
Abstract
Embryonic stem cells (ESCs) are pluripotent and can differentiate into all somatic cell types. ESCs are an alternative solution to hard tissue regeneration and skeletal tissue repair to treat bone diseases and defects using regenerative strategies. Parthenogenetic ESCs (PESCs) may be a useful alternative stem cell source for tissue repair and regeneration. The defects in full-term development of this cell type enable researchers to avoid the ethical concerns related to ESC research. Moreover, in female patients, if the PESCs are derived from oocytes, then they will have that patient's genetic information. Here, we present data demonstrating that osteogenic differentiation of PESCs can be promoted by insulin-like growth factor 2 (IGF2). PESCs were plated onto Petri dishes with ESC culture medium supplemented with or without IGF2, followed by culturing of the cells for 1 week. PESCs formed floating aggregates called embryoid bodies (EBs). An osteogenic lineage was induced from the EBs by incubating them in medium containing serum, ascorbic acid, β-glycerophosphate, and retionic acid, with or without IGF2, for 20 days. Gene expression of specific osteoblastic markers such as osteocalcin, osteopontin, osteonectin, bone sialoprotein, collagen type-I, alkaline phosphatase, and Runx2 (Cbfa-I) was analyzed by real-time polymerase chain reaction. The expression level of osteocalcin, osteopontin, osteonectin, and alkaline phosphatase was twofold higher in IGF2-treated PESC derivatives than IGF2-naive PESC derivatives. In vivo experiments were also performed using a critical-sized calvarial defect mouse model. Ten weeks after cell transplantation, more bone tissue regeneration was observed in the IGF2-treated PESC transplantation group than in IGF2-naive PESC transplantation group. Both our in vitro and in vivo data indicate that IGF2 induces osteogenic differentiation of PESCs. Addition of IGF2 may reactivate imprinting genes in PESCs that are only expressed in the paternal genome and are normally silent in PESCs. Our findings provide insights into the mechanisms of skeletal tissue repair and the imprinting mechanisms active in stem cells.
Introduction
In a previous study, we demonstrated that parthenogenetic ESCs (PESCs) in mice can be induced to differentiate into osteogenic cells in vitro. 14 PESCs are pluripotent, and if derived from an oocyte of a female patient, these cells have that patient's genetic information, thus preventing immune rejection responses in females. In humans, females are more susceptible to osteoporosis and other bone-related diseases than males. 15 Therefore, PESCs obtained from a patient's oocyte can be potentially used for organ or tissue regeneration therapy. PESCs have been shown to form three germ layers in a teratoma study. 16 This indicates that PESCs can contribute to the various tissues and organs of a developing embryo. Moreover, because the parthenote cannot develop to term, the use of PESCs avoids the ethical issues associated with human ESCs. PESCs may also improve organ transplantation efficiency by reducing the risk of major histocompatibility complex mismatches.17,18 PESCs have been generated from various animals such as mice,17–19 monkeys,20–22 rabbits, 23 buffalos, 24 and humans25–28 for basic research. However, the use of PESCs in the field of regenerative medicine is only beginning to be explored. Although PESCs have many advantages compared with ESCs, these cells have some defects in their differentiation potential, especially differentiation into endodermal and mesodermal lineages. 29
Various proteins have been implicated in the formation of the three primary layers in the mouse embryo. These include growth factors and their receptors, cell adhesion and extracellular matrix molecules, and transcription factors. Studies in various mouse models have provided evidence that members of the fibroblast growth factor family, the Wnt family, the insulin-like growth factor (IGF) family, and the transforming growth factor β superfamily, such as activins and bone morphogenetic proteins, have mesoderm-inducing roles.29–31 IGF2 is a very important factor for mesoderm formation in mouse embryonic development, 29 and it may cause the biased determination of primitive ectoderm cells toward mesoderm cells or promote the selective proliferation of already determined mesoderm cells. 32 IGF2 is known to act as both a mitogen and a differentiation factor by triggering different signaling pathways at the same time.33,34 Therefore, IGF2 may well cause both the determination and the proliferation of mesoderm cells. It has been also reported that IGF2 and IGF1 produced locally may modulate both osteoblast–osteoclast interactions and osteoblast formation and thereby play an important role in bone remodeling.35,36 Bone homeostasis depends on the balanced action of bone resorption by osteoclasts and bone formation by osteoblasts. Therefore, IGF2 may induce the osteogenic differentiation of stem cells.
The expression of imprinting genes related to growth and organ formation such as IGF2 is abnormal in parthenogenetic embryos, as these genes are only expressed by the paternal genome, which is absent in parthenotes. As an imprinting gene that is only expressed in paternal-oriented genomes, IGF2 have been shown to play an important role in regulating placental development and fetal growth.32,37–40 In the postimplantation embryonic period in mice, IGF2 mRNA and protein are produced in the primitive endoderm at embryonic day 6.5 (E6.5) and then appear in the extra-embryonic mesoderm cells at E7.0 and in the anterior-proximal and lateral embryonic mesoderm cells (E7.5). Later, IGF2 becomes abundant in mesoderm derivatives such as the developing heart (E8.0) and somites (E8.5). 41 Before E13.5, IGF2 signaling is transduced by the IGF1 receptor. 37 The IGF2 receptor is involved only in IGF2 degradation. 42 Deletion of IGF2 leads to placental and fetal growth restriction, especially in the early stages of gestation.37,43,44 Similarly, overexpression of IGF2 leads to placental and fetal overgrowth.38,45 IGF2 enhances growth via paracrine and autocrine actions that stimulate cell proliferation and survival.32,39 IGF2 appears to be induced by placental lactogens and high concentrations of peptides and mRNA in utero, suggesting that IGF2 is important in fetal metabolism. 46 Thus, fetal and placental IGF2 appear to play an important role in regulating the relationship between fetal and placental growth and the placental capacity to transport nutrients, which occurs by facilitated and active transport. 32 In humans, contrasting to mice, IGF2 expression is maintained postnatally. The significance of this continued expression of IGF2 in humans is unknown. 47
Poly-
In the present study, we evaluated the effects of IGF2, which is important for organ formation in embryonic development, on in vitro osteogenic differentiation of PESCs and in vivo bone regeneration in a critical-sized calvaria mouse defect model.
Materials and Methods
Animals and chemicals
All inorganic and organic compounds were obtained from Sigma-Aldrich Korea unless otherwise stated. Six-week-old C57BL6X DBA2 F1-hybrid (B6D2F1) female mice were used as sources for the oocytes. All media for handling and culture of oocytes and parthenotes were based on Chatot-Ziomek-Bavister medium (CZB) and potassium simplex optimized medium (KSOM).56,57
Preparation of parthenogenetic blastocysts for PESC generation
The procedure of oocyte recovery, parthenogenetic activation, and in vitro culture were performed as previously described.14,58 Briefly, 5–7-week-old female B6D2F1 were superovulated with 5 IU equine chorionic gonadotropin followed by a second injection of 5 IU human chorionic gonadotropin (hCG) 48 h later. Oviducts were excised 15 h after hCG injection, and an average of 40 oocytes per mouse were obtained. Hyaluronidase (1 mg/mL) was used to remove cumulus cells, and oocytes were washed with HEPES-buffered CZB and exposed to an activation medium, consisting of 10 mM SrCl2 with 5 μg/mL cytochalasin B in calcium-free CZB for 5 h. The activated oocytes were developed to the blastocyst stage in KSOM. The oocytes were incubated for 4 days at 37.5°C under 5% CO2 in air. After in vitro culture, zona pellucida of the expanding blastocyst was removed by the treatment of acid-Tyrode's solution.
Establishment and culture of ESC lines from parthenogenetic murine embryos
To generate PESCs, zona-free parthenogenetic blastocysts of B6D2F1 were transferred onto a feeder layer of STO cells in gelatinized tissue culture plates (Nunc) containing ESC medium consisting of Dulbecco's modified Eagle's medium (DMEM; Life Technologies) supplemented with 10% fetal bovine serum (FBS), 1% nonessential amino acids, 0.1 mM β-mercaptoethanol, and 1000 U/mL murine LIF (Chemicon). After 5 days of culture, the outgrown clumps derived from the ICM were counted under a stereomicroscope. The cell clumps were further trypsinized with 0.05% trypsin–EDTA and seeded onto new gelatinized four-well dishes with fresh ESC medium to generate PESCs. When the colonies appeared, they were considered to be at passage 0 and were propagated gradually under stringent culture conditions with careful monitoring and medium changes to ensure that the cells remained undifferentiated. The PESCs were cultivated on a feeder layer of mitomycin C-treated STO cells in DMEM supplemented with 10% FBS, 2 mM
Formation of EBs
To induce EB formation, ESCs or PESCs on the culture plates were trypsinized for 2 min at 37°C. Trypsinization was terminated by addition of DMEM containing 10% FBS. After a brief centrifugation, cells were resuspended in medium followed by medium supplementation with 10% FBS, 2 mM
Photos of an embryoid body (EB).
In vitro osteogenic induction of EBs
To induce osteogenic differentiation of EBs, we used a modified version of the osteogenic induction protocol of Buttery et al. 4 Briefly, EBs were resuspended in α-modified Eagle's medium containing 10% FBS, 50 U/mL penicillin, and 50 mg/mL streptomycin and allowed to adhere to six-well culture plates at a density of three EBs/well. The medium was then additionally supplemented with 50 mg/mL ascorbic acid and 10 mM β-glycerophosphate, with or without 50 ng/mL IGF2. The EBs were maintained in culture for 20 days and the medium was replaced every 3 days.
mRNA extraction and cDNA synthesis
RNA was extracted from osteogenically induced cells of each group for 20 days using Trizol reagent (Invitrogen). The induced cells of each group (6.0×105/mL) were harvested by centrifugation, resuspended in 1 mL of Trizol reagent by vortexing, and then incubated for 15 min in ice to lyse the cells. Then, 0.2 mL of chloroform was added, mixed by smooth shaking, and incubated for 15 min. After centrifugation at 12,000 rpm for 15 min at 4°C, the colorless upper aqueous phase was transferred to new tubes containing 0.5 mL of isopropanol by smooth shaking and incubated for 15 min. Total RNA pellet was obtained by centrifugation at 12,000 rpm for 10 min at 4°C, air-dried, and resuspended in diethyl pyrocarbonate-treated water. The isolated RNA samples were used for real-time polymerase chain reaction (PCR) analysis. For the synthesis of cDNAs, reverse transcription was performed for 1 h at 42°C in a final reaction volume of 25 μL containing total RNA, 5 μL of 5×reaction buffer (Promega), 5 μL of dNTP (each 2.5 mM), 2.5 μL of 10 μM synthesis primer, 0.5 μL of RNasin Plus RNase Inhibitor (40 U/mL; Promega), and 1 μL of M-MuLV reverse transcriptase (20 U/μL; Roche). cDNAs were diluted by the addition of 50 μL of RNase-free ultrapurified water.
Real-time PCR
Expression levels of osteogenic cell-specific genes, such as osteocalcin, osteonectin, bone sialoprotein, osteopontin, collagen type-I, alkaline phosphatase, and Runx2, were measured by real-time PCR in three groups (the IGF2-treated PESC group, the IGF2-naive PESC group, and the ESC group). Real-time PCR primers were designed using Primer Express software (Applied Biosystems). Real-time PCR was performed using the ABI PRISM 7500 system and SYBR Green PCR Master Mix (Applied Biosystems). The primers list of target genes is shown in Table 1. The extraction of mRNAs and the synthesis of cDNAs were performed twice, and all samples were run in triplicate to obtain technical replicates. In each run, 1 μL cDNA was used as a template added to 5 μL double-distilled water, 2 μL forward and reverse primers (20 pmol/mL), and 10 μL SYBR Green PCR Master Mix. The following amplification procedure was employed: denaturation stage (95°C for 10 min), amplification and quantification stage repeated 40 times (94°C for 15 s, 60°C for 1 min with single fluorescence measurement), and dissociation curve stage (temperature increments of 0.1°C per 30 s from 60°C to 95°C with fluorescence measurement). Gene expression was always related to expression of murine Gapdh as housekeeping gene, which is known to be a good reference gene for normalization of target genes expression levels. Quantification was performed using the ΔΔCT method. Nontemplate control was used as the negative control. The paired samples t-test was performed to compare the differences among the experimental groups.
Preparation of scaffolds and cells for calvarial implantation
The PLLA scaffolds were prepared as described in a previous report with some modifications. 59 In brief, the PLLA was dissolved in a mixed solvent (dioxane:dimethylcarbonate; 8:2) to make a 5% (w/v) solution. A solid–liquid phase separation technique and a subsequent solvent sublimation process were used to generate the porous PLLA scaffolds. The PLLA/dioxane solution was cooled to −20°C for 2 h and transferred to −80°C for additional 24 h. The frozen mixtures were freeze-dried at −10°C to −5°C of ice/salt bath for 7 days and then stored in a desiccator. The PLLA disks with a diameter of 4 mm and thickness of 1 mm were prepared. The scaffolds were sterilized with ethylene oxide gas. After sterilization, scaffold samples were soaked at 37°C in general growth medium (DMEM, 10% FBS, and 1% penicillin/streptomycin) for 24 h. Then, scaffold samples were soaked in serum that had been extracted from B6D2F1 mice. After soaking, scaffold samples were washed with PBS. In preparation for implantation, IGF-2-naive PESC and IGF2-treated PESC derivatives were seeded onto the scaffold. Following trypsinization, 10,000 cells were resuspended in one sample volume of general growth medium (8.0×104 cells/cm2) and seeded directly onto the scaffold. The same amount of medium without cells was used as an empty scaffold control. Before implantation, cell-seeded scaffolds were submerged at 37°C in medium and incubated for 24 h. One day after seeding, a couple of samples from PESC-derived seeded groups were embedded in paraffin, cut into 5-μm sections, and stained with hematoxylin and eosin (H&E) to ensure the state of seeded cells.
Scanning electron microscopy
One day after seeding, four samples in each group including the scaffold-only control were washed twice with PBS and fixed using 2.5% glutaraldehyde and 2% paraformaldehyde in PBS (pH 7.2) for 24 h. After fixing, the samples were washed twice with PBS and postfixed using 1% osmium tetroxide for 1 h and followed by washing twice with distilled water. Then, the specimen was dehydrated by dipping it in increasing concentrations of ethanol and then by critical point drying. After drying for 24 h, the specimens were sputter-coated with gold–paladium and observed by scanning electron microscopy (SEM) at 15 kV (FE-SEM Hitachi S-4700).
Surgical procedures
Sixteen mice of B6D2F1 strain (25–30 g) were used as study subjects and four animals in each of four groups were implanted. All animal experiments including animal management and surgical procedures were approved and performed under the guidelines of the Institutional Animal Care and Use Committee of Seoul National University (approval number: SNU-061023-1). The animals were anesthetized with a subcutaneous injection of a mixture of Zoletil™ and xylazine (30 and 10 mg per kg, respectively). The scalp covering the calvarial vault was shaved and scrubbed with betadine solution. An incision was made along the midline. Full-thickness skin and the periosteum were raised to expose the calvarial bone surface. Careful drilling with a 4-mm-diameter trephine bur was performed around the sagittal suture, and a standardized, round, segmental defect was made. During drilling, the area was irrigated with saline solution and the underlying dura mater was maintained intact. A PLLA scaffold, with or without cells, was placed in the defect. The periosteum and skin were closed in layers with adsorbable 5-0 chromic catgut (WRHI) nonabsorbable 4-0 black silk (Ethicon) sutures, respectively. Mice were sacrificed at 10 weeks after the implantation. Calvarial bone was excised with careful trimming. The specimen was fixed in 10% neutral buffered formalin solution at 4°C for >12 h.
Microcomputed tomography
Soft X-ray of the excised calvarial specimens was taken with a condition of 30 kV, 1.5 mA, 40 s of exposure, and 25 cm of distance. Then, the calvarial bone specimens were examined using a microcomputed tomography (micro-CT) machine (Skyscan 1072; Skyscan). Specimens were placed on a cylindrical sample holder with the coronal aspect of calvarial bone in a horizontal position to ensure parallel scanning conditions. The pixel size was 17.99 μm. Image files were reconstructed using a modified Feldkamp algorithm, which was created using microtomographic analysis software (TomoNT; Skyscan). After the three-dimensional visualizing process, bone volumes were measured in the region of interest. In addition, micro-CT scan was also performed in animals (blank control) that did not receive any implant, including the scaffold. Data were presented as average and standard error of means. The one-way ANOVA test was performed to compare the differences among the experimental groups. The number of samples in each group was 4.
Histologic evaluation
After micro-CT scanning, three specimens in each group except the blank control were decalcified in 10% EDTA in 0.2 M NaPO4, pH 7.4, for 7–10 days. The decalcified specimens were embedded in paraffin and cut into 5 μm sections, and then H&E staining was performed for histomorphological analysis to evaluate hard tissue formation in the bone defects.
Results
Bone-related gene expression in differentiated cells
Expression levels of all the genes analyzed were significantly higher in IGF2-treated PESC derivatives than IGF2-naive PESC derivatives (Fig. 2). Expression levels of osteopontin (p=0.017), osteonectin (p=0.021), osteocalcin (p=0.042), and Runx2 (p=0.032) were also significantly higher in IGF2-treated PESC derivatives than ESC derivatives, whereas the levels of the other genes were similar to those in ESC derivatives.
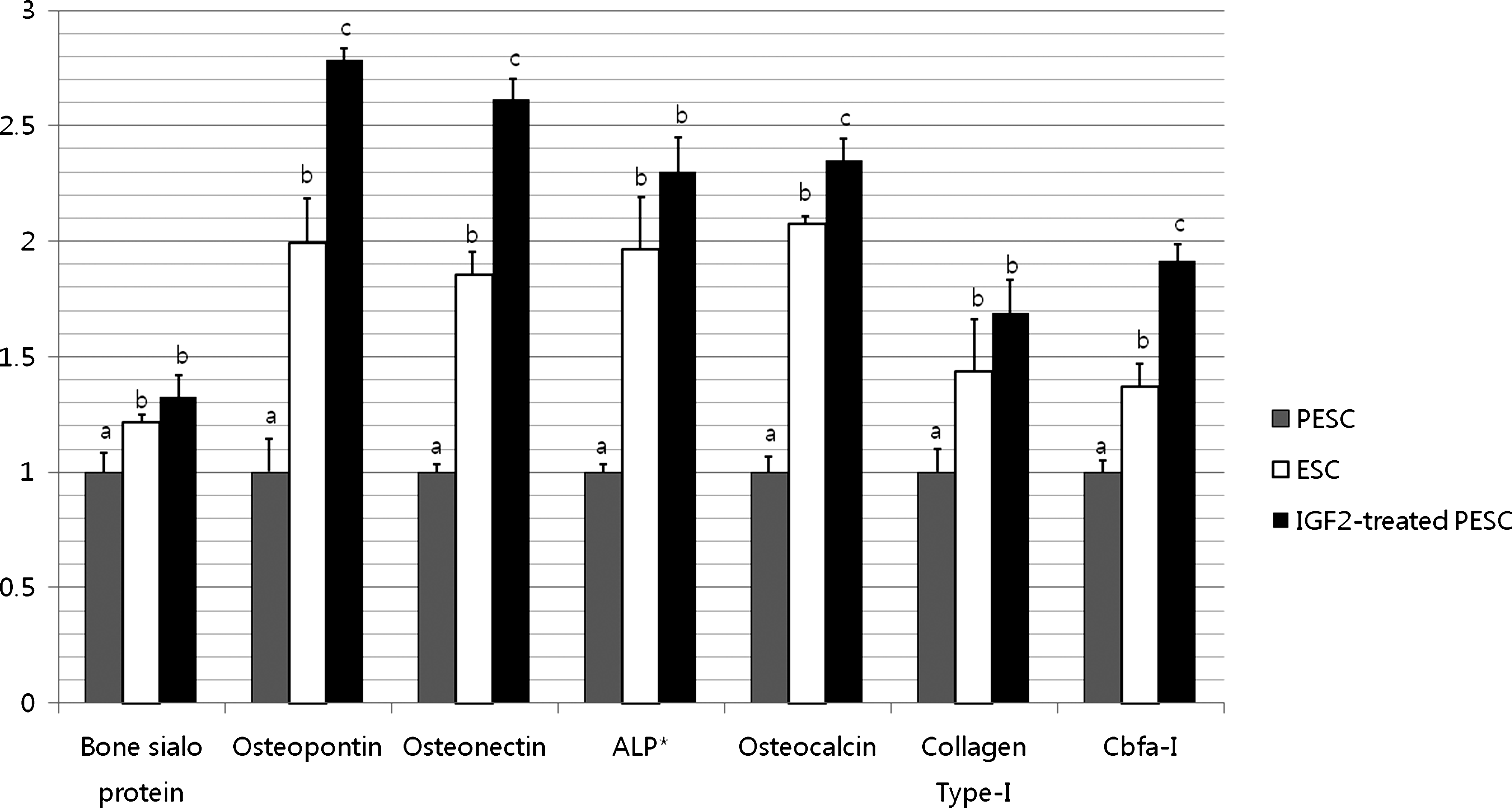
Osteogenic-specific gene expression in PESCs, ESCs, and IGF2-treated PESCs after osteogenic induction. IGF2-treated PESCs were treated with IGF2 for the entire osteogenic induction period. ESCs were used as the control. The extraction of mRNAs and the synthesis of cDNAs were performed twice, and all samples were run in triplicate to obtain technical replicates. Values with different superscripts are significantly different (a,b,cp<0.05, paired samples t-test). Expression levels of osteopontin (b,cp=0.017), osteonectin (b,cp=0.021), osteocalcin (b,cp=0.042), and Runx2 (b,cp=0.032) were significantly higher in IGF2-treated PESC derivatives than the other groups. ALP* is alkaline phosphatase. IGF2, insulin-like growth factor 2.
Bone regeneration potential in murine calvarial defects
The internal structure of scaffolds was evaluated by SEM. The morphologies of cultured cells in the PLLA scaffolds were evaluated by SEM and histological staining on day 1. As shown in Figure 3A, the prepared PLLA scaffolds were highly porous. The irregularly shaped pores were interconnected, and the size of individual pores ranged from several tens to two hundreds micrometer (67±22 μm, number of pores measured=176). One day after seeding, cells appeared well attached to the scaffold pores (Fig. 3B, C). H&E staining confirmed the live status of cells at the time of staining (Fig. 3D). After implantation of PLLA scaffolds containing IGF2-treated or IGF2-naive PESC derivatives in critical-sized calvarial bone defects, greater bone regeneration was evident in soft X-ray images in the defects treated with PLLA scaffolds containing IGF2-treated PESC derivatives than those treated with PLLA scaffolds containing IGF2-naive ones (Fig. 4). These results were confirmed by micro-CT measurements. New bone formation was about twofold higher in the group treated with IGF2-treated PESC derivative than in the group treated with IGF2-naive ones (Fig. 5), based on micro-CT measurements at 10 weeks after implantation. Histological analyses showed new bone regeneration in the calvarial bone defect regions of defects treated with scaffolds containing IGF2-treated cells (Fig. 6). In the scaffold-only (control) group, the defect region was filled with fibrous tissues because of foreign body reactions. Hard tissue and bone regeneration were not detected in the control group. The histological features of implantation sites treated with IGF2-naive PESC derivatives were similar to those seen in the control group, although small foci of dystrophic calcification were observed in some IGF2-naive PESC-treated specimens.
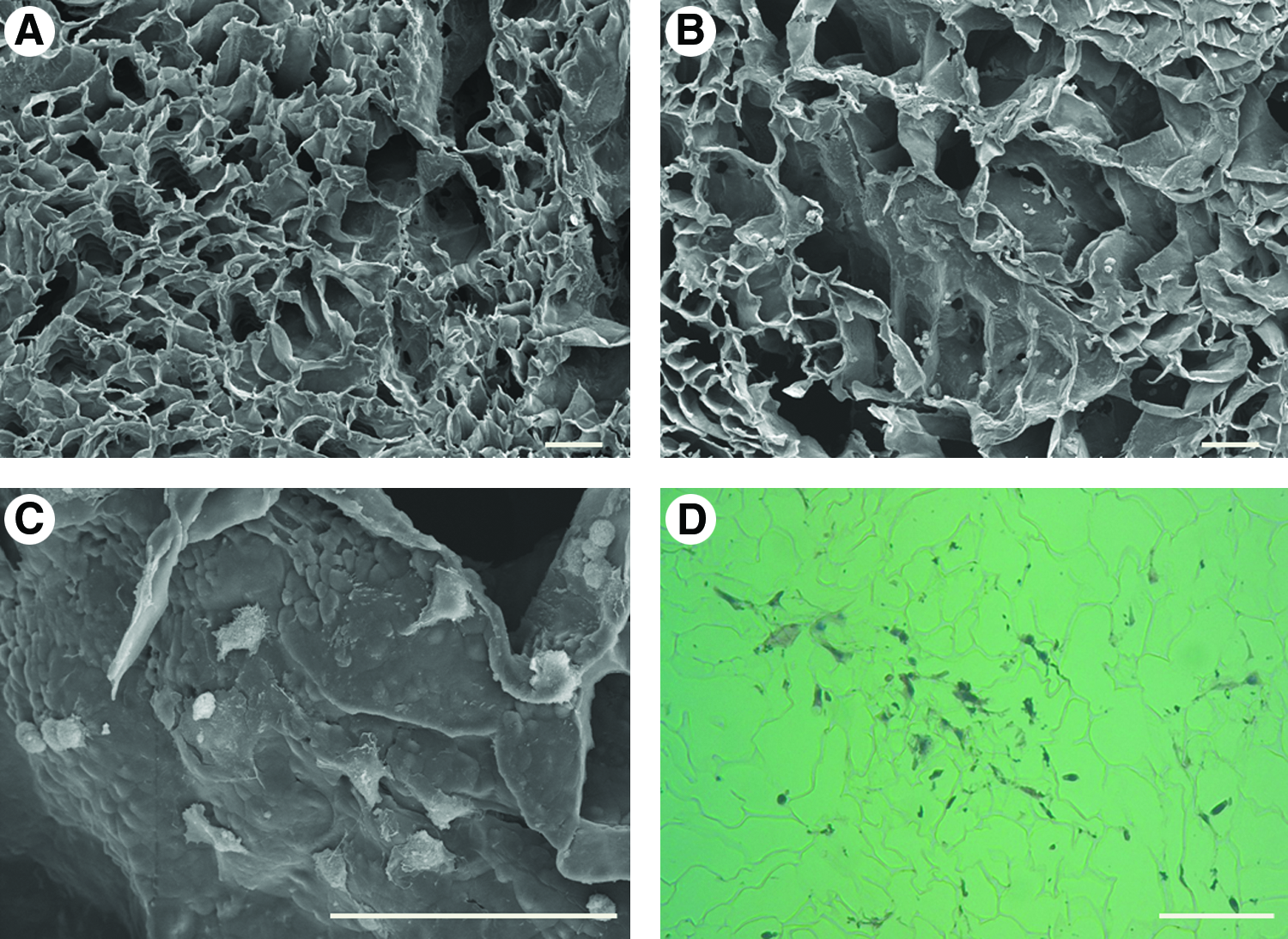
The morphologies of the PLLA scaffold and cultured PESC derivatives in the PLLA.

Soft X-ray images of the in vivo implantation site in critical-sized calvarial defects in mice. The circles indicate the original defect regions. The white dots within the circles may be calcification materials. (Left) Control group in which the PLLA scaffold only was implanted. (Center) Group implanted with PESCs. Before implantation, cells were induced to differentiate into an osteogenic lineage without IGF2 treatment for the entire induction period. (Right) Group implanted with IGF2-treated PESCs. Cells were treated with IGF2 for the entire induction period before implantation. More bone masses were observed in calvarial defects treated with PLLA scaffolds containing IGF2-treated cells than calvarial defects treated with scaffolds containing IGF2-naive cells. Scale bar: 5 mm. Color images available online at www.liebertonline.com/tea
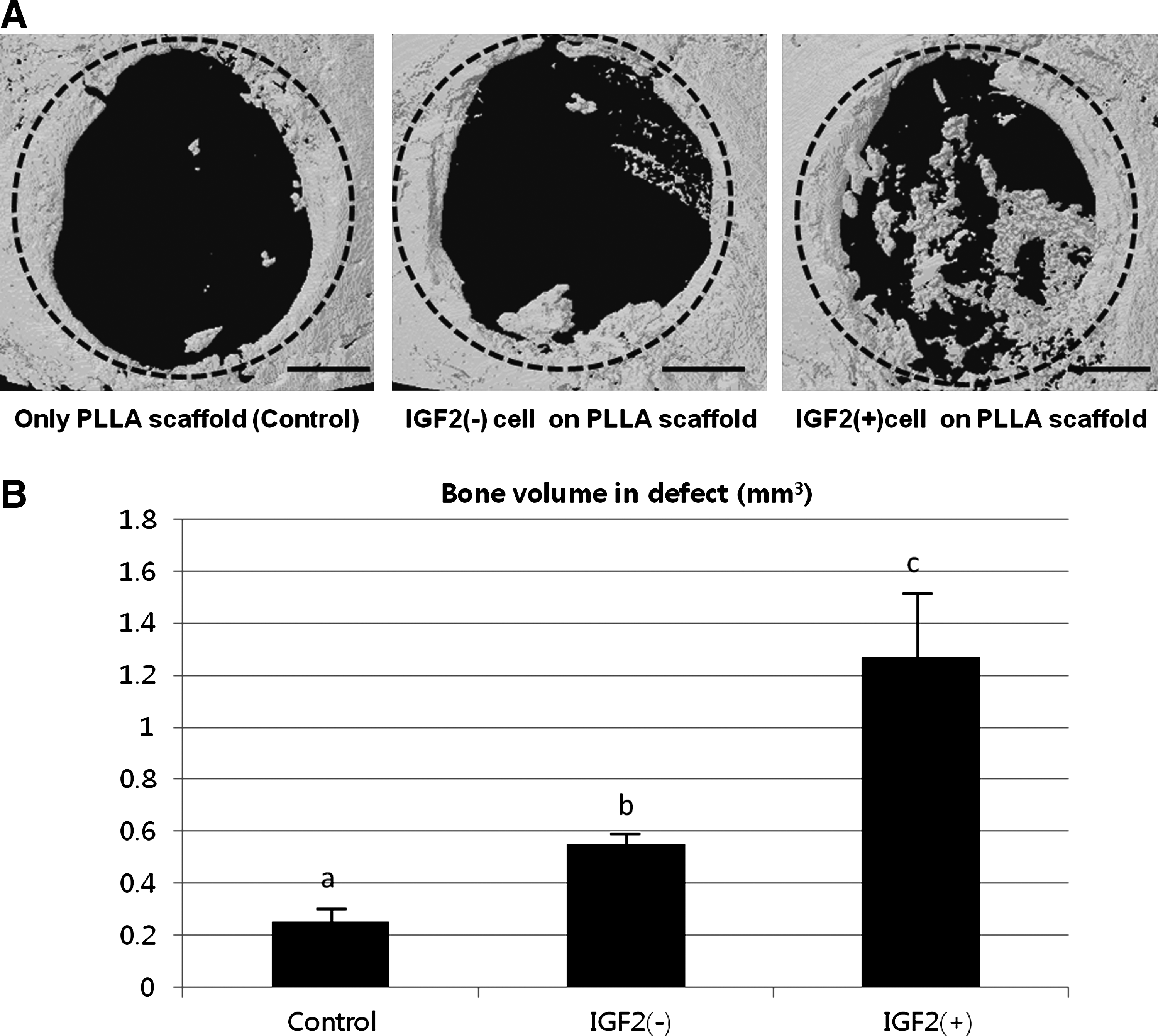
Microcomputed tomography (micro-CT) results of bone formation in critical-sized calvarial defects in mice.

Histological images after H&E staining.
Discussion
ESCs have been induced to differentiate the wide variety of cell types by supplementing the culture medium with specific factors. Cultures of ESCs in defined media containing various proteins and small molecules have been used as a strategy to investigate ESC differentiation in vitro.60–62 In particular, culturing cells under defined conditions in medium containing several factors is a good strategy for discovering factors that are critical for inducing or improving specific cell lineages. We have previously shown that PESCs, which are derived from parthenogenetic blastocysts, can be induced to differentiate into an osteogenic lineage by supplementation of the culture medium with defined induction factors. 14 However, the differentiation potential of PESCs is limited compared with that of ESCs.16,29 The limited differentiation potential of PESCs relative to ESCs may be related to the abnormal expression of imprinting genes in PESCs. We hypothesized that the addition of a soluble factor (IGF2) that is silenced in PESCs 63 could potentially enhance the osteogenic cell differentiation of PESCs. PESCs do not express IGF2, and our results demonstrate that the lack of endogenous IGF2 expression can be compensated for by exogenous supplementation with this factor, resulting in osteogenic cell differentiation. As mentioned earlier, IGF2 is a paternally expressed imprinting gene that is therefore not expressed in PESCs. We demonstrated using both in vivo and in vitro experiments that IGF2 enhanced the osteogenic differentiation potential of PESCs. Our in vitro study results reveal that IGF2-treated PESC derivatives differentiated into an osteogenic cell lineage better than PESCs not exposed to IGF2. The osteoblast-specific gene expression of IGF2-treated PESC derivatives was more similar to that of ESC derivatives than IGF2- naive PESC derivatives. Bone is a mineralized connective tissue that consists mainly of collagen type-I and other fiber or nonfiber matrix proteins, such as marker genes analyzed in this study and proteoglycans. 64 Osteopontin, a noncollagenous bone matrix molecule, is associated with osteogenic cell adhesion and is abundantly expressed during the early stages of osteoblast differentiation in the mouse. The expression of intermediate/late osteogenesis markers such as Runx2 (Cbfa-I), bone sialoprotein, and osteocalcin confirms the existence of a fully differentiated osteogenic cell population in addition to osteogenic progenitors.13,65,66 Therefore, in vitro results indicate that IGF2 promoted the osteoblastic differentiation of PESCs.
To confirm this in vitro result, an in vivo transplantation experiment was performed using a calvarial defect mouse model. We performed this in vivo study to determine whether the IGF2-treated PESC derivatives possessed actual bone formation capacity and could be potentially used to stimulate hard tissue regeneration. Ten weeks after cell transplantation, soft X-ray, micro-CT, and histology results confirmed that the IGF2-treated PESC derivatives had greater bone regenerative potential than IGF2-naive ones. Mesenchymal stem cells (MSCs) are widely used in bone regeneration research. They have the capacity to express markers of various type of tissues including muscle, 67 nerve, 67 bone, 68 and cartilage. 68 Implantation of MSCs is known to have the potential to enhance healing of bone 69 and cartilage. 70 In particular, bone regeneration experiments show ∼10%–20% healing efficiency in calvarial defect murine model experiment.71,72 Healing efficiency in the present study is comparable to previous reports that used MSCs as the cell source. With IGF2 treatment during osteogenic induction, bone differentiating capacity of PESCs reached a similar level as MSCs. As shown in Figure 6, samples in all experimental groups show minor bone healing evidence around the periphery of the defect region that originates from a natural bone healing process and not from the osteogenic regeneration derived from PESCs. In contrast, the sample of IGF2-treated PESC derivatives shows well-defined bone regeneration from the center of scaffold, indicating that bone regeneration originated from PESC derivatives. Although there is evidence of central bone regeneration in IGF2-naive PESC derivatives, the regeneration efficiency is markedly lower than the IGF2-treated group. Regardless of IGF2 treatment, we observed that bone regeneration generally occurred in the center regions of the scaffolds, which may be related to the cell seeding methods. The cells with medium tended to be pipetted onto the center of the scaffold during the seeding, enabling more cells to be established in the center of the scaffold than in the periphery.
At present, autogenous graft is considered as the standard in regeneration therapy protocols,73–75 although it still has some disadvantages such as concern for harvesting affordable amount of cells and the need for more surgery. Application of biomaterials such as PLLA can be an alternative to autogenous graft.73,74 PLLA is becoming an environmentally sustainable alternative to petrochemical-derived products because of its biodegradable characteristics and the renewable nature of its feedstock, 76 in addition to its nontoxic and noncarcinogenic properties. 54 It has been reported that implantation of PLLA is supportive for bone regeneration in in vivo models. 77 However, in this study, implantation of PLLA without osteogenic cells shows very limited bone regeneration capacity when compared with the implantation results of PLLA with IGF2-treated or naive PESC derivatives. Thus, implantation of the scaffold with cells having osteogenic potential may be still crucial for successful bone regeneration, clinically.
In conclusion, the results suggest that supplementation of the culture media of PESCs with exogenous IGF2 induces these cells to differentiate into an osteogenic lineage. Although there are many remarkable reports in the regenerative medical literature of successful tissue engineering using cellular supports, few of these studies investigated PESCs and their derivatives, despite the fact that by using PESCs, the ethical concerns associated with ESC research can be avoided. 78 If osteogenic cells from PESCs could enhance bone regeneration in humans, PESCs could be potentially used to treat female patients with irreversible osteoporosis or bone loss problems, although, for autogenous graft, this system may be limited to the patients who can provide their own oocytes. PESCs are therefore a potentially viable alternative therapy for hard tissue regeneration and skeletal tissue repair.
Footnotes
Acknowledgments
This study was supported by grants from the National Research Foundation of Korea (NRF) funded by the Ministry of Education, Science, and Technology (MEST; 2011-0027807), Mid-career Researcher Program of MEST (2010-0000049), and a grant from the Technology Development Program for Agriculture and Forestry, Ministry for Food, Agriculture, Forestry, and Fisheries (MIFAFF; 109020-3), Republic of Korea.
Disclosure Statement
No competing financial interests exist.