
Abstract
Electrical stimulation (ES) is a promising technique for axonal regeneration of peripheral nerve injuries. However, long-term, continuous ES in the form of biphasic electric current (BEC) to stimulate axonal regeneration has rarely been attempted and the effects of BEC on Schwann cells are unknown. We hypothesized that long-term, continuous ES would trigger the activation of Schwann cells, and we therefore investigated the effect of BEC on the functional differentiation of primary human mesenchymal stromal cells (hMSCs) into Schwann cells, as well as the activity of primary Schwann cells. Differentiation of hMSCs into Schwann cells was determined by coculture with rat pheochromocytoma cells (PC12 cell line). We also investigated the in vivo effects of long-term ES (4 weeks) on axonal outgrowth of a severed sciatic nerve with a 7-mm gap after retraction of the nerve ends in rats by implanting an electronic device to serve as a neural conduit. PC12 cells cocultured with hMSCs electrically stimulated during culture in Schwann cell differentiation medium (Group I) had longer neurites and a greater percentage of PC12 cells were neurite-sprouting than when cocultured with hMSCs cultured in growth medium (control group) or unstimulated hMSCs in the same culture conditions as used for Group I (Group II). Group I cells showed significant upregulation of Schwann cell-related neurotrophic factors such as nerve growth factor and glial-derived neurotrophic factor compared to Group II cells at both the mRNA and protein levels. Primary Schwann cells responded to continuous BEC with increased proliferation and the induction of nerve growth factor and glial-derived neurotrophic factor, similar to Group I cells, and in addition, induction of brain-derived neurotrophic factor was observed. Immunohistochemical investigation of sciatic nerve regenerates revealed that BEC increased axonal outgrowth significantly. These results demonstrate that BEC enhanced the functional activity of Schwann cells via the induction of neurotrophic factor release and guide-increased axonal outgrowth in vivo. The effectiveness of long-term ES highlights the feasibility of a BEC-based therapeutic device to accelerate nerve regeneration of severed peripheral nerve injuries with a gap.
Introduction
For injury recovery, axonal outgrowth from the proximal site into the distal region of the injury site is essential and closely related to Schwann cells, which ensheath axons via myelination. The PNS has an inherent capacity for regeneration via a cascade called Wallerian degeneration.17–19 This multistep process involves Schwann cells losing myelin, and then being stimulated to proliferate and produce a variety of neurotrophic factors, cytokines, and cell adhesion molecules, thereby regenerating axons across the lesion.20,21 Upregulation of neurotrophic factors during the nerve regeneration process after injury is critical. Nerve growth factor (NGF), brain-derived neurotrophic factor (BDNF), and glial-derived neurotrophic factor (GDNF) are representative examples of neurotrophic factors.22–25 They promote sprouting of axons from the proximal end of the cut nerve into the denervated nerve stumps. 26 Further, these factors affect Schwann cells in the distal nerve stump, and promote Schwann cell migration. 27 Thus, the delivery of neurotrophic factors in a controlled fashion through direct injection or coupling to an artificial nerve conduit is considered a promising tool for peripheral nerve regeneration.28–30
ES is thought to stimulate PNS regeneration by upregulating BDNF and trk receptor expression in motor/sensory neurons.31,32 However, these previous studies focused on axotomized neuron responses to ES both in vivo and in vitro. Despite the major role that Schwann cells play in neuronal regeneration, few studies have addressed the effects of ES on the differentiation or activities of Schwann cells with respect to proliferation or neurotrophic factor expression. We hypothesized that biphasic stimulation could be used to stimulate Schwann cells to produce axonal outgrowths, based on our previous findings that BEC stimulates proliferation and induces the expression of cytokines such as vascular endothelial growth factor (VEGF) and bone morphogenetic protein-2 from Schwann cells. 33 In the present study, we designed experiments to investigate whether biphasic stimulation could activate Schwann cell-related cellular mechanisms by characterizing the responses of primary Schwann cells to ES, and the differentiation of human mesenchymal stromal cells (hMSCs) exposed to ES into Schwann cells. BEC was delivered to cells using a stimulator-integrated circuit (IC) chip in an in vitro culture system. The functional capacity of differentiated hMSCs (dhMSCs) was evaluated by measuring neurite outgrowth of cocultured rat pheochromocytoma cells (PC12 cell line); in the presence of NGF, these cells produce neurite outgrowths. Cellular and molecular responses to BEC were confirmed by evaluating cell proliferation, neurotrophic factor release, and the expression of Schwann cell markers. We also explored the in vivo effects of biphasic stimulation on axonal outgrowth using an electronic nerve conduit connected to an implantable stimulator in a 7-mm rat sciatic nerve defect model.
Materials and Methods
Preparation and culture of hMSCs
Bone marrow was obtained from the iliac crest of a single human donor (female, 30 years-of-age) who provided informed consent. Procedures were approved by the local ethics committee (IRB at Seoul National University Dental Hospital, No. CRI05008) according to the legal regulations for human tissue and organs in Korea. Culture of hMSCs from bone marrow was performed as described previously. 34 Nucleated cells concentrated at the interface after a Ficoll-Paque (Amersham Biosciences) treatment were collected and washed with phosphate-buffered saline (PBS). Collected cells were plated at a density of 2×106 cells/100 mm culture plate and cultured in an expansion medium containing low-glucose Dulbecco's modified Eagle's medium (DMEM; Welgene, Inc.), 100 units/mL penicillin, 100 μg/mL streptomycin, and 10% heat-inactivated fetal bovine serum (HIFBS) under a humidified atmosphere of 5% CO2 at 37°C with medium changes every 3 or 4 days. Cells were passaged when they reached 70% confluence, and re-seeded in a new culture plate at a density of 3×105 cells/cm2. Second to fourth passage cells were used for all experiments.
The ability of hMSCs to differentiate into osteoblasts, chondrocytes, or adipocytes was confirmed according to previously published protocols.34,35 Osteogenic studies were carried out by plating hMSCs at a density of 1.7×104 cells/cm2 in a differentiation medium supplemented with high-glucose DMEM, 10% HIFBS, 50 μg/mL ascorbic acid, 10 mM β-glycerophosphate, and 100 nM dexamethasone (Sigma-Aldrich) at 1 or 2 days postplating when the cells were confluent. This differentiation medium was replenished every 3 or 4 days. Adipogenic induction of hMSCs was performed by culturing the cells in media containing 1 μM dexamethasone, 50 μM 3-isobutyl-1-methylxanthine (Sigma), and 10 μg/mL insulin (Sigma) for 20 days after plating at a cell density of 1.7×104 cells/cm2. Chondrogenic differentiation was investigating by culturing high-density cell pellets for 3 weeks in high-glucose DMEM supplemented with 1% insulin-transferrin-selenium (Sigma), 1 mM sodium pyruvate, and 50 μM L-proline (Sigma). 36 The medium was changed every 3 days. After the differentiation processes were complete, cultures were stained for osteoblasts, chondrocytes, or adipocytes using Alizarin red, Toluidine blue, and Oil red O, respectively, as described previously.34,35
Isolation of primary Schwann cells
Dorsal root ganglia (DRG) were harvested from the spinal cords of newborn Sprague-Dawley rats as described previously. 37 DRG were harvested and cut into small pieces, followed by enzymatic digestion using 0.25% collagenase at 37°C for 40 min. After centrifugation at 1300 rpm for 5 min, the cell pellets were suspended in 0.25% trypsin–EDTA, incubated at 37°C for 20 min, and then dispersed using a cell strainer. The filtrate was centrifuged for 5 min at 1000 rpm. Cells were washed three times with (4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid) (HEPES), and plated at a density of 2×106 cells/100 mm culture plate in the RPMI (Welgene) medium containing 10% HIFBS and 5% penicillin/streptomycin. Schwann cells from these plates were purified with a differential anti-mitotic agent (Cytosine-D-Arabino-Furanoside; Sigma) and a cold jet method was used to remove fibroblasts.
Fluorescence-activated cell sorting
A total of 105 first-passage hMSCs were incubated with FITC-conjugated anti-human CD105 (Ancell Co.), CD90 (BD Biosciences), CD45 (Ancell), or anti-mouse IgG antibody for 30 min or 90 min in the dark after being washed with washing buffer (PBS buffer containing 1% bovine serum albumin). Cells were washed twice with washing buffer and suspended in washing buffer containing 0.1% paraformaldehyde (Sigma) for fluorescence-activated cell sorting (FACS) analysis. Labeled and non-treated cells were sorted using the BD FACS Aria Cell Sorting System (BD Biosciences).
Differentiation of hMSCs into Schwann cells
hMSCs were plated at a density of 2500 cells/cm2 in the growth medium containing DMEM and 10% HIFBS. The differentiation process was initiated the next day by replacing the growth medium with DMEM containing 1 mM β-mercaptoethanol (BME; Sigma) for 24 h. Cells were then incubated for 72 h with the growth medium containing 35 ng/mL all-trans-retinoic acid (Sigma). Three days later, cells were finally transferred to DMEM containing a neurotrophic cocktail composed of 100 μM forskolin (Sigma), 10 ng/mL basic fibroblast growth factor (R&D Systems), 5 ng/mL platelet-derived growth factor-AA (R&D Systems), and 200 ng/mL Heregulin-β 1 (EGF Domain; Sigma), and cultured for 10 days with medium changes every 72 h to establish differentiated cultures.37–39 These treated hMSCs are hereafter referred to as dhMSCs.
Experimental design for BEC stimulation
The BEC stimulator and in vitro culture system were the same as those described previously. 33 This system, which uses a stimulator IC, can deliver different durations, amplitudes, and frequencies of current with precise resolution. All components of the in vitro culture system were handled as described in our previous study. To determine the optimal conditions to activate hMSCs during differentiation to Schwann cells, different amplitudes (5, 7.5, 10 μA/cm2) or durations of current (25, 125, 250 μs) at a pulse rate of 100 pulses/s were tested, resulting in a total charge that ranged from 187.5 to 1875 (×10−12 C/cm2). During Schwann cell differentiation, hMSCs were continuously exposed to BEC at day 4, after culture in a neurotrophic cocktail for 3 days. Primary Schwann cells were plated at a density of 2500 cells/cm2 in the RPMI medium and stimulated with BEC daily in continuous mode using the optimal parameters used during differentiation of hMSCs into Schwann cells. Cells were seeded on gold plates at the beginning of culture without ES. For primary Schwann cells culture, the lower gold plates were precoated with collagen solution to stabilize the attachment of Schwann cells. Gold plates coated with 0.15 mg/mL of type I collagen/well (Cellmatrix) were dried overnight under UV light irradiation for sterilization.
An implantable, electronic nerve conduit was designed for the in vivo rat sciatic nerve experiments and is described in detail elsewhere. 40 This device is composed of two parts: a polyimide base integrated with electrodes that serves as a tube interposed into a nerve gap, and a stimulator IC that generates and delivers BEC to the polyimide-based conduit. Both electrodes (anode and cathode) are parallel and positioned with 10 mm of one another on a 15 mm polyimide base, allowing the biphasic current flow to be bidirectional. A film-type electrode was changed to a conduit-type electrode by rolling and heating at high temperature, resulting in an electronic nerve conduit with a diameter of 1.5 mm. The stimulator IC was connected to a conduit-type electrode and packaged using a silicon elastomer with a 3 V coin type battery as a power source for 4 weeks. The ES parameters used were a current of 20 μA, a pulse duration of 100 μs, and a pulse rate of 100 pulses/s.
Functional bioassay of dhMSCs via coculture with PC12 cells
PC12 cells were maintained in the growth medium containing RPMI and 10% HIFBS, and neurite formation after treatment with NGF (10 ng/mL; R&D Systems) was confirmed. Two days before the completion of Schwann cell differentiation of hMSCs, PC12 cells were labeled with fluorescent dialkylcarboncyanine (Dil) dye (250 ng/mL; Molecular Probes, Inc.) to identify PC12 cells. After 10 days of culture the in differentiation medium, undifferentiated hMSCs (uhMSCs), dhMSCs, or electrically stimulated-dhMSCs were plated at 500 cells/cm2 in the growth medium containing DMEM and 10% HIFBS. The next day, the same number of Dil-labeled PC12 cells was cocultured on a coverslip containing seeded hMSCs. After 3 days of incubation, cocultured cells on the coverslips were fixed in 4% paraformaldehyde (1 h, room temperature) and immunostained with FITC-conjugated anti-β-tubulin III (monoclonal, 1:100; Serotec). The coverslips were mounted in crystal mounting solution (Biomeda Corporation) and observed under a confocal microscope (Olympus BX60). Neurite outgrowth was assessed using two independent parameters: the percentage of neurite-sprouting PC12 cells, and the length of the longest neurite, using Spot software (Diagnostic Instruments, Inc.). Four or 10 independent coculture experiments were carried out and neurite outgrowth was assessed. Cells bearing neurites longer than the cell body diameter were counted as neurite-sprouting cells.
Quantitative real-time reverse transcription–polymerase chain reaction
Total RNA was extracted by adding 0.5 mL of TRIzol® reagent (Invitrogen–Life Technologies) to cell pellets. One microgram of RNA from each sample was subjected to cDNA synthesis using SuperScript™ Reverse Transcriptase II (Invitrogen) and oligo 12–18 primer (Invitrogen) in a 20 mL reaction volume according to the manufacturer's instructions. RNA complementary to the cDNA was removed using Escherichia coli RNase H (Invitrogen). One microliter of cDNA was then subjected to PCR with the following amplification profile: predenaturation at 95°C for 40 s, amplification (denaturation at 95°C for 40 s, annealing at 60°C for 40 s, and extension at 72°C for 1 min) for 26–30 cycles, and a final extension step at 72°C for 10 min. PCR was performed in a DNA thermal cycler (model PTC-200; MJ Research). Once the thermal cycling was complete, the integrity and size of the resulting amplicons were assessed by electrophoresis (50 V, 90 min) on a 1.5% agarose gel with bands observed using a gel documentation system (Vilber Lourmat). The human- and rat-specific primer sequences used for real-time reverse transcription–polymerase chain reaction (RT-PCR) are listed in Table 1.
BDNF, brain-derived neurotrophic factor; GDNF, glial cell-derived neurotrophic factor; GFAP, glial fibrillary acidic protein; NGF, nerve growth factor; PMP22, peripheral myelin protein 22.
For quantitative RT-PCR (qRT-PCR), total RNA isolation and cDNA synthesis were carried out as described above. SYBR®Green PCR Master Mix (Applied Biosystems) was used to detect accumulation of PCR product during cycling on an ABI Prism 7700 sequence detection system (Applied Biosystems). The thermocycling conditions were as follows: predenaturation at 95°C for 10 min, followed by 40 cycles of denaturation at 95°C for 15 s and annealing and extension at 60°C for 1 min, followed by a final dissociation cycle at 95°C for 15 s, 60°C for 1 min, and 95°C for 15 s. qRT-PCRs were carried out in triplicate in at least three independent experiments (n>3). Oligonucleotide primers for qRT-PCR were designed using qPCR system sequence detection software v1.3 (Applied Biosystems), and primers were purchased from Bionics. Fold differences in levels of each gene were calculated for each treatment group using CT values normalized to transcript levels of the housekeeping gene β-actin according to the manufacturer's instructions.
In vivo rat sciatic nerve defect model
Male Sprague-Dawley albino rats (n=16), aged 9 weeks, were used for the animal experiments. All animals were randomly divided into two experimental groups. In the control group (n=8), an electronic conduit without a stimulator was implanted into nerve defects. In the ES group (n=8), a conduit connected to a stimulator packaging a 3 V coin-type battery as a power source for 4 weeks was implanted into the nerve defects (Supplementary Fig. S1; Supplementary Data are available online at www.liebertonline.com/tea). All operations were performed with general anesthesia using intraperitoneal injection of Zoletil (20 mg/kg, Ketaras; Yuhan Corporation) mixed with xylazine (10 mg/kg, Rompun; Bayer Korea Ltd.). After disinfection of the hindlimb skin with 10% Betadine (Potadines; Sam-Il Pharm.), the right sciatic nerve was exposed at the middle of the thigh, and the middle point of the nerve was sharply transected, creating a 7-mm defect after retraction of the nerve ends. The defect was bridged with an electronic conduit with a length of 15 mm and a diameter of 1.5 mm (Supplementary Fig. S2). Both the proximal and distal stumps of the nerve were anchored into the conduit electrode by inserting ∼3–4 mm of the nerve inside each end of the conduit with three stitches each using 9-0 nonabsorbable suture (Black Nylon, Ailee CO., Ltd.), resulting in a 7-mm gap. ES at an amplitude of 20 μA, a duration of 100 μs, and a frequency of 100 Hz was delivered continuously to both stumps via the conduit electrode for 4 weeks. All of the animals were treated and handled in accordance with the “Recommendations for Handling of Laboratory Animals for Biomedical Research” compiled by the Committee on the Safety and Ethical Handling Regulation for Laboratory Experiments of the School of Dentistry at Seoul National University.
Immunohistochemical staining
Schwann cell markers in in vitro cultured dhMSCs or primary Schwann cells were detected after 6 days of culture in the Schwann cell differentiation medium at an average cell density of 3000 cells per coverslip. Cells were washed three times with PBS, fixed in 4% paraformaldehyde for 30 min, and blocked in a blocking solution for 30 min. Cells were incubated with S100b (anti-rabbit; Dako Cytomation) (1:100), GFAP (anti-rabbit; Dako) antibody (1:100), or peripheral myelinated protein 22 (PMP22, mouse polyclonal, 1:100; Santa Cruz Biotechnology) antibodies for 1 h at room temperature, washed twice with PBS, and then incubated with secondary antibody labeled with FITC (anti-rabbit IgG; (Biomeda) [1:400]) for 40 min. Primary antibody controls were processed in parallel using only the secondary antibody. Slides were washed in PBS for 10 min and incubated with PBS-buffered 4′,6-diamino-2-phenylindole (DAPI) solution (1 μg/μL; Santa Cruz Biotechnology) using a dilution ratio of 1:1000 for nuclear staining. Section images of stained cells were captured using an Olympus Fluoview FV300 confocal laser scanning microscope and Fluoview software (Olympus Optical Co. Ltd.). Immunostaining of the stem cell marker, Stro-1 (anti-mouse, 1:100; BD BioSciences), in hMSCs was performed using the same protocol as described for Schwann cell markers.
In vivo expression of neuronal and Schwann cell markers was assessed 4 weeks after surgery. Each animal was killed using the same anesthesia as used for the initial surgery. The sciatic nerve regeneration conduit was taken out for paraffin sectioning as follows. The electronic conduit containing the regenerated nerve was washed with water after fixation in 10% formalin for 1 week. Followed dehydration in 70% ethanol, the conduit base material wrapping the regenerative nerve was removed during embedding in paraffin. After cutting the narrowest middle part, half of the regenerative nerve at the proximal side was embedded in paraffin for fixation, and then, sections with a thickness of 1.5 μm were cut (Supplementary Fig. S2). The resulting sections were mounted on HistoBond (Paul Marienfeld GmbH & Co. KG), and were blocked with horse serum for 40 min, followed by incubation with primary antibodies to PMP22 (mouse polyclonal, 1:100; Santa Cruz Biotechnology) at room temperature for 1.5 h. After washes with PBS, sections were incubated with FITC-conjugated β-tubulin III (1:100; Serotec) at room temperature in the dark for 2 h. After washes with PBS, sections were incubated with secondary antibodies conjugated to Alexa 546 (goat anti-mouse, 1:200; Invitrogen) for PMP22 at room temperature in the dark for 40 min. Following further PBS washes, sections were mounted in crystal mounting solution and observed under a confocal microscope. After capture of representative 400×fields for whole sections of each group, the numbers of β-tubulin III-positive neurites were counted using ImageJ software, and the neurite density (neurites number/mm2) was calculated.
Statistical analysis
In vitro neuronal outgrowth was evaluated using phase-contrast images and Spot software (version 4.3). Results represent an average of three independent experiments from which measurements were taken in three separate microscopic fields. All data are presented as mean±standard error of mean. Statistical analyses were performed using SAS 9.1.3 software (SAS Institute, Inc.). One-way ANOVA and post hoc multiple comparison Tukey's tests were applied to determine the significance of differences among experimental groups. The unpaired, two-tailed Student's t-test was used to determine statistical significance when comparing in vivo axonal outgrowth changes between control and ES-rats. Results were considered statistically significant at p<0.05.
Results
Identification of stem cell markers in hMSCs
In vitro-cultured hMSCs were isolated from bone marrow from one donor to ensure experimental consistency. Ninety-five percent of hMSCs showed homogeneity for the stem cell surface marker CD105 and 99% for CD90, and all hMSCs were negative for the hemopoietic stem cell surface marker CD45 based on flow cytometry (Fig. 1A). The multipotency of hMSCs was tested by their ability to differentiate into chondrocytes, osteoblasts, or adipocytes under appropriate culture conditions. Differentiation of hMSCs into chondrocytes was confirmed by staining of sulfated proteoglycans with Toluidine blue, whereas differentiation of hMSCs into osteoblasts or adipocytes was confirmed by Alizarin red S staining and Oil red O staining of lipids, respectively (Fig. 1B). uhMSCs showed positive staining for Stro-1, a stem cell marker of almost all hMSCs (Fig. 1C).

Characterization of subcultured human mesenchymal stromal cells (hMSCs).
Characterizing the differentiation of hMSCs into Schwann cells
Before investigating whether biphasic stimulation of hMSCs resulted in Schwann cell differentiation, the phenotypic progression of hMSCs into Schwann cells was investigated by morphological observations and determining the gene expression patterns of Schwann cell-specific markers such as S100b and GFAP. During a series of treatments of hMSCs with reducing agents and a combination of trophic factors, cells underwent phased changes in their shape from flat to round-shaped cells at the beginning of BME treatment, and then to spindle-like cells after treatment with retinoic acids; this spindle-shaped morphology was maintained during subsequent culturing (Fig. 2A). The presence of Schwann cell marker gene transcripts was assessed by RT-PCR (Fig. 2B). S100b and GFAP expression were highly upregulated after all-trans-retinoic acid treatment and after 4 days of treatment with trophic factors, but declined at day 8, whereas these Schwann cells markers were expressed at low levels throughout culture of hMSCs in the growth medium. Using immunofluorescence with DAPI dual staining, we found that dhMSCs expressed three markers characteristic of Schwann cells: S100b, GFAP, and PMP22 (Fig. 2C). Staining results showed that S100b and GFAP were expressed by almost all dhMSCs, whereas PMP22 was expressed in ∼50% of dhMSCs. uhMSCs did not stain for these Schwann cell marker proteins.
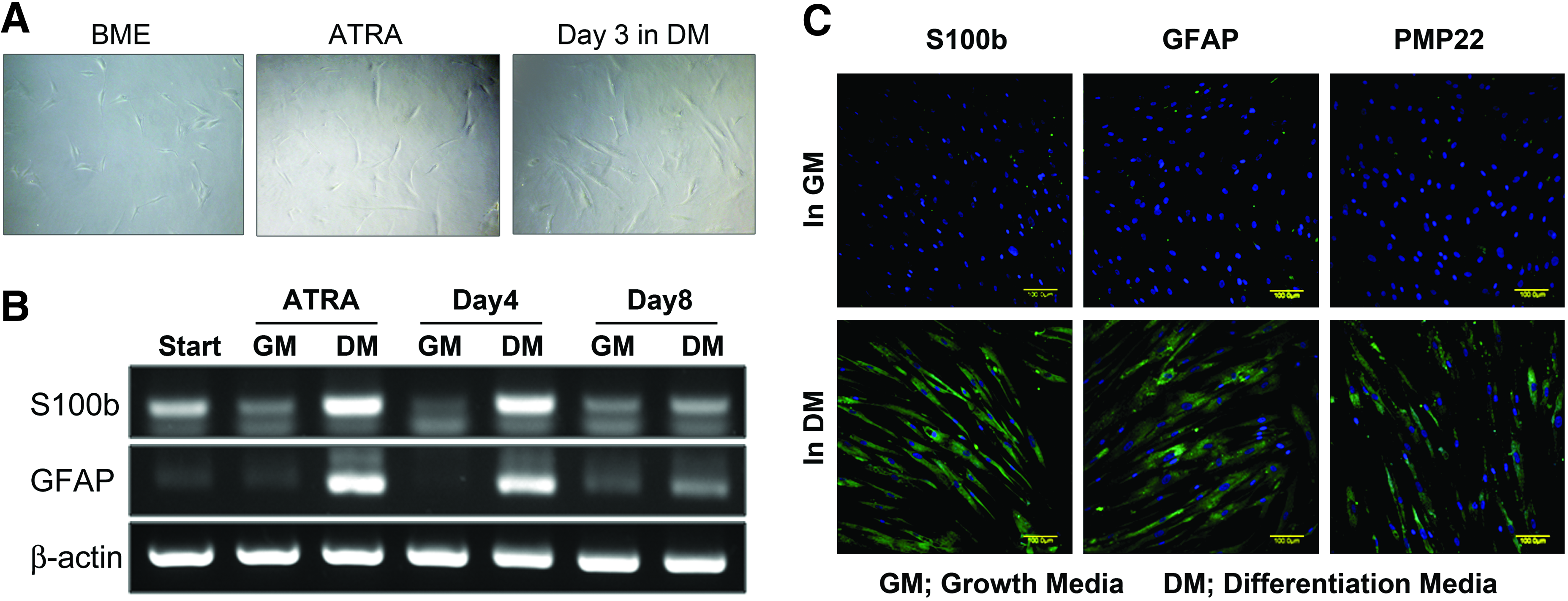
Differentiation of hMSCs into Schwann cells.
Screening of optimal BEC parameters for Schwann cell differentiation of hMSCs
HMSCs were exposed to BEC on a daily, continuous basis, as this has been shown to facilitate differentiation of hMSCs into osteoblasts. 33 Electric currents of different magnitudes with the duration or amplitude varied at a constant frequency of 100 Hz were tested, because cell responses can differ according to cell type (Fig. 3A). hMSCs were exposed to BEC at day 4 after 3 days of treatment with a combination of trophic factors. After stopping biphasic stimulation, hMSCs were further cultured in the differentiation medium for 3 days until functional assays were performed. The optimal electrical parameters were determined by assessing the length of the longest neurites after coculture of stimulated hMSCs with PC12 cells that form neurites after NGF treatment (Fig. 3B). Previous studies that used biphasic and balanced pulses reported that a short pulse width (about 100–150 μs) and frequencies between 2 to 150 Hz resulted in in vivo peripheral nerve regeneration.8,41,42 Because cells in vitro might respond to ES differently from cells in vivo, we varied the parameters by using durations of 25, 125, and 250 μs for amplitudes of 5, 7.5, and 10 μA at a fixed frequency of 100 Hz. In the first screening, three different durations (25, 125, and 250 μs) were tested at an amplitude of 7.5 μA. The length of the longest neurites in dhMSCs cultured only in differentiation medium was significantly increased by 2.2-fold compared to that in uhMSCs cultured in the growth medium. ES of dhMSCs increased the neurite length by 1.7-fold (p<0.05) at 25 μs, by 1.9-fold (p<0.05) at 125 μs, and by 2.2-fold (p<0.05) at 250 μs compared to dhMSCs. There was little difference among the electrically stimulated groups. The next screening step was performed at three different amplitudes (5, 7.5, and 10 μA) for a duration of 125 μs. The second screening revealed large differences among electrically stimulated groups. The largest increase was found in the 10 μA-stimulated group (12-fold, p<0.01), followed by the 5 μA-stimulated group (5.9-fold, p<0.01), and then the 7.5 μA-stimulated group (1.9-fold, p<0.05) compared to dhMSCs (Fig. 3C). PC12 cells were labeled with Dil dye before coculture with dhMSCs to identify neurite-forming cells. Immunofluorescence revealed positive β-tubulin III staining in Dil-labeled cells (PC12 cells) but not in dhMSCs, verifying that PC12 cells formed neurites because of coculture with dhMSCs (Fig. 3D). The optimal BEC parameters chosen were as follows: an amplitude of 10 μA, a pulse duration of 125 μs, and a pulse frequency of 100 Hz.

Determination of optimal parameters for biphasic stimulation of hMSCs during differentiation into Schwann cells.
Characterization of electrically stimulated dhMSCs
After selection of the optimal BEC parameters, all subsequent experiments were performed using these parameters (10 μA amplitude, 125 μs pulse duration, and 100 Hz pulse frequency) to characterize the effects of biphasic stimulation on the differentiation of hMSCs into Schwann cells. The functional capacity of dhMSCs was determined by coculture with PC12 cells by quantifying the length of the longest neurites and the percentage of PC12 cells that sprouted neurites. 43 PC12 cells cocultured with uhMSCs showed little neurite outgrowth compared with PC12 cells cocultured with electrically stimulated dhMSCs or unstimulated dhMSCs (these treated hMSCs are hereafter referred to as Group I and Group II cells, respectively). Notably, PC12 cells cocultured with Group I cells showed an increase in the length of the longest neurites by 7.6- and 22.3-fold compared to Group II cells and uhMSCs, respectively (Fig. 4A). In addition, the percentage of neurite-bearing cells induced by coculture with Group I cells was 1.8- and 5.4-fold greater than that observed with coculture with Group II cells and uhMSCs (p<0.05), respectively (Fig. 4B). Similar to the length of the longest neurites, the percentage of PC12 cells sprouting neurons was largest after coculture with Group I cells. qRT-PCR results showed that biphasic stimulation had a significant influence on the expression of neurotrophic factors, especially NGF and GDNF, but not BDNF expression, compared to nonstimulation with ES (Fig. 4C). NGF and GDNF expression showed a greater increase at day 6 when ES was stopped than at day 10, four days after stopping ES (4.79-fold at day 6 vs. 2.33-fold at day 10 for NGF, and 9.66-fold at day 6 vs. 4.36-fold at day 10 for GDNF). There was no change in BDNF expression at any of the time points evaluated. A slight increase in the expression of Schwann cell markers, including S100b, PMP22, and BDNF, was detected in Group I cells, but only GFAP expression was significantly greater (55%) in Group I cells than in Group II cells at day 6 when ES was stopped. ELISAs for NGF, GDNF, and BDNF were performed using the conditioned medium from cells cultured for 3 days before sampling at the same time point as samples were collected for qRT-PCR. NGF and GDNF levels were 51% (p=0.207) and 119% (p=0.009) higher in Group I cells than in Group II cells at day 6, and 241% (p=0.005) and 80% (p=0.01) higher in Group I cells than in Group II cells at day 10, respectively. BDNF release was not altered, consistent with the qRT-PCR results (Fig. 4C).
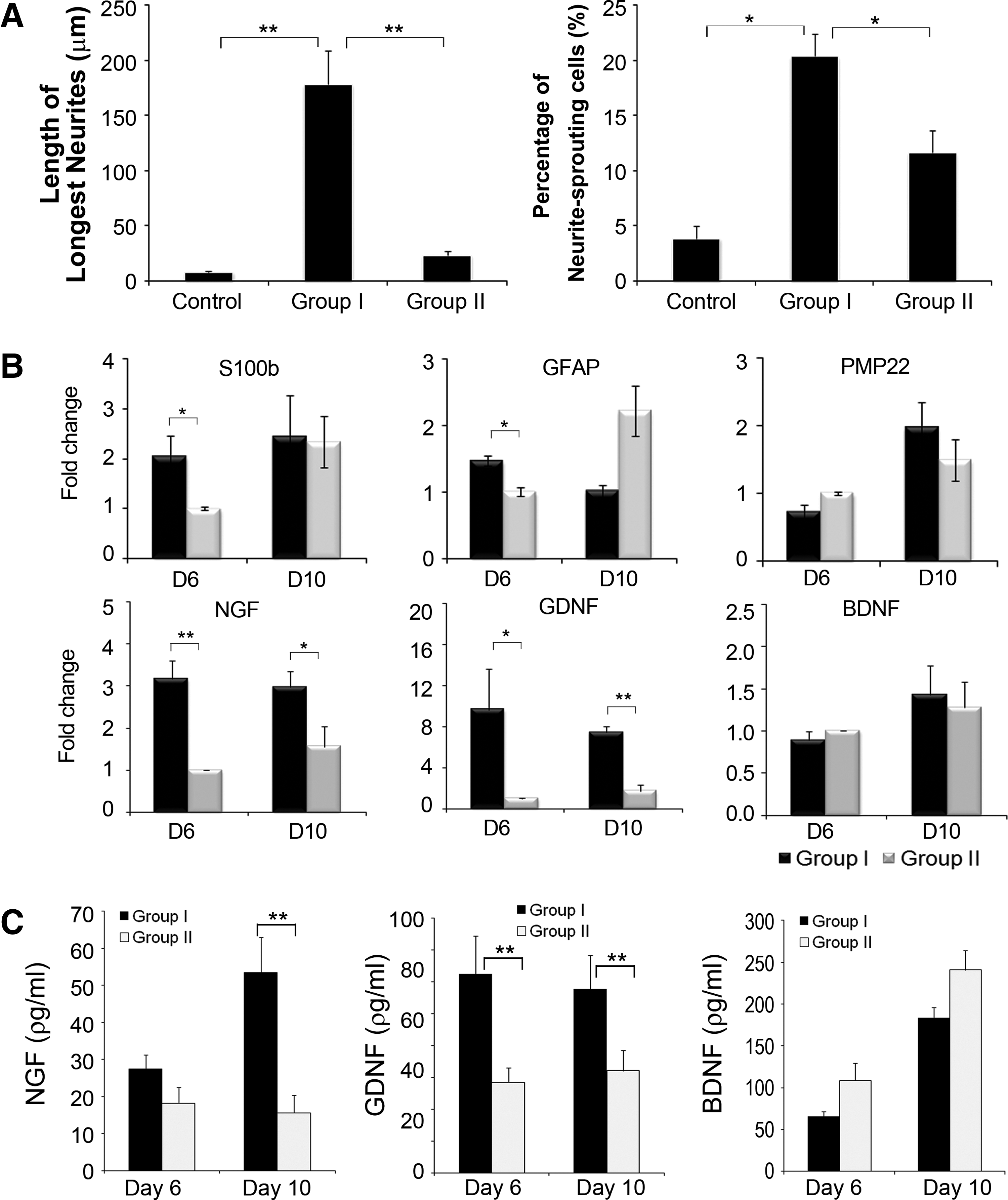
Characterization of ES-dhMSCs using optimal BEC parameters. hMSCs were electrically stimulated at the chosen optimal parameters (10 μA, 125 μs, 100 pulses/s).
The effects of optimal BEC parameters on primary Schwann cells
Primary Schwann cells were isolated from DRG harvested from newborn Sprague-Dawley rats. Cultured Schwann cells had long, spindled shape and displayed the whorling pattern of growth commonly seen in Schwann cell cultures (Fig. 5A). 44 Primary Schwann cells stained with a DAPI dual stain showed positive expression of three markers characteristic of Schwann cells, namely, S100b, GFAP, and PMP22 (Fig. 5B). The effect of BEC on these cells was investigated by assessing the proliferation of these cells and the gene expression of Schwann cell markers using the optimal BEC parameters used to stimulate hMSCs during differentiation into Schwann cells. Electrically stimulated Schwann cells showed a slight increase in proliferation until 3 days after stimulation and a fivefold increase in proliferation after 7 days of continuous stimulation compared to unstimulated Schwann cells (Fig. 6A). Consistent with the effect of biphasic stimulation on Schwann cell differentiation of hMSCs, biphasic stimulation increased the gene expression of neurotrophic factors after 6 days treatment; NGF, GDNF, and BDNF levels were 4.4-fold (p<0.01), 7.0-fold (p<0.05), and 3.9-fold (p<0.05) higher in electrically stimulated primary Schwann cells than unstimulated Schwann cells, respectively (Fig. 6B). Even PMP22 expression showed a slight increase by 1.9-fold in the stimulated group versus the unstimulated group. The ELISA results for NGF, GDNF, and BDNF reflected the qRT-PCR results (Fig. 6C). ELISAs were performed both on days 3 and 7 during continuous stimulation. NGF protein levels in the stimulated groups increased 5.7-fold at day 3 and 2.3-fold at day 7 compared to the unstimulated group. Although GDNF and BDNF levels remained unchanged at day 3, they increased significantly by 2.63- and 1.13-fold, respectively, in electrically stimulated Schwann cells compared to unstimulated cells.

Identification of primary SCs.
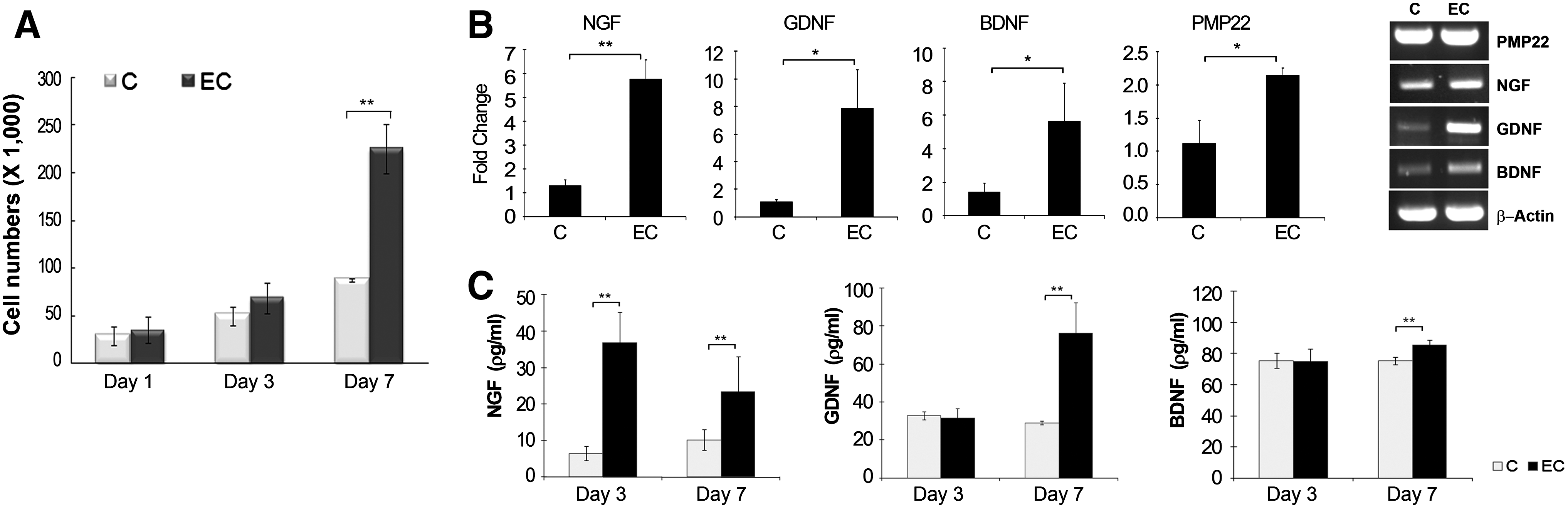
Responses of primary SCs to biphasic stimulation. Primary SCs were plated at a density of 2500 cells/cm2 in the RPMI medium and stimulated with BEC daily in continuous mode at the same parameters as in Figure 4.
In vivo peripheral nerve regeneration using an electronic nerve conduit
The entire conduit was exposed to continuous biphasic stimulation during the observation period. In vivo BEC parameters were as follows: an amplitude of 20 μA, a pulse duration of 100 μs, and a pulse rate of 100 pulses/s, similar to the optimal in vitro parameters. In a previous report, we verified the effects of this device and its efficacy using walking track assessment, electrophysiological analyses, and immunohistochemistry for PMP 22. Because we did not examine axonal outgrowth in our previous study, we examined neurite formation in the present study. Histological examination was performed by immunostaining for β-tubulin III as an index of neurite formation 45 and counter-staining for PMP22 as an indicator of myelin formation. 46 Inspection of transverse axial sections near the proximal ends showed little difference in the density of myelin between control and ES groups due to abundant formation of myelin, similar to that seen for normal nerves, in both groups (data not shown). However, when transverse axial sections of the central part of the nerve were investigated, there were clear differences in the number of regenerated axons, axon thickness, and organic compartmentalization of myelin and neurites between normal and regenerated nerves, whereas the regenerated nerve trunks selected from control and ES groups displayed a similar ultrastructural organization (Fig. 7A). Myelin in normal nerves forms a highly ordered, organically linked frame with a honeycomb-like structure. Moreover, axons are surrounded by the PMP22 protein, indicating stable myelination. However, in both the ES and control groups, most of the axons were not surrounded by PMP proteins, indicating that they were not ensheathed by myelin. Axonal regeneration was determined by counting β-tubulin III-positive neurites in transverse axial sections of the central portion of the regenerated nerve. Neurites that stained positive for β-tubulin III occurred at a density of 1141.09±98.30 neurites/mm2 in the ES group compared to a density of 696.15±207.23 neurites/mm2 in the control group, showing that biphasic stimulation induced a significant increase in neurite density of 64% (Fig. 7B). In our previous study, detailed analysis of evoked nerve action potentials and the number of myelin sheaths demonstrated the functional recovery of severed nerves with a 7-mm gap in the ES group. 40
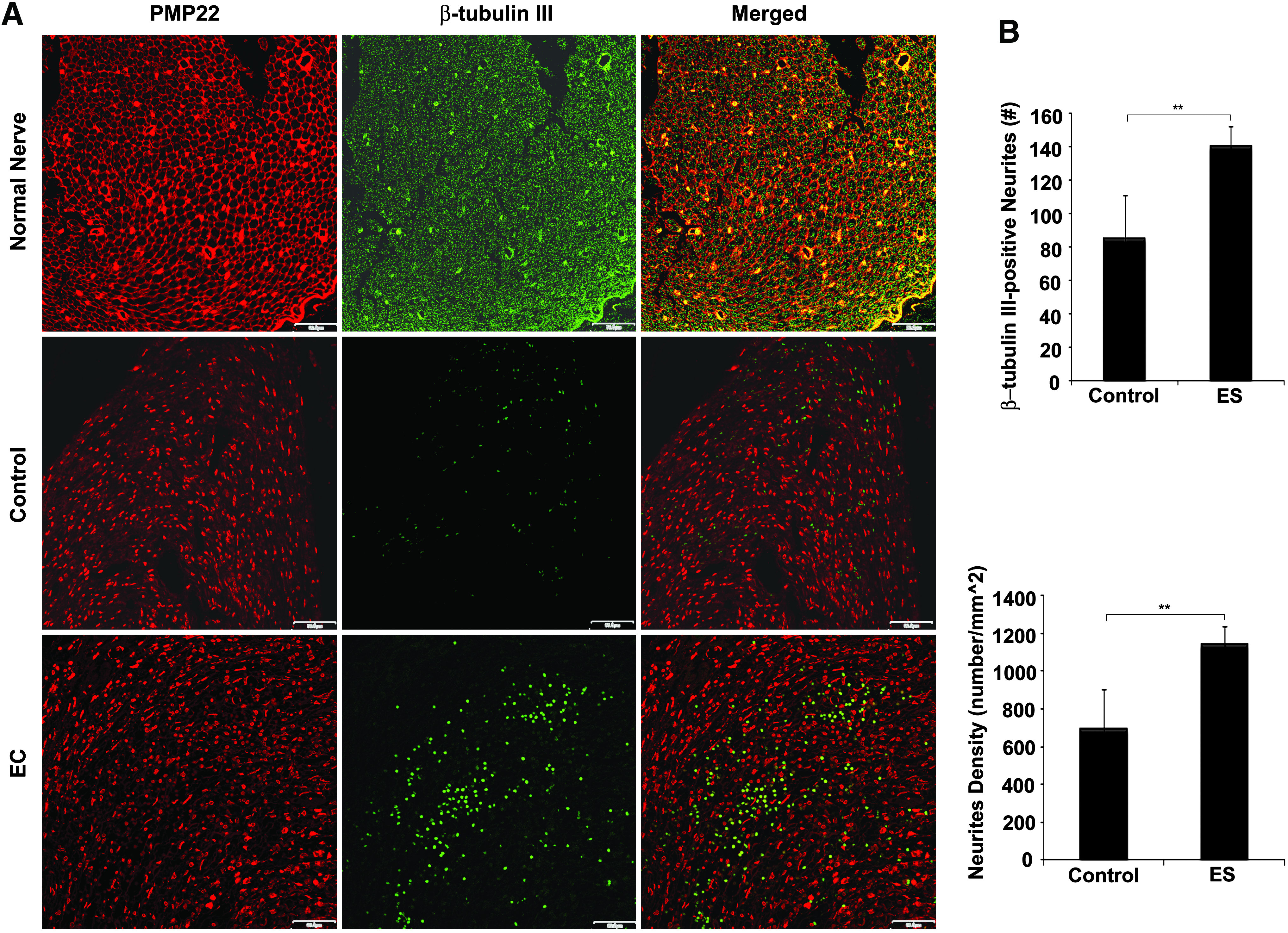
Fluorescent micrographs of regenerated nerve tissue within an electronic nerve conduit. A rat sciatic nerve defect with a 7-mm gap was bridged with a conduit-type electrode and continuously exposed to BEC for 4 weeks. The control group did not have a stimulator attached to the conduit-type electrode, whereas the electrically stimulated group (ES group) was stimulated for 4 weeks.
Discussion
Recent studies have documented that ES can regenerate peripheral nerves and be used to treat spinal cord injuries in rodent models.47,48 In this study, we focused on characterizing the effects of ES on Schwann cells in the context of peripheral nerve regeneration. In this report, we demonstrated that (1) isolated and expanded hMSCs can be induced to differentiate into Schwann-like cells in vitro, (2) continuous biphasic stimulation enhances functional differentiation of hMSCs into Schwann cells more efficiently than biochemical treatments alone, and (3) BEC increases cell proliferation and the expression of neurotrophic factors from primary Schwann cells. Moreover, an acellular electronic nerve graft bridging a 7-mm-long sciatic nerve defect significantly increased axonal outgrowth after 4 weeks of continuous biphasic stimulation, most likely due to increased Schwann cell activity. BEC stimulated Schwann cells and dhMSCs to upregulate the expression of NGF, BDNF, and GDNF. The present findings are consistent with the results from our previous studies using BEC; long-term biphasic stimulation enhances tissue regeneration due to stimulation of proliferation and the secretion of cytokines.
Our in vitro culture system is a suitable model to investigated cellular responses to BEC stimulation because a range of parameters, including the amplitude, duration, and pulse frequency of the electrical current, can be controlled. In a previous study, we demonstrated that BEC induced the expression of cytokines, and of these cytokines, VEGF induction was continuous, whereas the expression of other cytokines was transient. 33 However, BEC itself was not osteoinductive, but supported the osteoblast differentiation of hMSCs by stimulating proliferation and inducing the expression of osteogenesis-related cytokines (VEGF, insulin-like growth factor-1, and bone morphogenetic protein-2). We predicted that BEC would have a similar effect in nerve tissue. In light of this hypothesis and previous studies that reported that biphasic pulses have a stimulating effect on motor/sensory neurons,9,49 we investigated the effects of biphasic stimulation on Schwann cells given the critical role that Schwann cells play in peripheral nerve regeneration. We evaluated different parameters to determine the optimal BEC parameters for stimulation of Schwann cell activity. However, expression of a few Schwann cell markers does not indicate that the cells bearing these “Schwann cell markers” are capable of all the complex functions of a Schwann cell. 39 Thus, the capacity of dhMSCs to function as Schwann cells was determined by measuring the length of the longest neurites of PC12 cells, because a primary function of Schwann cells is to guide axonal growth and stabilize neuronal structures through myelination. A current of 10 μA and a pulse duration of 125 μs resulted in a total charge number (1250×10−12 C/cm2) greater than that used in osteoblast differentiation (1.5 μA/250 μs; 375×10−12 C/cm2). Nevertheless, biphasic stimulation using these values enhanced the activity of primary Schwann cells and increased differentiation of hMSCs into Schwann cells. This result highlights the fact that the optimal effective charge and ES parameters are different for different cell and tissue types. 50
The gene expression of Schwann cell marker and immunofluorostaining results are strongly indicative that a series of treatments with a reducing agent, retinoic acid, and a combination of trophic factors induces a phenotypic change in dhMSCs along Schwann cell lineage, consistently with previous studies.37,39 To validate the effect of ES on the cell function of dhMSCs, we tested the functional potential of dhMSCs to elicit neurite outgrowth from PC12 cells because PC12 cell is known to differentiate into neuronal-like cell in the presence of NGF or in the treatment of Schwann cell conditioned media.43,51,52 Our experiment was designed through coculture of PC12 cells with dhMSCs, and resulted in consistent tendency with the other methods aforementioned. As like stated that the effects of conditioned media were not as strong as NGF, 51 PC12 cells cocultured with dhMSCs showed less efficiency than NGF treatment, but gave a demonstrable neurite outgrowth induced by dhMSCs. Schwann cells secrete several neurotrophic factors such as BDNF, GDNF, and NGF,20,53 but do not induce neurite outgrowth in PC12 cells excluding NGF. 53 Our results indicate that ES enhanced the functional activity of Schwann cell-like dhMSCs, and increased expression of NGF or GDNF confers benefits on neurite outgrowth.
Under conditions of injury, Schwann cells transiently increase the production of neurotrophic factors, including NGF, BDNF, and GDNF, as well as adhesion and extracellular matrix molecules that are permissive substrates for neurite growth. 24 However, denervated Schwann cells in the distal nerve pathways (chronic denervation) cannot secrete sufficient neurotrophic factors to maintain the survival of neurons, resulting in the progressive failure of functional recovery after nerve injuries, even when surgical repair is performed. 29 Therefore, approaches that deliver neurotrophic factors have been shown to improve the axonal regeneration of injured peripheral nerves.26,29,30 The potential of ES to serve as an effective tool for the repair of injured peripheral nerves is highlighted by the upregulation of neurotrophic factor expression observed in our study and those of others. Gordon et al. reported that short-term ES of less than 1 h in duration at a low frequency of 20 Hz greatly accelerated the expression of BDNF and its high-affinity trkB receptor in regenerating motoneurons, which probably contributed to the observed acceleration in axonal regeneration.8,31,49 A recent report of the effect of ES on Schwann cells demonstrated that temporary ES for less than 1 h induced the release of NGF from cultured rat Schwann cells via calcium-dependent pathways involving calcium influx through T-type voltage gated calcium channel (VGCCs), internal calcium mobilization from IP3-sensitive stores and caffeine/ryanodine-sensitive stores, and a calcium-triggered exocytosis mechanism. 54 However, neither of these studies investigated the effects of continuous long-term ES (duration of several days) on Schwann cells, even though knowledge of the long-term effects of continuous ES is important to determine the regenerative capacity of implantable electric devices designed to heal nerve injuries with a gap.
In the present study, we demonstrated that biphasic stimulation resulted in a significant increase in the expression of the neurotrophic factors NGF, GDNF, and BDNF, as well as the expression of PMP22 by primary Schwann cells. ES also facilitated the differentiation of hMSCs into Schwann cells with increased expression of NGF and GDNF, but with no notable changes in the expression of Schwann cell markers such as PMP22 or S100b. NGF and BDNF are members of the neurotrophin family of proteins that are essential for PNS development in terms of neuron survival and the regulation of peripheral myelination. NGF is secreted by neurons and activates Schwann cell myelination 55 and enhances peripheral myelination during development and remyelination after injury. 56 NGF also induces proliferation and enhances fiber regeneration in oligodendrocytes. 57 GDNF, a member of the TGF-β family of growth factor-encoding genes, was initially identified as a survival factor for midbrain dopaminergic neurons.57,58 GDNF prevents motoneuron degeneration in mice and rats after axotomy. Therefore, induction of NGF and GDNF expression by BEC can explain the enhanced neurite-forming activity of dhMSCs exposed to ES and the increase in SC proliferation after BEC. Our results suggest either an autocrine or paracrine signaling role for NGF, GDNF, or BDNF secreted by Schwann cells in axonal regeneration, resulting in an increase in both axonal outgrowth and Schwann cell population size, as shown in our recent report. 40 Further, the induction of neurotrophic factors in dhMSCs exposed to ES was sustained for several days after ES was stopped. This is the first report of the effects of long-term, continuous ES on primary Schwann cells and the differentiation of hMSCs into Schwann cells. Our findings indicate that ES can potentially be used to address the decreased expression of NGF and GDNF by axotomized neurons and denervated Schwann cells, which limits the repair of nerve injuries.
Whether monophasic or biphasic current is better for peripheral nerve regeneration is still an issue of debate. 42 Biphasic pulses are not regarded as suitable for peripheral nerve regeneration because they do not have electrophoretic effects, as opposed to monophasic currents that can orientate cell membrane proteins toward the cathode, creating an “electric cue” for regeneration.59,60 However, BEC has several advantages over monophasic EC, even though BEC does not induce electrophoretic effects. These advantages include biocompatibility, charge balance, and maintenance of a constant current and pH, making BEC less toxic than monophasic current after prolonged exposure. 61 Despite the properties of BEC, however, experiments in an in vivo animal nerve injury model revealed that even a brief exposure (less than an hour) to a biphasic pulse resulted in the functional recovery of a transected nerve or crush injury. Long-term stimulation using BEC is not recommended for the purposes mentioned above, because results were largely similar for a range of exposure times ranging from 1 h to 2 weeks. 8 In the current study, we used an electronic device as a neural conduit for the repair of a nerve injury with a 7-mm-long gap to deliver continuous BEC to both stumps of the defect. We have demonstrated previously that use of this modified retinal electrode system resulted in the successful recovery of evoked nerve action potential as well as a significant increase in myelin formation after a prolonged 4-week stimulation period. 40 In the current study, we investigated the effects of biphasic stimulation on axonal outgrowth by staining for β-tubulin III in transverse axial sections of the middle areas of regenerated nerve tissue. Significantly greater neurite formation was observed in the ES group than the control group. Moreover, PMP expression was greater in the ES group than in the unstimulated group. However, axon bundles in the ES group were not fully ensheathed by myelin compared to normal nerves.62,63 It may be that more time is required to completely ensheath nerves. Nevertheless, it is very interesting that ES alone was sufficient to regenerate a nerve defect without the addition of neurotrophic factors or MSCs. This in vivo evidence supports our in vitro findings that BEC not only upregulated the expression of NGF, GDNF, BDNF, and PMP22, but also increased the number of Schwann cells. This study suggests that an implantable electric device has potential to improve functional recovery after peripheral nerve damage.
In conclusion, continuous biphasic current stimulation signaled Schwann cells to proliferate and increase the expression of the neurotrophic factors NGF, GDNF, and BDNF. Exposure to BEC during Schwann cell differentiation of hMSCs enhanced the Schwann cell-like functional behavior of dhMSCs; these Schwann cell-like dhMSCs stimulated neurite outgrowth in PC12 cells to a greater extent than dhMSCs treated with biochemical factors alone. The cytokine-inducing and proliferative effects of biphasic stimulation of Schwann cells are likely key mechanisms by which axonal outgrowth and in vivo peripheral nerve regeneration are enhanced. The present in vitro and in vivo findings suggest that BES may be a viable therapeutic treatment option to repair nerve injuries in the PNS.
Footnotes
Acknowledgment
This work was supported by the National Research Foundation of Korea (NRF) grant funded by the Korea government (MEST) (No. 2010-0000207).
Disclosure Statement
The authors confirm that there are no known conflicts of interest associated with this publication, and there has been no significant financial support for this work that could have influenced its outcome.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.