
Abstract
Improving vascularization of tissue-engineered oral mucosa (TEM) is a major challenge in the field of plastic surgery. Hypoxia is a stimulator of angiogenesis through a number of mechanisms. Therefore, hypoxia is a critical parameter that can be controlled in an effort to improve angiogenesis. In the present study we studied the secretion of a number of angiogenic factors during hypoxia exposure and evaluated the effect of TEM conditioned medium on endothelial cells. TEM was constructed by seeding human oral mucosa keratinocytes and fibroblasts on acellular human donor skin. TEM was exposed to hypoxia during 6, 12, and 24 h. Cellular hypoxia was assessed by immunolocalization of the hypoxia-inducible factor-1α. Secretion of vascular endothelial growth factor, placental growth factor (PlGF), tissue inhibitors of matrix metalloproteinases-1 and -2, and the activity of matrix metalloproteinase-9 significantly increased during hypoxia exposure. Moreover, conditioned medium from hypoxic TEM strongly enhanced endothelial cell proliferation and migration. In vitro exposure of TEM to hypoxia improves its capacity to support endothelial cell proliferation and migration, which suggests that hypoxia preconditioning of TEM potentially improves angiogenic responses for in vivo implantation.
Introduction
Tissue-engineered oral mucosa (TEM), using keratinocytes from different sites of the oral cavity5,6 and a variety of scaffolds,7,8 is a promising technique for reconstruction of oral defects. Recently, we and others have shown that substitutes, composed of oral mucosa keratinocytes and fibroblasts cultured on an acellular dermis, possess histological and immunohistochemical characteristics close to normal oral mucosa.7,9,10
However, so far, intraoral implantation of mucosal substitutes in animal models showed a relatively poor long-term viability resulting in loss of grafts. 11 The primary reason for graft failure appears to be the lack of adequate and timely graft vascularization.12,13 Usually, the survival of cells within the engrafted substitute is limited by diffusion of nutrients and oxygen from the underlying wound site, and in view of the typical graft size, this diffusion mechanism is inadequate for sustained survival. One approach to increase graft acceptance is to precondition the mucosal substitute to encourage rapid vascularization from the patient's wound bed.
The observation that low oxygen levels stimulate angiogenesis14,15 and increase the angiogenic capacity of stromal cells16,17 has resulted in a novel and relatively simple approach for inducing postimplantation blood vessel formation in tissue-engineered grafts by hypoxic priming before engraftment.
The aim of this study was to investigate whether hypoxia preconditioning of tissue-engineered mucosa increases the secretion of important angiogenic factors, and to evaluate whether these changes affect the proliferation and migration of endothelial cells in vitro.
Materials and Methods
Chemicals and culture media
Dulbecco's modified Eagle's medium (DMEM) 4.5 g/L glucose, Ham's F12 culture medium, human endothelial serum-free medium, penicillin, streptomycin, amphoceterin B, dispase, collagenase type I, and trypsin/ethylenediaminetetraacetic acid (EDTA) were purchased from Invitrogen. Fetal calf serum (FCS) was purchased from PAA Laboratories. Bovine serum albumin (BSA), epidermal growth factor (EGF), keratinocyte growth factor (KGF), and other chemicals were purchased from Sigma-Aldrich. Vascular endothelial growth factor (VEGF) and fibroblast growth factor (FGF-2) were purchased from Peprotech EC. Insulin was purchased from Eli Lilly.
Cell culture
Keratinocytes and fibroblasts were isolated from buccal biopsies that were obtained from three patients upon approval of the Medical Ethics Committee (# MEC+2007-282). Biopsies were rinsed in DMEM with 100 IU/mL penicillin, 100 μg/mL streptomycin, 50 μg/mL gentamicin, and 2.5 μg/mL amphoceterin B. The epidermis was separated from the dermis by overnight incubation in 2.5 mg/mL dispase solution. After rinsing the epidermal sheet with phosphate-buffered saline (PBS), keratinocytes were isolated from the epidermis using 0.25% trypsin-EDTA and the single cell suspension was seeded onto lethally irradiated 3T3 fibroblast feeder layers, according to Rheinwald and Green. 18 The keratinocyte culture medium consisted of a 3:1 mixture of DMEM and Ham's F12 medium with 5% FCS, 1 μM hydrocortisone, 1 μM isoproterenol, 0.1 μM insulin, 1% penicillin/streptomycin, and 1 ng/mL EGF.
Fibroblasts were isolated by mincing the dermis followed by an incubation in collagenase/dispase (1.5 mg/mL/2.5 mg/mL, respectively) solution for 2 h at 37°C. Afterward, a single-cell suspension was obtained by filtering the collagenase/dispase solution containing the dermis, with a 100 μm cell strainer (Sigma-Aldrich). The isolated fibroblasts were cultured in DMEM containing 10% FCS and 1% penicillin/streptomycin. Passages two to five were used for the experiments.
Preparation of acellular dermis
Human cadaver skin, cryopreserved in 85% glycerol, and cytomegalovirus, human immunodeficiency virus and hepatitis B negative, was obtained from the European Skin Bank. The epidermis was removed from the dermis after incubating in PBS with 1% penicillin/streptomycin, 100 μg/mL gentamicin, and 5 μg/mL amphoceterin B for 3 weeks at 37°C. 19 The dermis was divided into 1.0 cm2 sections and stored in DMEM supplemented with 1% penicillin/streptomycin, 0.5% gentamycin, and 5 μg/mL amphoceterin B until use.
Tissue-engineered mucosa
TEM was prepared as described previously.9,20 Briefly, 5×104 fibroblasts were seeded onto the acellular dermis using a centrifugal seeding technique. 21 Subsequently, 1×106 keratinocytes were seeded into a stainless steel ring placed on top of the dermis and incubated overnight in keratinocyte culture medium. Subsequently, the cultures were fed with the same keratinocyte medium containing 2.4×10−5 M BSA, 1 μM hydrocortisone, 1 μM isoprotenerol, 0.1 μM insulin, 1×10–5 L-carnitine, 1×10–2 L-serine, 1 μM D-L-tocopherol-acetate, and a lipid supplement containing 25 μM palmitic acid, 15 μM linoleic acid, 7 μM arachidonic acid, 1% penicillin/streptomycin, 1 ng/mL EGF, 4 ng/mL KGF, and cultured under submerged conditions for an additional 2 days. Thereafter, the cultures were lifted to the air/liquid (A/L) interface and cultured for 14 days in the same medium except that serum was omitted, the concentration of linoleic acid was increased to 30 μM and 100 mg/mL ascorbic acid phosphate, 1 ng/mL EGF and 4 ng/mL KGF were added. The medium was changed twice a week.
Oxygen systems
The standard oxygen level was defined as the pO2 that exists in a standard, conventional, humidified tissue culture incubator at 37°C (20%). The low (1.5%) oxygen system was established in a humidified environmental chamber set at 37°C. This incubator uses an oxygen analyzer to monitor and maintain the selected chamber oxygen concentration. This oxygen concentration was maintained with a calibrated gas mixture consisting of 95% nitrogen and 5% carbon dioxide.
After 2 weeks of culture at the A/L media, in normoxic conditions, the media were changed to A/L media without EGF and KGF and incubated for 24 h in normoxic conditions, before continuing incubation under hypoxic conditions for 6, 12, and 24 h. Parallel tissues were maintained for identical time periods under normoxia as controls. In another experiments, hypoxia exposure of TEM was extended up to 48 h.
Collection of TEM-conditioned media and histological preparation
Conditioned media from TEM exposed to hypoxia/normoxia for 6, 12, and 24 h were collected, centrifuged at 400 g for 5 min at 4°C, and stored at −80°C until further analysis. Hereafter, TEM was harvested and prepared for histological analysis. For histological determinations, samples were snap-frozen with liquid nitrogen for cryosectioning. Sections (6 μm) were cut and stained with hematoxylin and eosin (Klinipath). Stained sections were viewed using a light microscope with an Olympus eyepiece micrometer (A×0071, 20.4 mm2; Olympus).
Immunohistochemistry
For Ki-67 and hypoxia-inducible factor 1α (HIF-1α) staining, cryosections were fixed for 10 min with acetone, washed with 3 changes of PBS, and blocked with 10% normal goat serum and 10% normal human plasma (Sanquin). Tissue sections were incubated with anti-Ki67 (1:200) (DAKO) or anti HIF-1α (1:100) (Novus Biologicals) in PBS with 10% normal human plasma (Sanquin) for 1 h at room temperature. Slides were washed and incubated with goat anti-mouse or goat anti-rabbit biotin-labeled antibodies (DAKO) in 2% normal goat serum, 2% normal human plasma, and 5% BSA (Sigma) in PBS for 30 min at room temperature, followed by incubation with Streptavidin-ABC-HRP (DAKO) in PBS for 30 min at room temperature. For observation of the Ki-67- and HIF-1α-positive cells, the slides were incubated for 5 min at room temperature with a substrate which consisted of 5% 3,3′-Diaminobenzidine tetra hydrochloride hydrate (Sigma-Aldrich), PBS, and 30% H2O2. After washing the slides thoroughly with tap water, sections were counterstained with hematoxylin for background observation and coverslipped with Vectamount mounting media and examined with a light microscope. Control slides were incubated with an irrelevant mouse IgG.
Apoptosis assay
To determine the number of apoptotic cells, The DeadEnd Colorimetric TUNEL assay kit (Promega) was used. Cryosections were fixed and stained according to the manufacturer's instructions. Sections were counterstained with hematoxylin for background observation and coverslipped with Vectamount mounting media and examined with a light microscope. Control sections were incubated with PBS.
Quantification of hypoxic cells, cellular proliferation, and apoptosis
Immunostaining for HIF-1α, Ki67, and apoptotic assay were performed on tissues from three individual experiments from each hypoxic and normoxic time point. Hypoxic cells were quantified by counting the number of HIF-1α-positive cells within 12 random microscopic fields (magnification 200×). The amount of HIF-1α-positive cells was expressed as a percentage of the total number of cells. For quantification of proliferative cells, the number of Ki67-positive basal cells within 12 random microscope fields (final magnification 200×) was manually counted. The number of Ki67-positive nuclei from the total number of basal cells (×100%) was used to determine the proliferation index. For quantification of apoptotic cells, the number of apoptotic nuclei found within the length of the entire epidermal (basal layer) tissue section was manually counted.
Two observers, who were blinded to the conditions, carried out the quantification independently.
ELISA assay of conditioned medium
Concentration of TEM-secreted angiogenic factors in the conditioned medium was measured using commercially available sandwich ELISA kits according to the manufacturer's instructions (VEGF, PlGF, basic FGF [bFGF], hematopoietic growth factor [HGF], tissue inhibitors of matrix metalloproteinase [TIMP]-1, and TIMP-2; R&D Systems). Results are expressed as ng or pg/cm2 tissue with each sample consisting of 4 mL supernatant derived from 1 cm2 tissue.
Zymography for matrix metalloproteinase-9 and matrix metalloproteinase-2
Gelatinolytic proteinases in TEM-conditioned medium were assayed by gelatin-substrate zymography. Aliquots of 15 μL of conditioned medium were diluted 1:1 with sample buffer (0.1 M Tris-HCl, 4% SDS, 20% glycerol, 0.005% bromophenol blue, and 10 mM EDTA) and electrophoresed through a 10% polyacrylamide gel containing 2% gelatin as substrate. Following SDS-PAGE, SDS was removed from the gels by 2.5% (v/v) Triton-X-100 washes (2×20 min), and the gels incubated in assay buffer (50 mM Tris-HCl, 1% Triton X-100, and 5 mM CaCl2). After an overnight incubation at 37°C, the gel was stained with 0.1% Coomassie Brilliant Blue and cleared with 7% acetic acid and 5% methanol. Matrix metalloproteinase (MMP)-2 and MMP-9 were observed as unstained bands. Gelatinolytic activity was detected as clear bands against the aqua-blue stained gelatin background. As a marker for electrophoretic mobility of gelatinases in zymograms, the pro- and active forms of MMP-2 and MMP-9 (Calbiochem) were used. Gels were scanned by Kodak Image Station 440CF (Kodak) and the relative intensity of each band was quantified using NIH ImageJ software (http://rsb.info.nih.gov/ij/).
Human umbilical vein endothelial cells proliferation assay
Isolated human umbilical vein endothelial cells (HUVEC) at passage 4 were seeded at a density of 3×104 cells/well in 48-well plates in endothelial growth medium (EGM) consisting of human endothelial-SFM supplemented with 20% FCS, 10% human serum, 20 ng/mL FGF-2, and 100 ng/mL EGF for 24 h. The next day the cells were starved with high-glucose (HG)-DMEM supplemented with 0.5% FCS for 24 h. Cells were washed with PBS and treated with conditioned medium from TEM exposed to hypoxia/normoxia (1:1 diluted with HG-DMEM supplemented with 0.5% FCS). Control HUVEC cultures received EGM and HG-DMEM supplemented with 0.5% FCS.
Cells were harvested after 48 h with 0.05% trypsin/EDTA, resuspended in medium, and stained with 0.4% trypan blue. Cell counts were done in triplicate in a Neubauer chamber. Each assay was performed in duplicate. Data from three independent experiments were pooled for statistical analysis.
HUVEC migration assay
Migration assays were performed in transwell plates (Costar) of 6.5 mm filters with a pore size of 8 μm. The filters were coated with growth factor-reduced Matrigel (Becton Dickinson Labware) for 30 min at 37°C. HUVEC at passage 4 were seeded at a density of 5×104 cells in the upper compartment in 100 μL of EGM. The same medium (600 μL) was added to the lower compartment. Cells were allowed to adhere for 2 h, and then the medium in the upper and lower compartment was replaced with HG-DMEM supplemented with 0.5% FCS. The inserts were transferred to new 24-well plates containing 600 μL of conditioned medium from TEM who has been exposed to hypoxia/normoxia; EGM and HG-DMEM supplemented with 0.5% FCS in the lower compartment. The cells were allowed to migrate for 6 h at 37°C. The nonmigrated cells were removed from the upper surface by scraping with a cotton swab. Migrated cells were fixed in absolute methanol for 2 min at room temperature and stained with Giemsa (1:20) (Sigma-Aldrich) for 15 min. Migrating cells on the lower surface of each filter were quantified by counting five random microscopic fields under a light microscope (final magnification 200). Each assay was performed in duplicate. Data from three independent experiments were pooled for statistical analysis. HUVEC proliferation and migration assays are schematically depicted in Figure 1.

Schematic representation of HUVEC proliferation and migration assays in the presence of conditioned medium (CM) obtained from TEM exposed to normoxia or hypoxia. HUVEC, human umbilical vein endothelial cell; TEM, tissue-engineered oral mucosa.
Effect of VEGF on HUVEC proliferation and migration
The role of VEFG on the enhanced proliferation and migration of HUVEC observed with conditioned medium from hypoxic TEM was analyzed. Therefore, proliferation and migration assays were performed using conditioned medium from TEM exposed to normoxia supplemented with 0.6 ng/mL, an amount equal to the maximum amount of VEGF measured in conditioned medium from TEM exposed to hypoxia for 24 h, conditioned medium from TEM exposed to hypoxia for 24 h, or EGM supplemented with 20 pM FGF and 10 pM VEGF
Statistical analysis
The data are presented as the mean±standard error of the mean. Statistical analyses were conducted using one-way analysis of variance using GraphPad Prism software. Statistical difference was defined as p≤0.05. Comparisons between group means were made with the Tukey-Kramer test for multiple comparisons.
Results
Confirmation of cellular hypoxia
To confirm that TEM metabolically responded to lowered oxygen conditions, we assessed whether cells in TEM exposed to 1.5% O2 activated HIF-1α, a central regulator of the cellular response to hypoxia and ubiquitously expressed in mammalian cells and degraded when exposed to normoxia. As illustrated in Figure 2A–D, HIF-1α-positive cells were detected in the hypoxic samples. Exposure to hypoxia for 12 and 24 resulted to a 2- and 2.6-fold increase in the number of HIF-1α-positive cells, respectively, as compared to the normoxic samples (Fig. 2E).
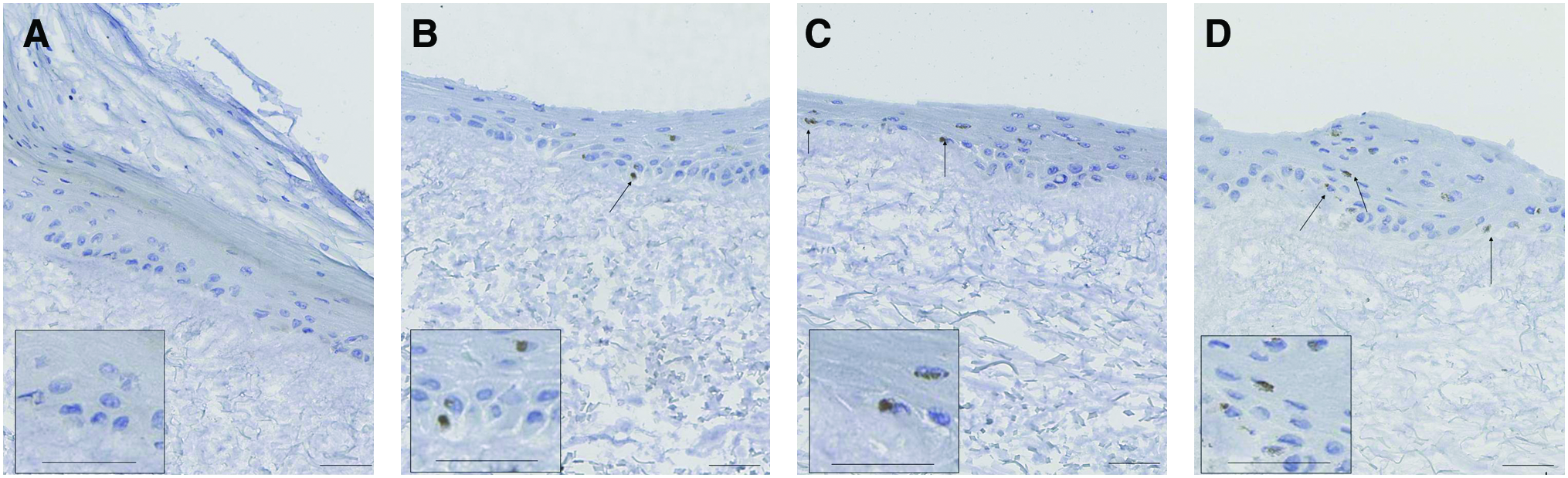
Confirmation of cellular hypoxia. TEM was exposed to
No obvious changes in the morphology of the epidermis were observed at the light microscope level (Fig. 3A, B). At 24 h of oxygen deprivation, a displacement of nuclei was observed in a number of cells within all living cells of the epidermis. Nuclear material was shifted to the perimeter of the nucleus, resulting in vacuoles.
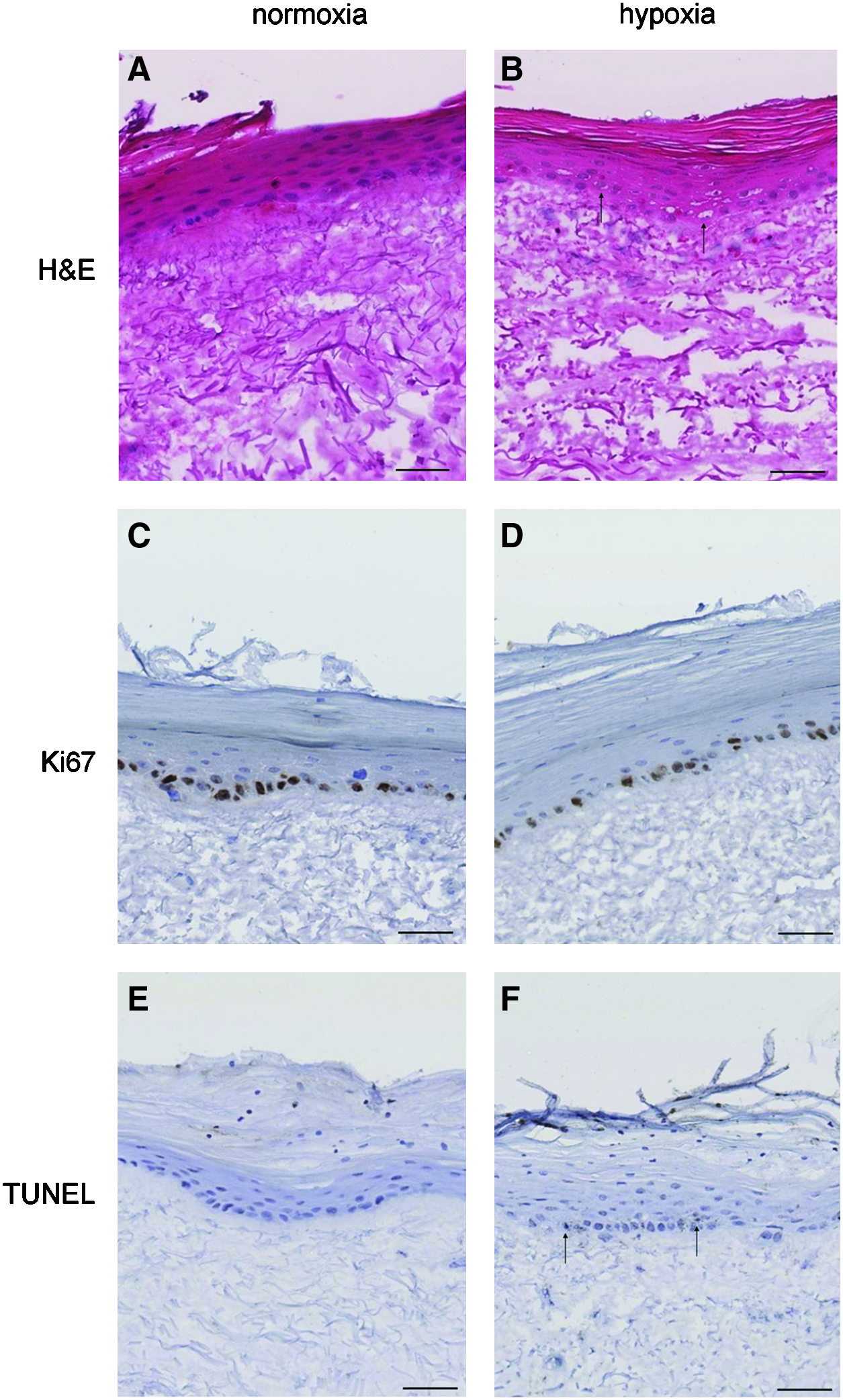
Histological appearance of TEM exposed to
In all samples, Ki67-positive cells (proliferative cells) were found in the basal layer (Fig. 3C, D). Compared with normoxic controls, no changes in keratinocyte proliferation were observed after tissues were exposed to hypoxia for up till 24 h (data not shown). As shown in Figure 3E and F few apoptotic cells were detected in TEM exposed to hypoxia. No differences in the number of apoptotic cells were found in TEM exposed to hypoxia compared to the normoxic controls (data not shown).
To analyze whether prolonged hypoxia exposure periods may affect the viability of TEM, in another experiment hypoxia exposure was extended for up to 48 h. Longer periods of hypoxia resulted in a 3.5-fold increase in the number of HIF-1α-positive cells compared with the normoxic controls (Supplementary Fig. S1A; Supplementary Data are available online at www.liebertonline.com/tea). In addition, TEM showed cells with pycnotic nuclei as well as alterations in epidermal attachment to the underlying connective tissue that was not found at 24 h of hypoxia. (Supplementary Fig. S1B). Exposure of TEM to hypoxia for 48 h resulted in a significant decrease in the number of proliferating cells compared to 24 h of hypoxia exposure (TEM exposed to hypoxia for 24 h, 36.8±2.3; TEM exposed to hypoxia for 48 h, 12.5±1.4; mean±SEM, p<0.001). (Supplementary Fig. S1C).
Assessment of angiogenic factors in TEM exposed to hypoxia
To study the effect of hypoxia on the angiogenic capacity of TEM, the secretion of a number of angiogenic factors was measured in the conditioned media of TEM. Hypoxia exposure of TEM for 12 and 24 h resulted in a 35% increase in the amount of secreted VEGF compared to the normoxic controls. Hypoxia also increased the secretion of PlGF (2.1-fold), TIMP-1 (1.6-fold), and TIMP-2 (2.0-fold) in TEM after 24 h of incubation (Fig. 4). If present, the amount of HGF and bFGF in the conditioned medium were below the detection limits of the ELISA used. None of the angiogenic factors were detected in unconditioned culture media (data not shown).
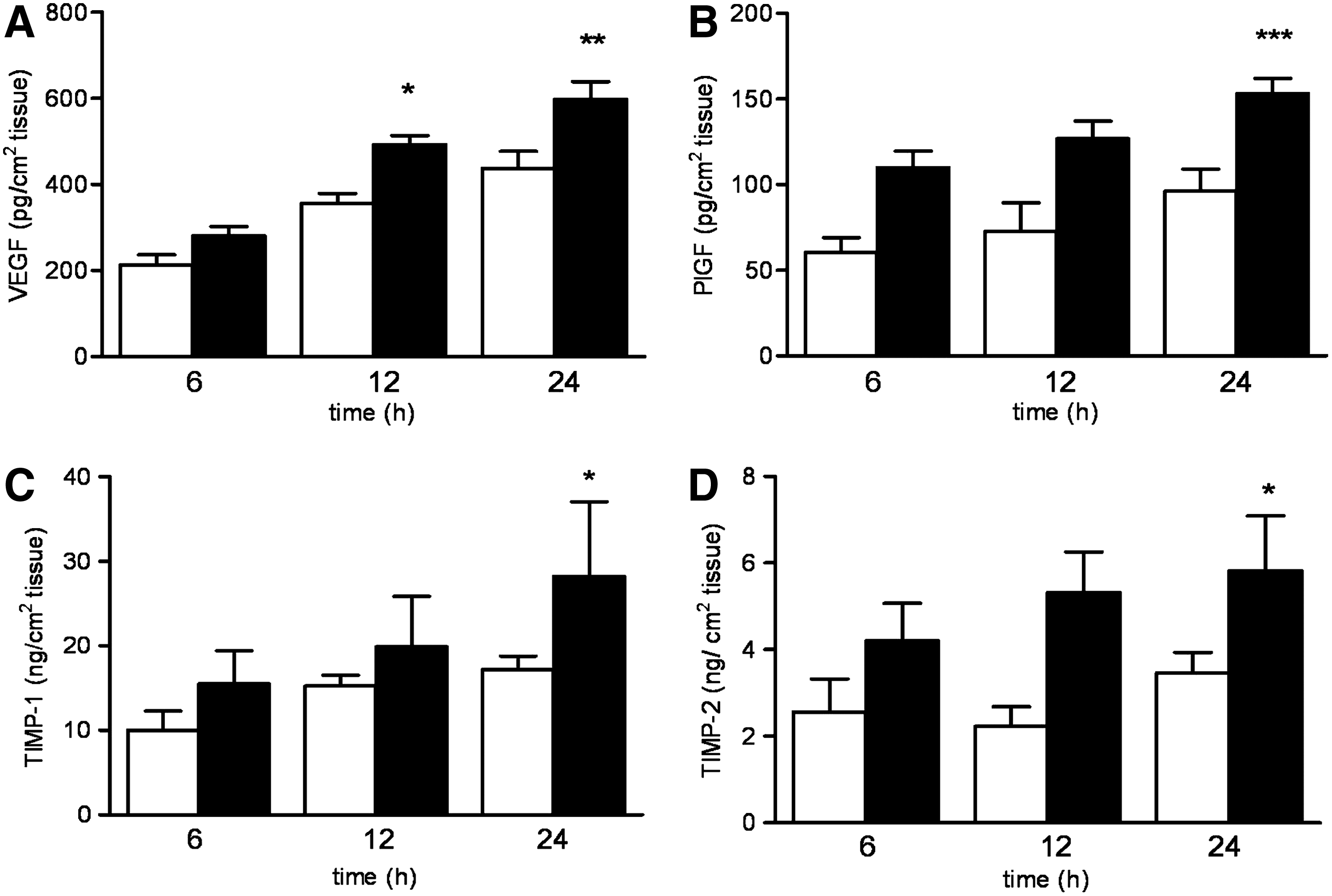
Release of angiogenic factors by TEM. The secretion of
Since longer hypoxia periods may influence VEGF production by TEM, the secretion of this growth factor was analyzed in conditioned medium obtained after 48 h of hypoxia. Exposing TEM to hypoxia for up to 48 h resulted in a slightly increase in the levels of secreted VEGF compared to the amounts of VEGF secreted at 24-h hypoxia exposure; however, these differences proved not to be statistically significant (secreted VEGF after 48 h of hypoxia, 262.6±38.6 pg/cm2 tissue; secreted VEGF after 24 h of hypoxia, 168.1±2.0 pg/cm2 tissue) (Supplementary Fig. S1D). In addition, HGF and bFGF were not detected after exposing TEM to 48 h of hypoxia (data not shown).
Given the elevated levels of TIMPs in the medium of TEM exposed to hypoxia, we also analyzed the activity of MMPs in our cultures by gelatin zymography. Figure 5A shows a representative zymography of conditioned medium from TEM exposed to normoxia/hypoxia, which are quantified in Figure 5B–E.
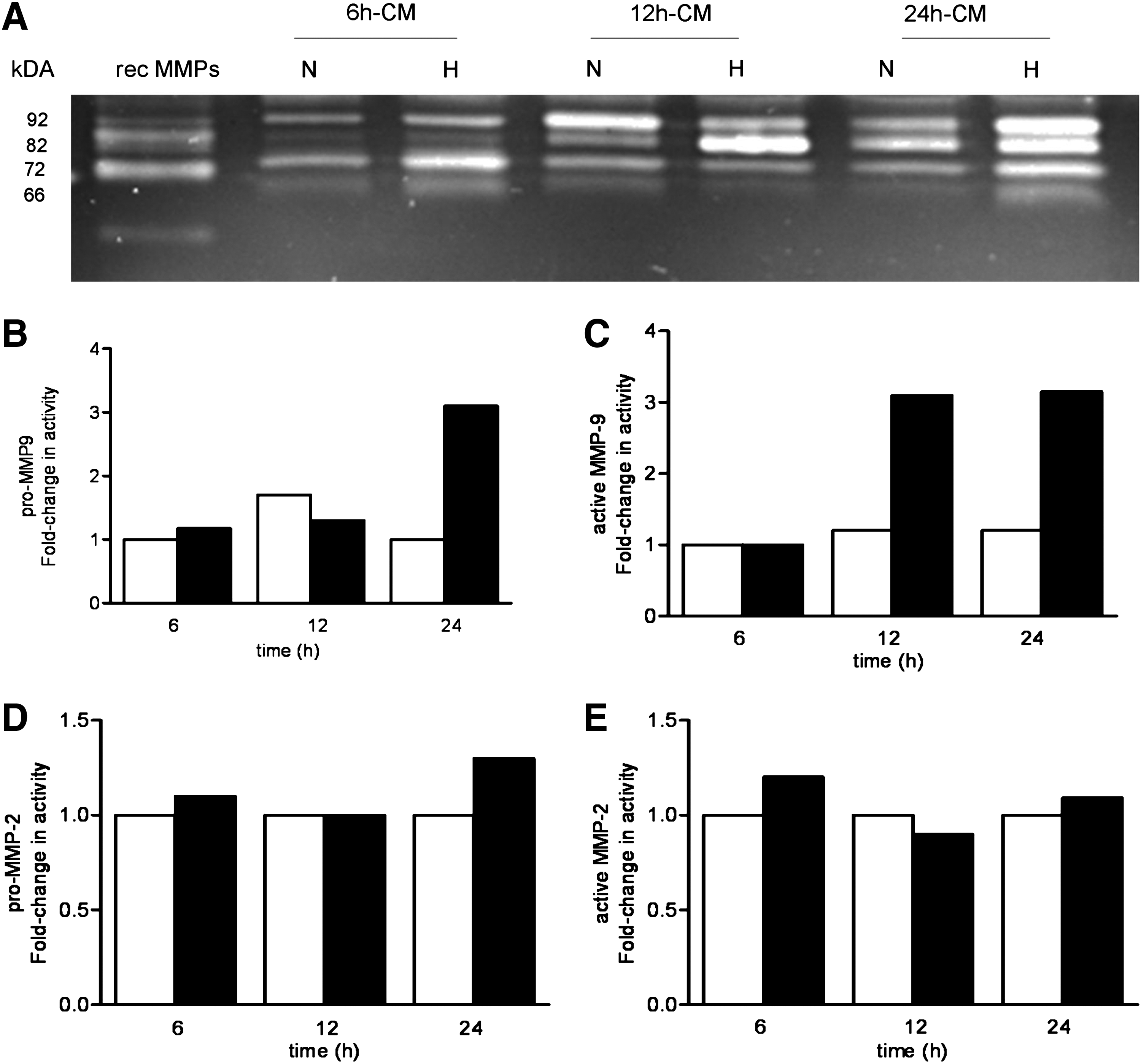
Increased MMP-2 and MMP-9 activity under hypoxic conditions TEM was exposed to normoxia (N) or hypoxia (H) for 6, 12, and 24 h. The medium was collected and MMP-9 and MMP-2 activities were measured by gelatin zymography.
Zymography gels show that TEM produces gelatinase activity in bands at 92, 82, 72, and 68 kDa (Fig. 5). The bands at 92 and 82 kDa correspond to the pro- and the active forms of MMP-9, respectively, and the bands at 72 and 66 kDa to the pro-and the active forms of MMP-2 (Fig. 5A). The stimulating effect of hypoxia on the level of MMP-9 activity was particularly evident in media from TEM exposed to hypoxia for 12 and 24 h (Fig. 5A, B). Higher levels of MMP-2 were also found in TEM exposed to hypoxia for 24 h compared to the normoxic controls. (Fig. 5D, E).
Effects of hypoxic TEM-conditioned medium on endothelial cell numbers and migration
To verify the biological activity of the factors secreted in the conditioned medium, the effects of conditioned medium derived from hypoxic TEM on human endothelial cells growth were analyzed. Conditioned medium from hypoxic TEM, collected at 6 and 12 h, equally stimulated endothelial cell growth. This in contrast to conditioned medium derived from 24 h hypoxic TEM that resulted in higher numbers of endothelial cells compared to medium conditioned from TEM exposed to normoxia (Fig. 6A). Besides proliferation, also migration of endothelial cells is an important aspect in angiogenesis. Therefore, we analyzed whether conditioned medium from hypoxic TEM was capable of attracting endothelial cells in vitro. Conditioned medium from TEM exposed to hypoxia for 6 and 12 h had no significant effect on the attraction of endothelial cells. However, conditioned medium from 24 h hypoxic TEM increased the migration of endothelial cells twofold compared to conditioned medium exposed to normoxia (Fig. 6B). Conditioned medium collected from TEM exposed to normoxia at the different time points did not affect endothelial cell migration.
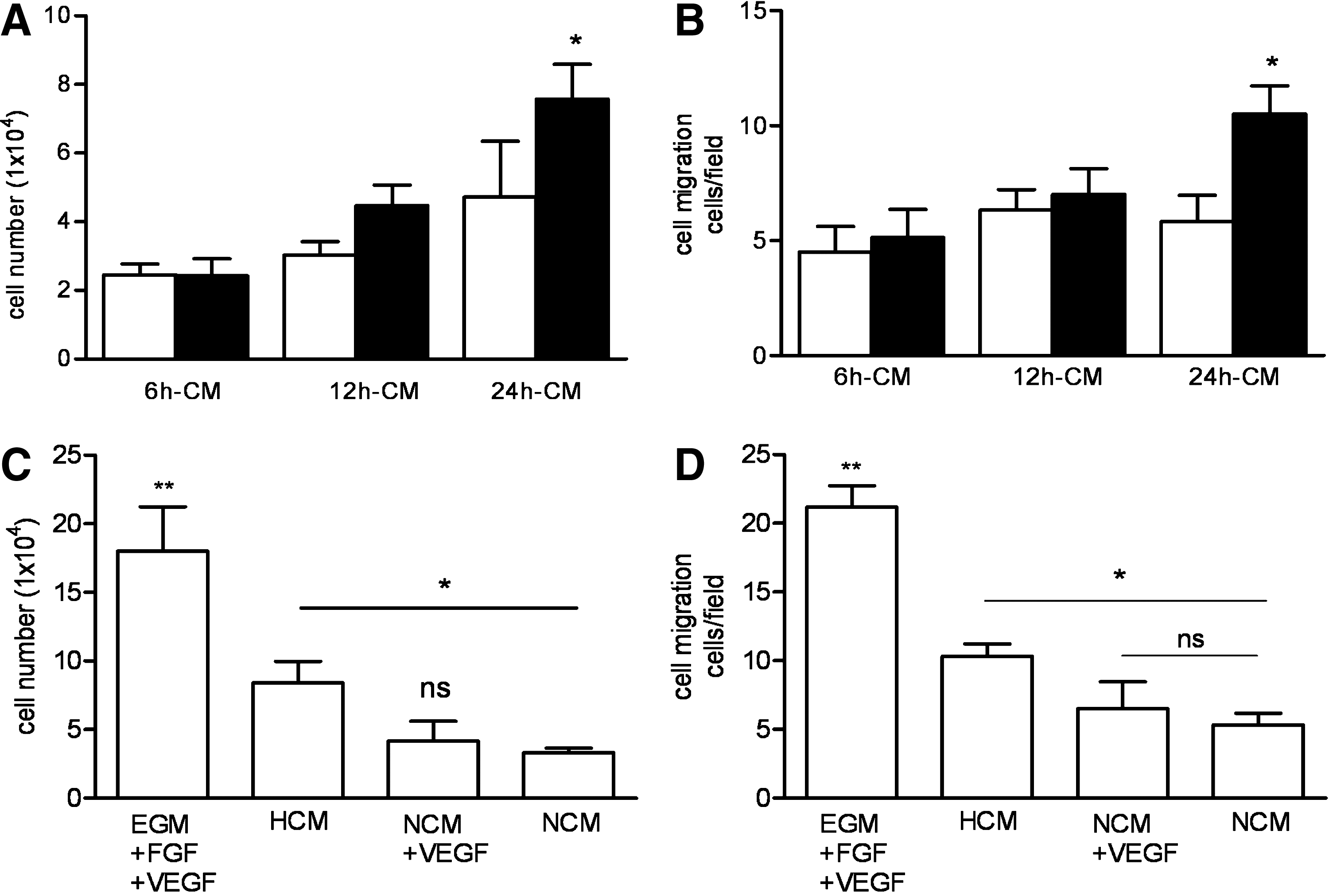
Since VEGF is thought to be the primary cytokine responsible for initiating angiogenesis, we examined whether the secreted VEGF levels of TEM exposed to hypoxia influenced HUVEC proliferation and migration by performing these assays with normoxic TEM-conditioned medium supplemented with VEGF and hypoxic TEM-/normoxic TEM-conditioned medium. When cultured with normoxic TEM-conditioned medium supplemented with VEGF, no additional effects on HUVEC proliferation or migration over the controls were observed (Fig. 6C, D). Figure 6C and D also shows that culturing with EGM supplemented with VEGF and FGF significantly increased the number of proliferating and migration HUVEC.
Discussion
In the present study, we demonstrated that hypoxia preconditioning of TEM enhances the secretion of angiogenic factors, increasing the proliferation and migration of endothelial cells in vitro. The normal morphology preserved by the construct and the absence of basal cell degeneration indicate that no profound structural changes occurred by exposing TEM to short periods of low oxygen concentrations.
This capacity to adapt to external hypoxic stress would be specifically important at sites of implantation, where the ingrowth of vessels into the engineered construct requires days to weeks; hence, an inadequate supply of oxygen to cells after transplantation must be assumed. 22
The secretion of the angiogenic factor VEGF by oral mucosal substitutes has been reported before.23,24 To our knowledge, however, this is the first study in which the secretion of angiogenic factors by tissue-engineered oral mucosa, other than VEGF, is analyzed. Several studies have shown that hypoxia strongly stimulates the secretion of angiogenic factors in a variety of cells such as adipose-derived stromal cells, smooth muscle cells, HUVECs, and endothelial progenitor cells.14,17,25 We therefore analyzed the secretion of several factors known to influence angiogenesis. We found significantly higher levels of VEGF, PlGF, TIMP-1, and TIMP-2 in the conditioned medium of TEM exposed to hypoxia compared to conditioned medium of TEM cultured under normoxia.
Both VEGF and PlGF are known to stimulate endothelial cell proliferation and induce angiogenesis,26,27 whereas TIMP-1 and TIMP-2 are known to regulate the activities of MMPs,28,29 and to modulate growth, differentiation, and migration of cells. 30 The fact that high levels of TIMP-1 and TIMP-2 were found in the conditioned medium of TEM exposed to hypoxia prompted us to investigate whether active MMPs were present in this media. Zymography analysis indicated increased MMP-2 and MMP-9 levels in conditioned media of TEM exposed to hypoxia compared to the normoxic controls. TIMPs and MMPs play an important role in angiogenesis since they activate and modify angiogenic growth factors and cytokines, remove matrix proteins needed for endothelial cell migration, and create space in the matrix to allow generation of endothelial cell tubules.31–33
The above-described results suggested that hypoxia- preconditioned TEM may have a higher angiogenic capacity than normoxic TEM. To investigate this further, we obtained conditioned medium from TEM exposed to hypoxia for 6, 12, and 24 h and found that 24 h hypoxic medium significantly increased the number of endothelial cells in vitro, compared to normoxic medium. Since not only proliferation of endothelial cells but also its migration is important for angiogenesis, we assessed HUVEC migration toward the medium conditioned by hypoxic and normoxic TEM. Our results indicate that conditioned medium obtained from TEM exposed to hypoxia for 24 h stimulated endothelial cells migration in vitro.
Experiments using the same concentrations of VEGF as the concentrations present in the hypoxic conditioned medium to induce HUVEC proliferation and migration showed lower numbers of proliferating and migrating HUVEC than the ones found by using hypoxic conditioned medium. This finding indicates that the increased secretion of VEGF upon hypoxia exposure did not only account for the increased HUVEC proliferation and migration and suggests that other factors, including PlGF, TIMP-1, and TIMP-2, may play an important role. These results are in line with previous work that showed that soluble factors derived from hypoxic fibroblasts, other than VEGF and bFGF, are necessary to induce angiogenesis in vitro. 34 Also, in agreement with Griffith and George, 34 bFGF seemed not to play a role in the enhanced HUVEC proliferation and migration observed in the presence of conditioned medium from hypoxic TEM since its production was not upregulated upon hypoxia.
We confirmed that TEM was exposed to hypoxia by the detection of HIF-1α protein, an ubiquitous expressed mediator of the cellular response to hypoxia in mammalian cells, which plays a pivotal role in the anaerobic metabolism, angiogenesis, erythropoyesis, and vasodilatation. 35 The HIF-1α protein is degraded when exposed to oxygen through its oxygen-dependent degradation domain. Under hypoxic conditions, the HIF-1α protein is translocated into the nucleus, where it dimerizes with HIF-1β to its active form.36–38 In our study, significantly higher numbers of abundant HIF-1α-positive cells were detected when TEM was exposed to hypoxia for 12 and 24 h. Interestingly, HIF-1α-positive cells were also observed in the normoxic samples. Oxygen concentrations have been seen to decline rapidly from the exterior to the interior of grafts39,40; thus, it is likely that at least some cells in the normoxic engineered mucosa construct are hypoxic.
Oxygen tension is known to influence proliferation of cells. 41 Incubation for up to 24 h under hypoxic conditions did not influence cell proliferation in TEM; however, longer incubation periods resulted in a decrease in proliferative capacity as well as in morphological alterations of the epidermis of the engineered mucosa construct. These observations are supported by previous studies culturing skin substitutes at low oxygen concentrations42,43 and indicate that 24 h is the maximal period to expose TEM to hypoxia. The oxygen tension we have used in our work was based on studies reporting increases in VEGF production in keratinocytes and fibroblasts that were exposed to low oxygen concentrations.44–46 It could be that the oxygen tension that was chosen for this study is not the most optimal; however, based on our results we can conclude that exposure of engineered mucosal substitutes to 1.5% of oxygen for 24 h upregulate the secretion of angiogenic mediators and stimulate endothelial proliferation and migration in vitro. Whether other oxygen concentrations may improve the angiogenic capacity of engineered mucosa constructs need to be clarified in future studies.
Taken together, our results show that engineered mucosa constructs produce soluble mediators that are upregulated under hypoxia conditions and that these mediators are responsible for angiogenesis related events in vitro. Since VEGF, PlGF, TIMP-1, TIMP-2, MMP-2, and MMP-9 were upregulated under hypoxia, it is likely that these mediators play a role in the enhanced proliferation and migration of endothelial cells observed in our study.
This is in agreement with studies directed at improving tissue regeneration in vivo. In these studies, hypoxia preconditioning of human mesenchymal stem cells promoted postimplantation blood vessel formation.47,48 Although in our study hypoxia exposure of TEM shows to be beneficial for endothelial cell proliferation and migration in vitro, higher effects were found when stimulating with endothelial medium supplemented with VEGF and bFGF. This observation has to be considered when pretreating engineered mucosa constructs with hypoxia prior implantation
Clinical applications of in vitro engineered oral mucosa tissue are still limited. For engineered mucosa tissue constructs to become a viable option for future clinical use, they must be able to survive in vivo. For this, the presence of an adequate blood supply is necessary. Several approaches to improve vascularization of tissue-engineered constructs have been used, including incorporation of dermal microvascular endothelial cells49–52 or adipose-derived stromal cells, 53 and overexpression of VEGF in modified engineered substitutes. 54 Combining such techniques with hypoxia preconditioning to hypoxia may improve the survival of oral mucosa engineered constructs in vivo.
Footnotes
Acknowledgments
We are grateful to Joost Rens (Department of Experimental Surgical Oncology, Erasmus MC) for providing the hypoxic culture facilities and to Dr. G. van Osch (Departments of Orthopaedics & Otorhinolaryngology, Erasmus MC) for her comments regarding this article. This work was supported by the Vanderes Foundation (Contract no. 155).
Disclosure Statement
No competing financial interest exist.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.