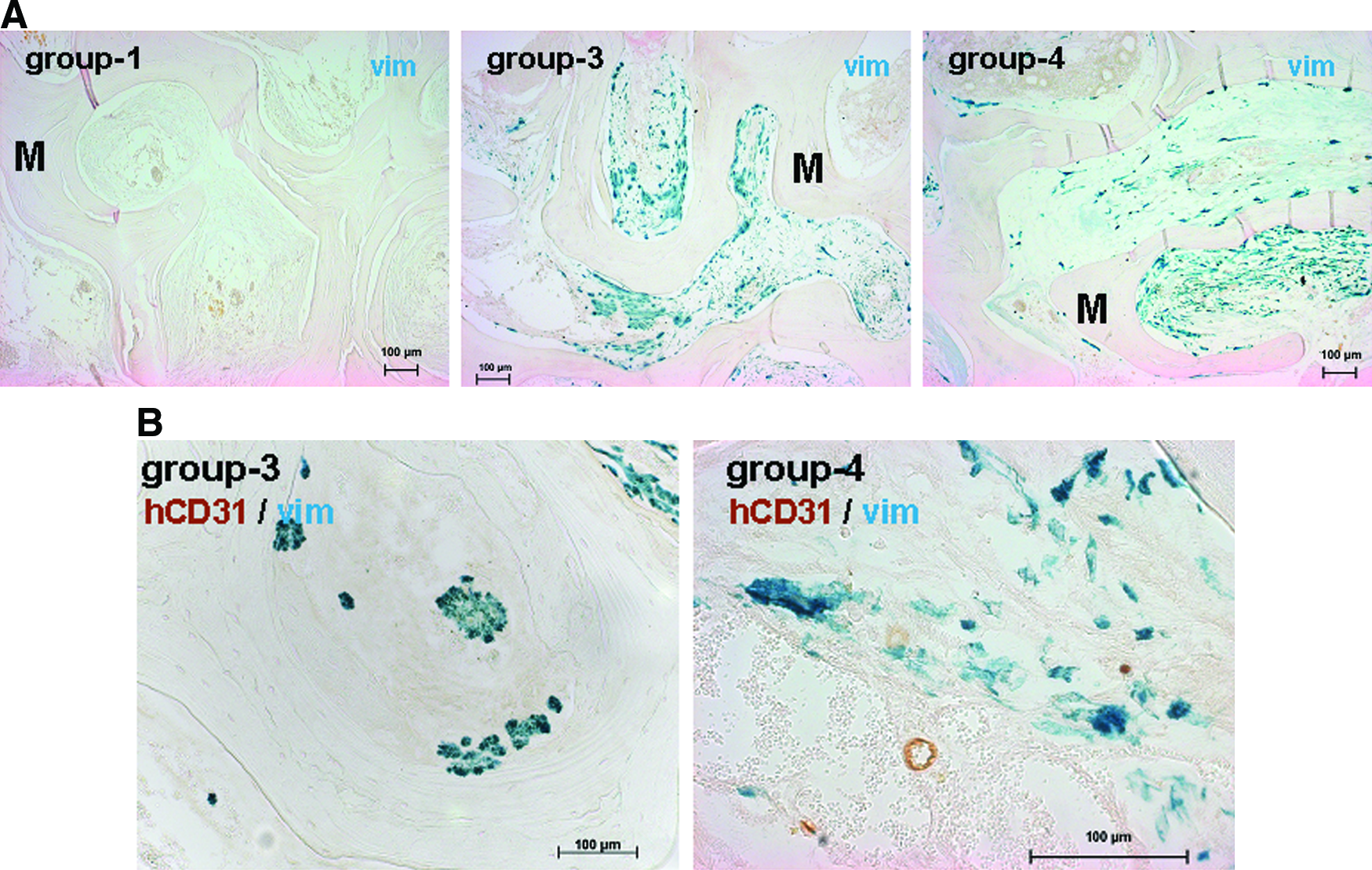
Distribution and viability of implanted human MSCs.
The 4.3-mm calvarial defects that were created in SCID mice were previously reported to represent critical-sized bone defects in adult mice, 19 which means that these defects are unable to heal by themselves. This was also proven in our particular model by the complete lack of bone formation in nonreconstructed empty defects as evidenced by von Kossa stainings (Fig. 5A). Instead, defects were partially filled with a layer of fibrous tissue. At higher magnification, a thin layer of host cells could be detected by hematoxylin/eosin staining (Fig. 5A). These cells were embedded in the fibrous tissue, which did not show any signs of neovascularization.
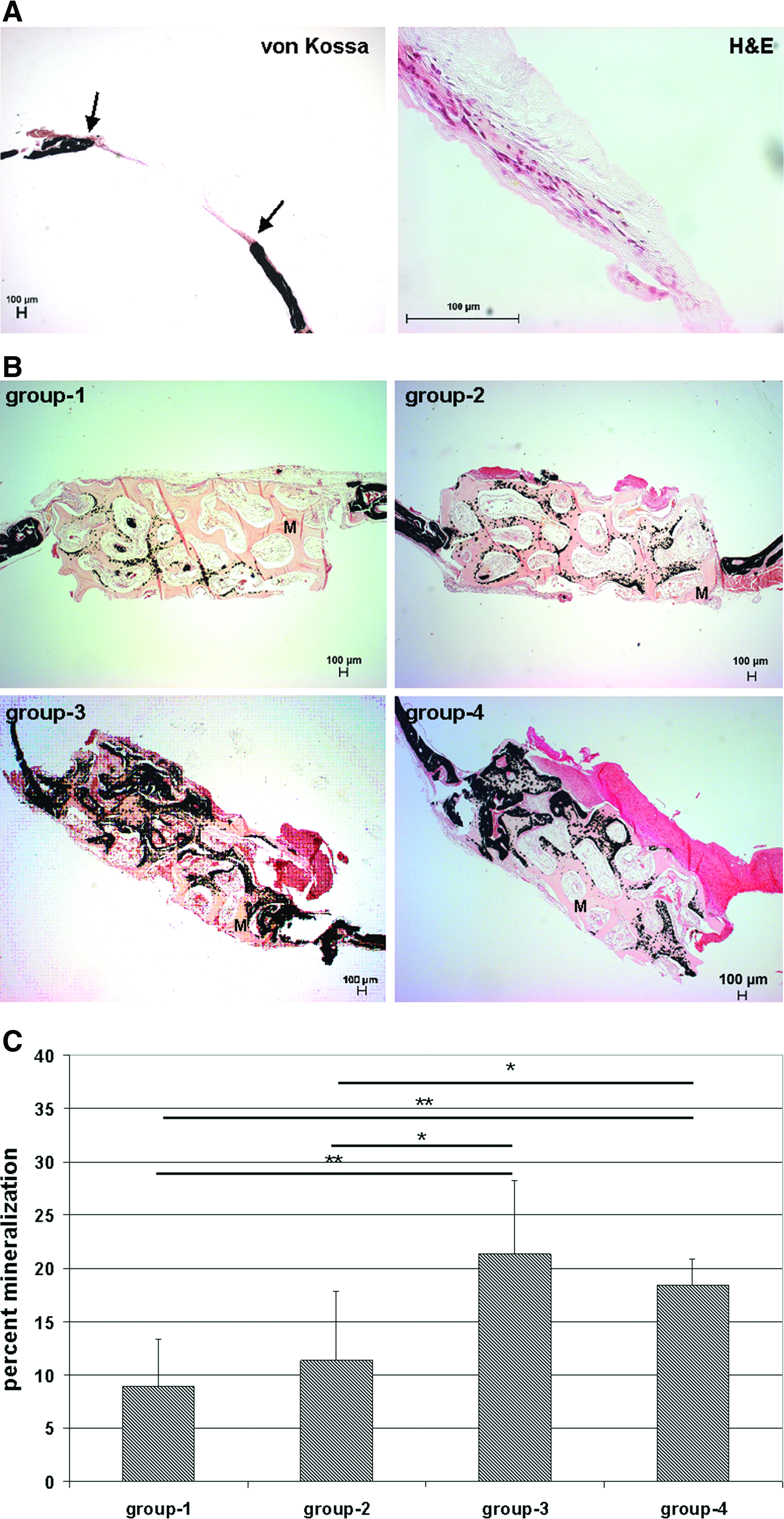
Quantification of bone formation.
To observe new bone formation in the reconstructed defects, von Kossa stainings were performed on calvarial specimens, 6 weeks after implantation. Sparse osteogenesis could be detected in the negative control (group 1) as well as in the grafts seeded with HUVECs only (group 2). In contrast, implants containing MSCs (groups 3 and 4) showed clearly more bone formation in relation to groups 1 and 2 (Fig. 5B). Interestingly, mineralization was most prominent at the interface between the fibrin/Matrigel loaded pores and the scaffold material. This distribution pattern of mineralization was detectable in all groups. To quantify the extent of osteogenesis in the explants, areas of new bone formation were calculated on basis of the von Kossa stainings and expressed as percentages with respect to total defect area (Fig. 5C). This quantification indicated that the constructs seeded with MSCs (groups 3 and 4) exhibited significantly more bone formation than the negative control (group 1) or the constructs seeded with HUVECs alone (group 2). However, no statistically significant difference could be detected between groups 3 and 4, indicating that there exists no positive correlation between bone healing and improved vascularity mediated by HUVEC implantation.
To rule out the possibility that the lack of improved bone formation in the cocultivation group may be due to differences in osteoclast activity, we performed TRAP stainings to observe osteoclasts. As shown in Figure 6, no osteoclast-specific staining could be detected within our experimental groups, suggesting that the observed extent of bone formation was not secondary modulated by osteoclast-mediated bone resorption.
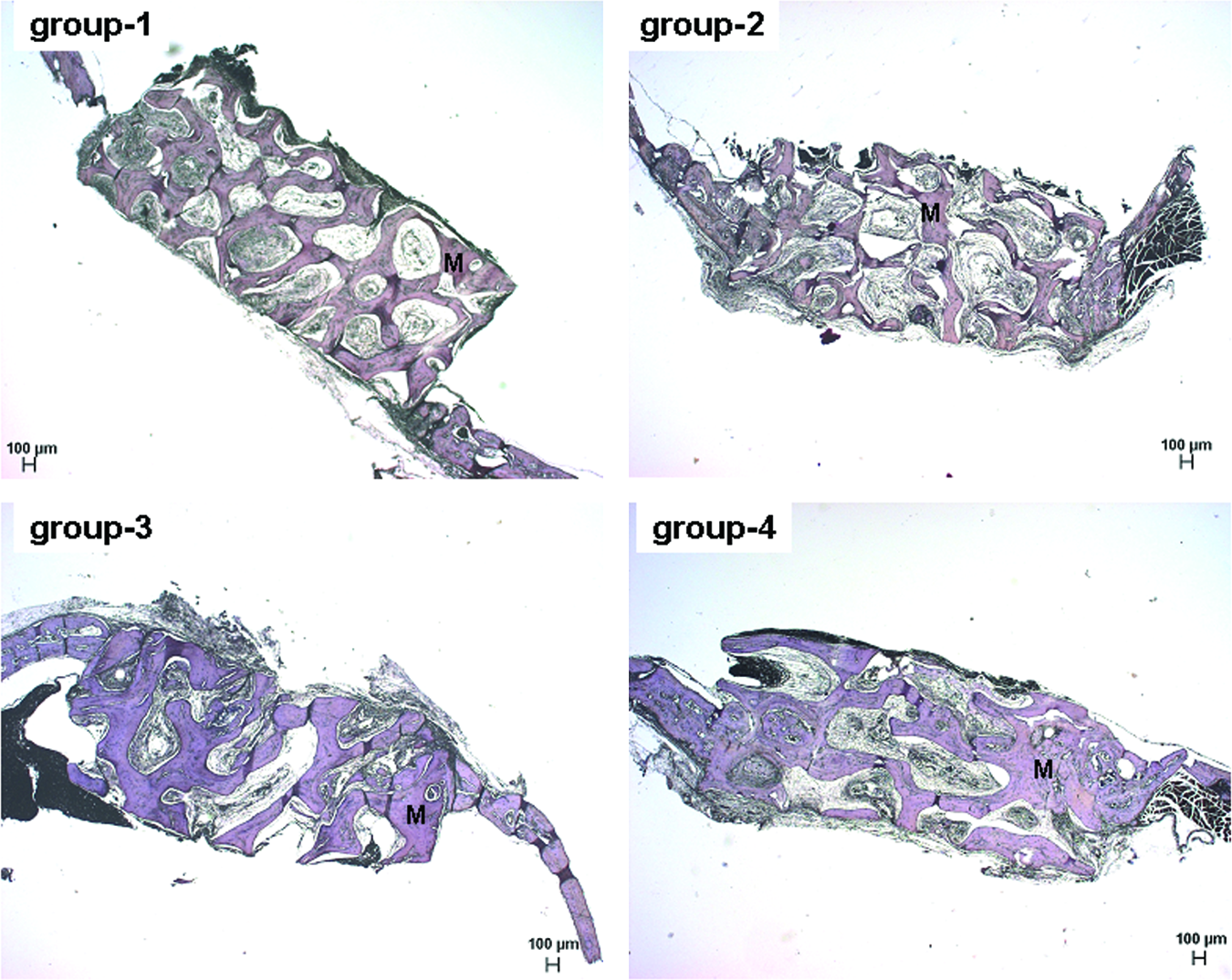
TRAP staining. TRAP staining was performed to determine the amount of osteoclasts within the explants from the reconstructed defects. No positive TRAP staining (should be indicated by red color) could be detected within the explants. Magnification: 25 × . Scale bar: 100 μm. M, PBCB matrix. Group 1, without cells; group 2, HUVECs alone; group 3, MSCs alone; group 4, HUVECs + MSCs. TRAP, tartrate resistant acid phosphatase. Color images available online at www.liebertonline.com/ten.
In summary, our results show that endothelial cells implanted alone or in combination with MSCs into PBCB-scaffolds form stable functional vessel networks. This effect was more pronounced in the cocultivation group, indicating that coimplanted MSCs support EC-triggered neovascularization. In addition, implanted MSCs effectively supported bone formation in calvarial bone defects. However, bone formation was not increased in the cocultivation group suggesting that the EC-derived neovasculature did not improve MSC-triggered bone regeneration in this particular model of bone healing.
Discussion
Nearly all tissues are dependent on an efficient blood supply to warrant delivery of nutrients and oxygen. This fact must be considered by the further development of tissue engineering concepts in terms of putative clinical applications, since implantation of voluminous avascular grafts will ultimately lead to the failure of the implanted constructs due to apoptosis and subsequent necrosis. 20 Multifarious strategies to induce therapeutic angiogenesis are described and comprise the use of recombinant angiogenic growth factors,21–23 gene therapeutic approaches,12,24 or cell-based therapies using endothelial cells.13–16
In the present study, we have employed a three-dimensional cocultivation system consisting of HUVECs and MSCs to investigate whether a HUVEC-derived vasculature might support osteogenesis mediated by coimplanted MSCs in a critical-sized orthotopic calvarial defect model of SCID mice.
Immunocompromised mice with the Prkdcscid mutation 25 represent a generally accepted in vivo model for xenograft research because the scid mutation, which affects the B as well as the T lymphocyte development, provides a wide tolerance to implantation of foreign tissues. 26 Moreover, calvarial bone defects of SCID mice have already been used in numerous studies to investigate the bone formation capacity of implanted MSCs.27,28 Therefore, the defects in B and T lymphocyte functions do not negatively interfere with bone development or regeneration, making this model suitable for the in vivo investigation of bone formation.
To study neovascularization and osteogenesis, fibrin/Matrigel-immobilized cells were seeded into a commercially available osteoconductive matrix (PBCB; Tutobone) and implanted into 4.3-mm calvarial defects. Implants were retrieved after 6 weeks and evaluated for neovascularization and bone formation. From these experiments, it became clear that implanted HUVECs were able to form stable perfused neovessels within the constructs. Moreover, the newly formed human vasculature was covered by α-SMA-positive mural cells, an absolute requirement for the formation of stable and long-lasting blood vessels.29,30 By quantification of the microvascular densities within the explants, we made the interesting observation that the number of EC-derived neovessels was much higher in constructs seeded with HUVECs and MSCs than in the constructs seeded with HUVECs alone. This effect may be explained by the fact that MSCs, like primary osteoblasts, secrete pro-angiogenic growth factors, thereby supporting neovessel growth.31,32 Another attractive explanation for the increased microvascular density in the coimplantation group, is based on the recently published observation that coimplanted bone-marrow-derived MSCs differentiated in vivo into pericytes and were able to efficiently stabilize nascent blood vessels. 33 In our experiments, we have subjected the MSCs in vitro to an osteogenic predifferentiation regime for a limited time period of only 3 days, because it was published previously that this short-term procedure enhances the osteogenic potential of MSCs upon implantation. 34 Although not investigated directly in our experiments, it seems to be very likely that not all MSCs were committed to the osteogenic lineage by this procedure and that presumably a subfraction of implanted MSCs differentiated into pericytes in vivo, thereby stabilizing HUVEC-derived neovessels.
In the context of bone healing of the generated calvarial defects, we have seen that MSCs implanted alone or in combination with HUVECs effectively supported bone regeneration. This finding was not unexpected, since MSCs have been reported to promote bone regeneration in numerous bone defect models.7–9 The extent of MSC-triggered bone regeneration in our calvarial defect model of SCID mice was similar to that reported on autologous MSCs used to regenerate calvarial bone defects of immunocompetent B6D2F1 mice. 19 To investigate whether bone formation in our experimental model was due to intramembranous ossification or whether it was accomplished via an intermediate step involving cartilage formation, we performed Safranin O stainings to observe chondrocyte-specific extracellular matrix formation (data not shown). From these experiments it became clear that all explants were devoid of any cartilage indicating that as expected, ossification has not taken place via an endochondral bone formation process.
It is generally accepted that bone healing is strongly dependent on neovascularization. Experimental inhibition of VEGF-mediated angiogenesis, by using a soluble neutralizing VEGF receptor, decreased neovascularization, bone formation, and callus mineralization in femural fractures as well as in tibial cortical bone defects, whereas exogenous recombinant VEGF-induced angiogenesis and supported osteogenesis. 35 In this context, it was surprising to note that MSC-mediated bone regeneration was not improved by the human EC-derived neovasculature in our experimental setting. This result was in contrast to other reports where EC-derived neovessels supported osteogenesis in various bone defect models.36–39 However, it is important to note that these groups have used long-bone defect models (femur and ulna) in combination with different scaffold materials and different types of endothelial or endothelial progenitor cells to engineer blood vessels. These differences render it very difficult to interpret the contradictory results of the studies. However, it is reasonable to assume that the actual implantation site and particularly the vascularity of the surrounding tissue may have an enormous effect on the outcome of the experiments. It is well accepted in the literature that physical stimuli play critical roles in the control of osteogenic differentiation of MSCs as well as in minearlized matrix production and fracture repair.40–42 In this context it is important to note that the cranial implantation sites used in our experiments were not restricted to any significant mechanical loading during the healing process, making it additionally difficult to make any comparison to long-bone defect models in terms of mechanical strain-dependent ossification.
It is also known from the literature that expression of osteogenic marker genes such as alkaline phosphatase is enhanced in osteoblasts and MSCs upon direct coculture with HUVECs, suggesting that HUVECs may have a direct effect on osteogenic differentiation of MSCs.43,44 Although we have coimplanted HUVECs and MSCs in group 4, the missing effect of HUVECs on MSC-mediated osteogenesis may be due to the endothelial cells not being in direct contact with implanted MSCs. This hypothesis is strengthened by our observation from the hCD31 stainings, implying that all detectable hCD31-positive HUVECs have been incorporated into human blood vessels with no HUVECs in single cell configuration being detectable in direct cell contact to MSCs. We assume that the nonincorporated HUVECs did not survive the 6 week observation period.
Also interesting in the context of neovascularization is the observation that all explants retrieved from our calvarial defect model contained mouse blood vessels originating from the implantation bed. The microvascular density of the mouse vessels was not very high, but uniform in all groups. Therefore, the endogenous neovascularization may have been sufficient to sustain osteogenesis.
In our experiments we have used a fibrinogen/Matrigel mixture as a matrix for seeding the cells into the PBCB scaffolds, because we have seen in previous experiments that the Matrigel component displayed a beneficial effect on the integrity of the matrix after subcutaneous implantation into SCID mice. 45 However, it is important to note that Matrigel is a product derived from the basement membrane of the Engelbreth-Holm-Swarm mouse sarcoma 46 containing a huge number of different growth factors and extracellular matrix components of ill-defined composition, which makes Matrigel unacceptable for clinical applications. Therefore, to transfer our experimental approach into a putative subsequent clinical setting, it would be indispensable to omit the Matrigel component. Since fibrinogen is already approved for the clinic, a very simple and attractive strategy would be to resuspend the cells directly into fibrinogen without the addition of Matrigel before seeding the cell/matrix suspension into PBCB scaffolds.
In summary, our experiments have shown that it is feasible to induce human blood vessels in calvarial defects by grafting human endothelial cells. The extent of vessel formation was strongly increased by coimplanted MSCs that may stabilize nascent vessels. In addition, implanted MSCs effectively supported bone healing in calvarial defects. The missing supportive effect of engineered vessels on MSC-mediated osteogenesis may be explained by sufficient endogenous neoangiogenesis originating from the surrounding murine tissue in this xenogenic model.
Footnotes
Acknowledgments
The authors thank Nadine Hünke von Podewils and Beate vom Hoevel for excellent technical assistance. The authors would like to thank Sandra Strassburg for critical reading of the article. This work was supported by funding through the Deutsche Forschungsgemeinschaft (FI 790/3-2).
Disclosure Statement
No competing financial interests exist.