
Abstract
In many studies, adult stem cells have been found in human periodontal ligament (PDL), but in most cases they were found in the permanent teeth. The aim of the present study was to characterize stem cells from the PDL of deciduous teeth (dPDLSCs) and compare them with those from the PDL of permanent teeth (pPDLSCs). Stem cell markers were examined by a flow cytometric analysis. The results of in vitro differentiation into adipogenic and osteogenic lineages were analyzed by histochemical staining and quantitative reverse transcription–polymerase chain reaction (RT-PCR). The results of in vivo transplantation were analyzed by histological staining, immunohistochemical staining, and quantitative RT-PCR. There were no significant differences in the proliferation rate, cell cycle distribution, expressions of stem cell markers such as Stro-1 and CD146, or in vitro differentiation. The pPDLSC transplants made more typical cementum/PDL-like tissues and expressed more cementum/PDL-related genes (CP23 and collagen XII) than did the dPDLSC transplants. Together, these results suggest that pPDLSCs are better candidates for use in reconstructing periodontium.
Introduction
Because deciduous teeth differ from permanent teeth with respect to their morphology, constituents, and life cycle, it is reasonable to assume that cells originating from deciduous and permanent teeth will behave differently. Some investigators have reported that SHED differ from DPSCs with regard to their proliferation rate (that of the former being greater than that of the latter) and the differentiation pattern (unlike DPSCs, SHED are unable to reconstitute a complete dentin/pulp-like complex in vivo).5–7 It was recently reported that the periodontal ligament (PDL) of deciduous teeth also contains adult stem cells,8,9 and it was found that the proliferation rate and potential to differentiate into adipogenic and osteogenic lineages of these stem cells were superior to those from permanent teeth. 9 However, in vivo transplantation has not yet been studied in this cell type.
There have been many attempts to use PDLSCs for tissue reconstruction, not only to replace destroyed periodontium in animal and human models,10–14 but also for other applications such as the formation of bone around prosthetic implants, 15 and even plastic reconstruction. 16 However, the application of stem cells from the PDL of deciduous teeth to tissue engineering has not yet been reported.
Stem cells obtained from deciduous teeth have some advantages as a source of stem cells in regenerative medicine. This is not because deciduous teeth can be obtained easily and noninvasively; rather, it is because the proliferation and differentiation activities are higher for cells isolated from patients at a younger age.7,17 PDLSCs obtained from the deciduous teeth can be cryopreserved, and later in the lifetime of the donor they could be used as a source of stem cells for reconstructing periodontium destroyed by periodontal diseases. In addition, on delayed replantation of avulsed teeth or on auto/allo-transplantation of teeth thawed from a tooth bank, PDL tissues engineered with those PDLSCs would be helpful for preventing replacement root resorption by odontoclasts/osteoclasts and for the successful reattachment to the alveolar bone. 18
Therefore, the aim of the present study was to determine not only in vitro, but also in vivo the characteristics of stem cells obtained from the PDL of deciduous teeth and permanent teeth, and thus to establish which would be most suitable for use in periodontal regeneration.
Materials and Methods
Cell cultures
PDL tissues were obtained from healthy permanent premolars (n=10) or anterior deciduous teeth (n=6) extracted for orthodontic reasons or space management from 15 healthy persons (four boys and three girls, aged 5–16 years). The experimental protocol was approved by the Institutional Review Board of the Dental Hospital, the Yonsei University, and informed consents to participate were obtained from all of the subjects and their parents (#2-2010-0011). PDLSCs were obtained by explant culture from the tissues. In brief, the tissues were gathered from the middle area of the root. The explants were covered with cover glass and incubated with a growth medium comprising the α-minimum essential medium (α-MEM; Invitrogen, Carlsbad, CA) containing 10% fetal bovine serum (FBS; Invitrogen), 100 U/mL penicillin and 100 μg/mL streptomycin (Invitrogen), 2 mM
Proliferation assay
The proliferation of the cells was measured using the Cell Counting Kit (CCK)-8 (Dojindo, Kumamoto, Japan), according to the manufacturer's instructions. This assay utilizes the property of WST-8 [2-(2-methoxy-4-nitrophenyl)-3-(4-nitrophenyl)-5-(2,4-disulfophenyl)-2H-tetrazolium, monosodium salt] being able to produce a water-soluble colored formazan by the activity of dehydrogenases in living cells. Briefly, the cells were plated in 96-well culture plates (BD Falcon, Franklin Lakes, NJ) at a density of 500 cells/well. At the test time points (1, 3, 5, 7, and 9 days), 10 μL of the CCK-8 solution was added and the cells incubated for a further 4 h. The absorbance at 450 nm was measured using a Benchmark Plus Microplate spectrophotometer (Bio-Rad Laboratories, Hercules, CA) to estimate the number of vital cells in each well.
Cell cycle analysis
The cells were harvested by trypsinization and then fixed in cold 70% ethanol for 1 h at 4°C. After washing twice with phosphate-buffered saline (Invitrogen), samples were incubated in 0.2 mg/mL RNase A (LaboPass, Sapporo, Japan) for 1 h at 37°C. The cells were stained with propidium iodide (40 μg/mL; Sigma) at 4°C for 30 min, and then subjected to a FACSCalibur flow cytometer (BD Biosciences, San Jose, CA) for a cell cycle analysis. The findings were analyzed with FCSExpress V3 software (De Novo Software, Los Angeles, CA).
Colony-forming unit-fibroblast assay
Single-cell suspensions of the dPDLSCs, pPDLSCs, and BMMSCs were seeded into six-well culture plates (480 cells/well) and incubated therein for 10 days. Cultures were then fixed with 10% buffered formalin (Sigma) for 1 h, and stained with 0.3% crystal violet (BD Biosciences) for 5 min. The number of colonies containing over 50 cells was counted with the aid of a light microscope.
Flow cytometry analysis
Single-cell suspensions were obtained by detaching monolayers of the cells with a cell dissociation buffer (Invitrogen). The cells were resuspended in the flow cytometry staining buffer (eBiosciences, San Diego, CA) and, in accordance with the manufacturer's instructions, incubated with an adequate amount of mouse monoclonal antihuman antibodies (fluorescein isothiocyanate-conjugated CD146, R-phycoerythrin [PE]-conjugated CD90, PE-conjugated CD105, PE-conjugated CD31, and PE-conjugated CD45; all supplied by eBiosciences) or 5 μg of antihuman Stro-1 (immunoglobulin M [IgM]; R&D Systems, Minneapolis, MN) per 1×106 cells for 1 h. For antihuman Stro-1 staining, the cells were additionally incubated with the PE-conjugated goat anti-mouse antibody (0.1 μg/1×106 cells; IgM; SouthernBiotech, Birmingham, AL) for 30 min. For negative controls, primary antibodies were omitted. All of the aforementioned procedures were performed in the dark at 4°C. The expression profiles were examined using the FACSCalibur flow cytometer and analyzed with FCSExpress V3 software. Positive expression was defined as a level of fluorescence >99% of the corresponding control.
Gene expression analysis using reverse transcription–polymerase chain reaction
When the cells reached subconfluency (i.e., they occupied 70%–80% of the culture dish), total cellular RNA was isolated using an RNeasy Mini Kit (Qiagen, Valencia, CA), according to the manufacturer's instructions. The extracted RNA was eluted in 30 μL of water, and the integrity and concentration were evaluated using a spectrophotometer (Nanodrop ND-1000; Thermo Scientific, Waltham, MA). One microgram of total RNA was reverse transcribed with a Maxime RT premix kit (Intron Biotechnology, Seoul, Korea) according to the manufacturer's instructions. Briefly, total RNA was reverse transcribed using an oligo d(T)15 primer for 1 h at 45°C, and the reaction was stopped by incubation for 5 min at 95°C. The polymerase chain reaction (PCR) amplifications were performed in a 20-μL reaction volume using a Maxime PCR premix Kit (Intron Biotechnology) and gene-specific primers (as listed in Table 1) in a thermal cycler (Swift MaxPro, ESCO, Singapore). The PCR conditions were 95°C for 2 min, followed by the appropriate number of cycles of 94°C for 20 s, 60°C for 10 s, 72°C for 20 s, and a final 5-min extension at 72°C. Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) amplifications were carried out as positive controls to assure the quality of the cDNAs used for this experiment. All PCRs were performed within the exponential amplification range. The PCR products were mixed with LoadingStar (DyneBio, Sungnam, Korea), separated on 2% agarose gels by electrophoresis, and then photographed under ultraviolet excitation with ChemiDoc XRS (Bio-Rad Laboratories).
Quantitative reverse transcription–polymerase chain reaction. Annealing procedures were performed at 60°C for all primers.
GAPDH, glyceraldehyde-3-phosphate dehydrogenase; PPAR γ2, peroxisome proliferator-activated receptor γ2; LPL, lipoprotein lipase; BSP, bone sialoprotein; ALP, alkaline phosphatase.
Quantitative reverse transcription–polymerase chain reaction
Total cellular RNA extraction and cDNA synthesis were performed as described above. A quantitative PCR assay was performed by monitoring in real time the increase in fluorescence of the SYBR Green dye on a Thermal Cycler Dice real-time system (Takara Bio, Otsu, Japan), according to the manufacturer's instructions. Each PCR assay was carried out in duplicate in a 20-μL volume using SYBR Premix Ex Taq (Takara Bio) for 10 s at 95°C for the initial denaturing step, followed by 45 cycles at 95°C (denaturation) for 5 s, 60°C (annealing) for 15 s, and 72°C (amplification) for 10 s. Amplification specificity was confirmed by visualizing PCR products on 1.5% agarose gels and by a melting-curve analysis (from 60°C to 95°C) after the completion of 45 cycles. The values for each gene were normalized to the expression levels of GAPDH, and relative quantification of studied genes was calculated by using the formula 2−ΔΔCt. 19 Specific primer sequences and product sizes for each gene are listed in Table 1.
In vitro differentiation
For adipogenic differentiation, the cells were seeded at a density of 1×104 cells/cm2 in the growth medium in 12-well culture dishes. When reaching confluence, the cells were treated for 10 days with the adipogenic induction medium (α-MEM containing 10% FBS, 100 U/mL penicillin, 100 μg/mL streptomycin, 1 μM dexamethasone [Sigma], 10 μg/mL human insulin [Sigma], 100 μM indomethacin [Sigma], and 500 μM 3-isobutyl-
For cementogenic/osteogenic differentiation, the cells were prepared in 12-well culture dishes as described above. When reaching confluence, cultures were treated with the osteogenic induction medium (α-MEM containing 10% FBS, 1% antibiotics, 0.1 M dexamethasone, 2 mM β-glycerolphosphate [Sigma], and 50 μM ascorbic acid 2-phosphate) for 5 weeks. As a control, the cells were cultured only in the growth medium without differentiation stimuli. After 5 weeks, calcification of the extracellular matrix was visualized using Alizarin Red S staining and quantified with cetylpyridinium chloride. Briefly, cells were fixed for 30 min with 10% natural-buffered formalin at 4°C and stained with 2% Alizarin Red S (pH 4.2; Sigma) for 10 min at room temperature. The Alizarin Red S dye was extracted by adding 0.5 mL of 10% cetylpyridinium chloride (Sigma) to each well for 30 min. The absorbance at 570 nm was measured using a Benchmark Plus Microplate spectrophotometer (Bio-Rad Laboratories). Changes in the gene expressions of alkaline phosphatase (ALP) and bone sialoprotein (BSP) after cementogenic/osteogenic differentiation were evaluated with quantitative RT-PCR.
In vivo transplantation
All animal procedures were performed in accordance with a protocol approved by the Institutional Animal Care and Use Committee of the Yonsei University (#10-056). Approximately 3×106 in vitro expanded cells were mixed with 40 mg of macroporous biphasic calcium phosphate (MBCP; Biomatlante, Vigneux de Bretagne, France) and then incubated at 37°C for 2 h. After centrifugation at 500 g for 5 min, the supernatant was removed. The cell pellets with MBCP were transplanted into the dorsal subcutaneous pockets of 5-week-old male immunocompromised mice (BALB/c-nu, SLC, Shizuoka, Japan), as described previously. 1 Briefly, midlongitudinal skin incisions were made on the dorsal surface of each mouse, and four subcutaneous pockets were made by blunt dissection. The BMMSC, pPDLSC, and dPDLSC pellets mixed with MBCP, and MBCP particles alone (used as a negative control) were placed in each pocket of the same mouse. After 8 weeks, the mice (n=25) were sacrificed and all transplants were retrieved.
For quantitative RT-PCR analysis, the specimens (n=15 for BMMSCs, 19 for pPDLSCs, and 17 for dPDLSCs) were homogenized in the RLT buffer from the RNeasy Mini Kit with a homogenizer (Bullet Blender, Next Advance, Averill Park, NY) immediately after retrieval. Total RNA was extracted and reverse transcription was performed as described above. The relative gene expressions of human BSP, human collagen XII, human osteopontin, human osteocalcin, and human CP23 for each transplant were evaluated by quantitative PCR. Values for each gene were normalized to the expression levels of human and mouse GAPDH, and the expression levels of the genes of concern in each transplant relative to those of carrier transplants (MBCP without any cells) were calculated.
For the histology and immunohistochemical analysis, specimens (n=9 for BMMSCs, 13 for pPDLSCs, and 14 for dPDLSCs) were fixed in 10% buffered formalin for 1 day, decalcified with 10% ethylenediaminetetraacetic acid (pH 7.4; Fisher Scientific, Houston, TX) for 2 weeks, embedded in paraffin, and sectioned at a thickness of 5 μm. Specimens were subjected to hematoxylin and eosin and Masson's trichrome staining, as well as immunohistochemical staining with antihuman BSP (ab78278, Abcam, Cambridge, UK; rabbit polyclonal, diluted 1:1500), antihuman osteocalcin (AB10911, Millipore, Temecula, CA; rabbit polyclonal, diluted 1:2500), and antihuman collagen XII (sc-68862, Santa Cruz Biotechnology, Santa Cruz, CA; rabbit polyclonal, diluted 1:1200). For antigen retrieval before osteocalcin staining, sections were pretreated with proteinase K (Dako, Carpinteria, CA); for BSP, sections were pretreated by boiling in 1% citrate buffer (pH 6.0). An endogenous peroxidase activity was quenched by the addition of 3% hydrogen peroxide. Sections were incubated in 5% bovine serum albumin to block nonspecific binding. The primary antibodies were diluted to give optimal staining and the sections were incubated overnight. The following secondary antibody was applied (as supplied) for 20 min: peroxidase-labeled polymer conjugated to goat anti-rabbit immunoglobulin (EnVision+System-HRP Labeled Polymer Anti-rabbit; K4003, Dako). Color development was performed using labeled streptavidin biotin kits (Dako), according to the manufacturer's instructions. In brief, sections were incubated with streptavidin peroxidase conjugate for 10 min. Color was developed 1 min after the addition of diaminobenzidine substrate. The sections were counterstained with Gill's hematoxylin (Sigma). Control sections were treated in the same manner, but without treatment with primary antibodies.
Statistical analysis
All of the experiments were repeated under at least three independent conditions. All data are presented as mean and standard deviation values. Any differences were determined by the Kruskal–Wallis test (p<0.05) followed by the Mann–Whitney test with Bonferroni correction (p<0.017) using SPSS software (version 17.0; SPSS, Chicago, IL).
Results
Proliferation assay and cell cycle analysis
The cells outgrown from the explants exhibited a typical spindle-shaped fibroblastic morphology; the morphology did not differ between pPDLSCs and dPDLSCs (Fig. 1A–D). In the proliferation assay, the optical density of dPDLSCs was similar to that of pPDLSCs at every time point examined, but was significantly higher than that of BMMSCs (Fig. 1E). The cell cycle analysis also showed that the percentage of G1-phase dPDLSCs (57.1%) was similar to that of dPDLSCs (57.3%), but was lower than that of BMMSCs (70.1%), as seen in Figure 1F.
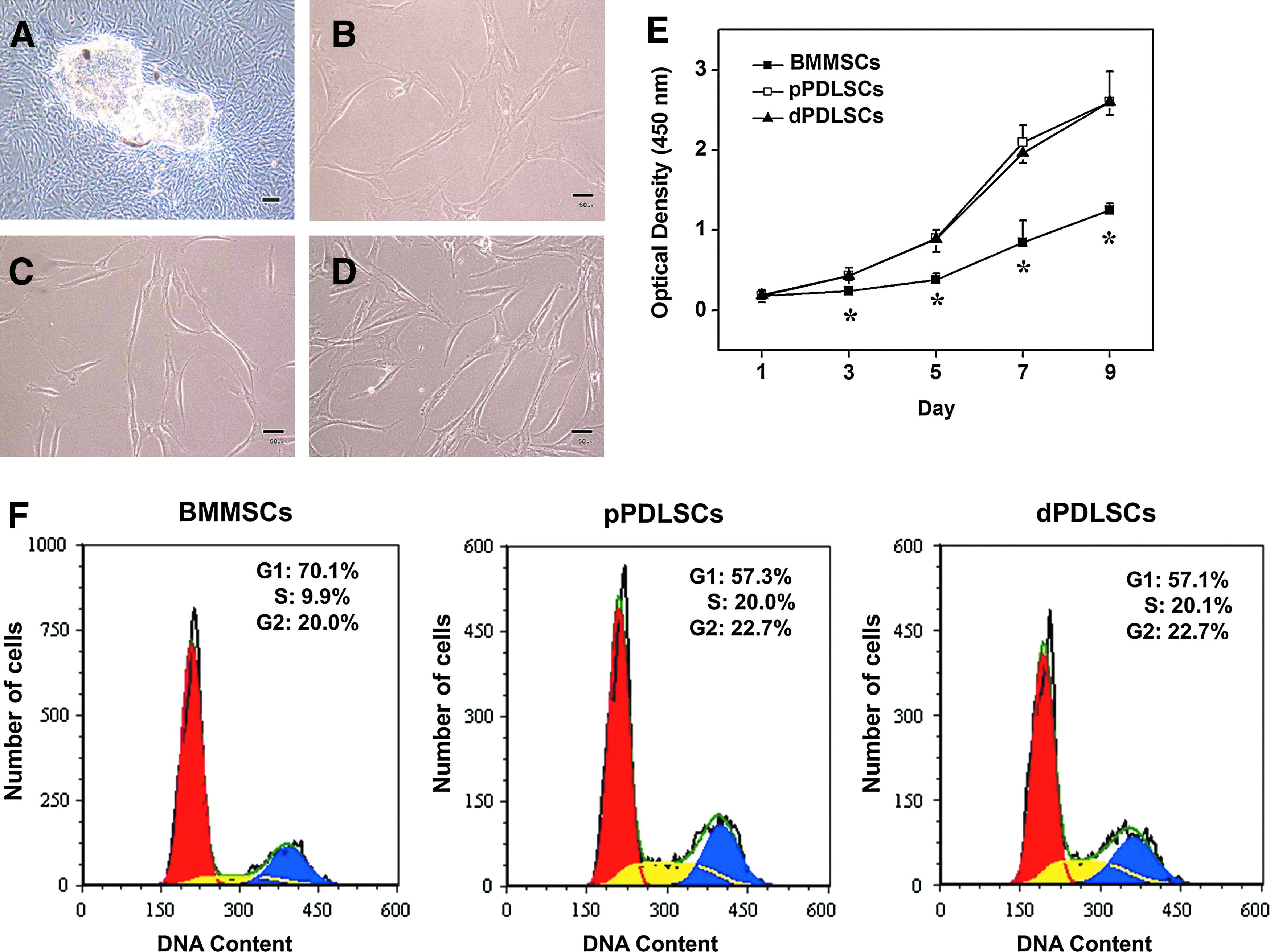
Morphologic characteristics and proliferation of BMMSCs, pPDLSCs, and dPDLSCs.
Colony-forming unit-fibroblast assay
All three cell types exhibited colony-forming ability (Fig. 2A). The number of colony-forming unit-fibroplasts (CFU-Fs) per 480 cells in dPDLSCs (42.3±7.3) was slightly lower than that in pPDLSCs (51.4±8.9), but the difference was not statistically significant. However, the BMMSCs had the significantly lowest number of CFU-Fs (20.7±2.2; p>0.05; Fig. 2B).
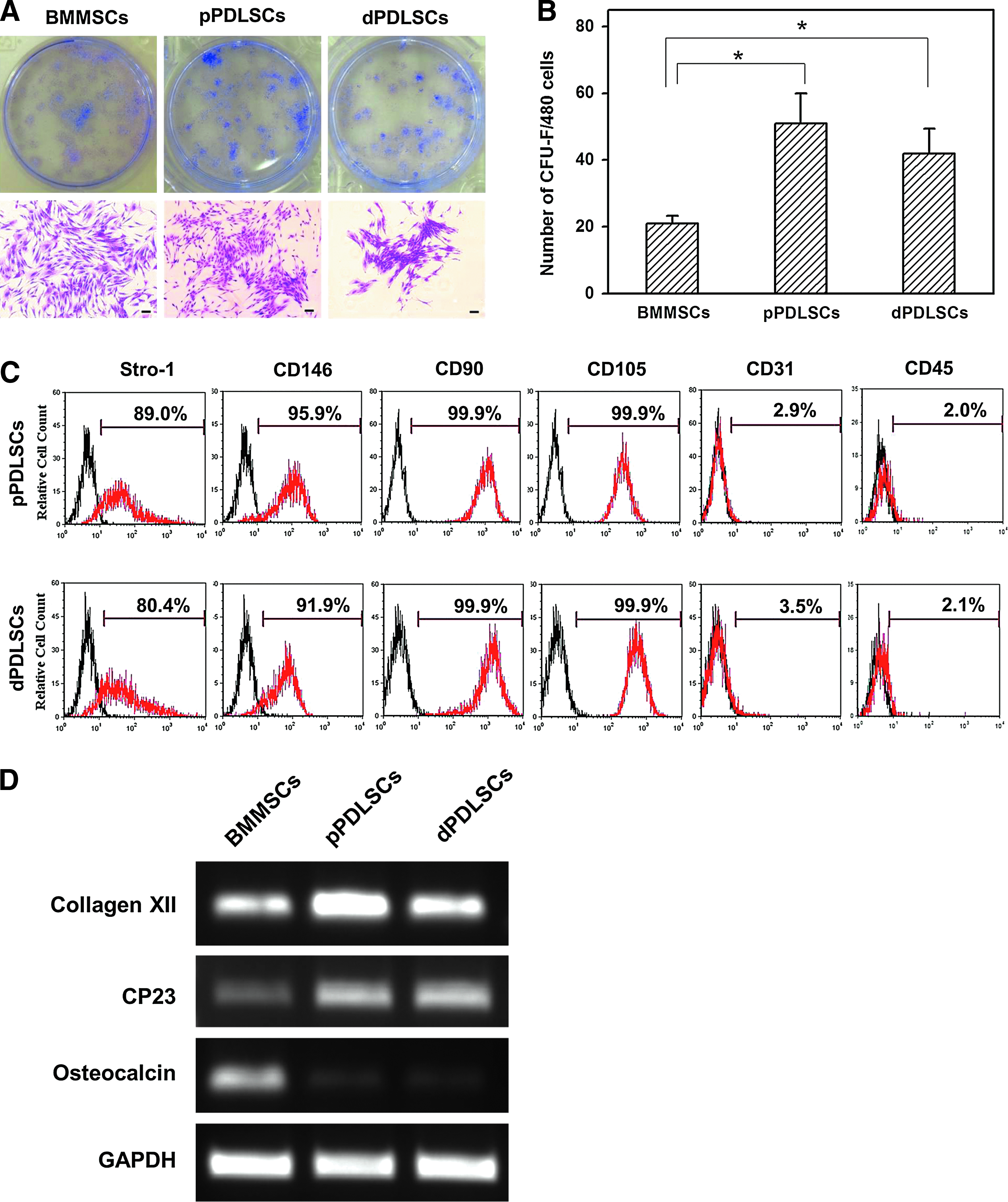
Colony-forming unit-fibroblast assay and flow cytometry analysis.
Flow cytometry analysis
The results of the flow cytometric analysis of markers related to mesenchymal stem cells are shown in Figure 2C. Almost all of the pPDLSCs and dPDLSCs expressed CD90 and CD105 (>99.9%). CD146 and Stro-1 were expressed in a considerable number of pPDLSCs and dPDLSCs (>80.4%). However, the expressions of CD31 (an endothelial stem cell marker) and CD45 (a hematopoietic cell marker) were low in both cell types (<3.5%).
Gene expression pattern by RT-PCR
The gene expression patterns of BMMSCs, pPDLSCs, and dPDLSCs are shown in Figure 2D. The expressions of collagen XII and CP23 (specific markers for periodontium) were higher while that of osteocalcin (a marker for matrix mineralization) were lower in both pPDLSCs and dPDLSCs than in BMMSCs.
In vitro differentiation
Under adipogenic stimuli, all three stem cells could differentiate into cells that had vacuoles containing lipids. Adipogenic differentiation occurred more in the BMMSCs than in both types of PDLSCs, as evaluated by Oil Red O accumulation (Fig. 3A) and quantitative RT-PCR analyses of changes in the expressions of PPAR γ2 and LPL (Fig. 3B). Under cementogenic/osteogenic stimuli, all three types of stem cells differentiated into cells that produced mineralized extracellular matrix. These findings were confirmed based on the Alizarin Red S accumulation (Fig. 3C) and upregulation of ALP and BSP gene expressions in the absence of any differences among the three types of stem cells (Fig. 3D). The extents of adipogenic and cementogenic/osteogenic differentiation were similar in the pPDLSCs and dPDLSCs.
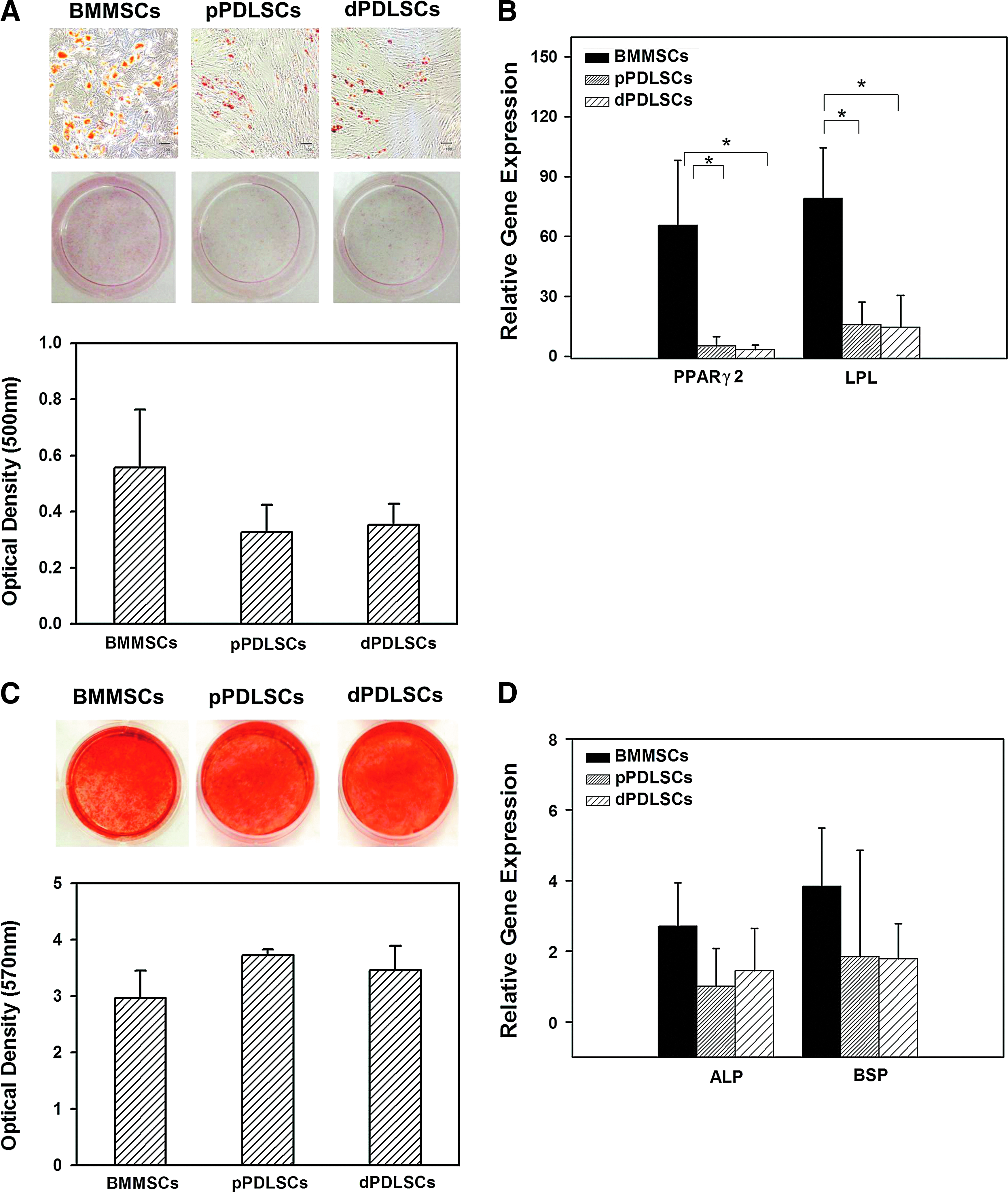
Adipogenic and cementogenic/osteogenic differentiation of BMMSCs, pPDLSCs, and dPDLSCs.
In vivo transplantation
Eight weeks after transplantation, all three types of stem cell could produce hard tissues at the periphery of the MBCP, but not in MBCP transplants (Fig. 4A–D). BMMSC transplants made bone-like tissues that exhibited a lamellar pattern of fiber alignment, lining of osteoblast-like cells, and outer dense fibrous tissues parallel to them in somewhere. In addition, there were embedded cells within the matrix, which exhibited reduced cytoplasmic space and condensed nuclei-like osteocytes in lacunae. In contrast, both pPDLSC and dPDLSC transplants formed cementum-like tissues with a fiber matrix that had a more irregular pattern or a linear alignment perpendicular to the lining cell layer and dense collagen bundles perpendicular to newly formed hard tissue resembling Sharpey's fibers. In some areas, the tissues contained embedded cementocyte-like cells resembling cellular cementum. Relative to the dPDLSCs, the pPDLSCs made more prominent Sharpey's fiber-like collagen bundles and PDL-like fibrous tissues (Fig. 4B, C, F, G). Immunohistochemical staining revealed that antiosteocalcin and anti-BSP antibodies reacted with the cells on the margin of the bone/cementum-like tissues—the signal was prominent on BMMSC transplants (Fig. 4I–P), whereas much more collagen XII was evident on the interstitial fibers and interface toward cementum-like tissues in the pPDLSC and dPDLSC transplants than in the BMMSC transplants (Fig. 4Q–T).

Histological and immunohistochemical staining of BMMSC, pPDLSC, and dPDLSC transplants.
After transplantation, the expression levels of genes related to mineralization (BSP, osteocalcin, and osteopontin) were lower in pPDLSC and dPDLSC transplants than in BMMSC transplants, while those related to cementum/PDL complex (CP23 and collagen XII) were higher in pPDLSC and dPDLSC transplants. pPDLSC transplants expressed slightly more genes than did dPDLSC transplants (Fig. 5).

Gene expression patterns of BSP, osteocalcin, osteopontin, collagen XII, and CP23 in the BMMSC, pPDLSC, and dPDLSC transplants. The gene expression level of MBCP transplants was set as the control (normalized to one). Data were obtained from five separate experiments (n=15 for BMMSCs, 19 for pPDLSCs, and 17 for dPDLSCs). Data are mean and standard deviation values. *Kruskal-Wallis test, p<0.05. OPN, osteopontin.
Discussion
Human beings have two kinds of teeth, deciduous and permanent, which differ in morphologic, histologic, and developmental characteristics. In addition, the proliferation activities and differentiation patterns differ between the cells isolated from the two types of teeth.5,7,25 In the present study, it was found to be difficult to distinguish between pPDLSCs and dPDLSCs simply according to their morphology and proliferation patterns. However, the finding that stem cells from the PDL tissue had a higher proliferation rate than BMMSCs concurred with the findings of some other studies.26,27 Many reports have stated that the cells isolated from the pulp tissues of deciduous teeth have a higher proliferation rate than those of permanent teeth.5,7,25 In the case of the PDL, Silverio et al. 9 reported that the PDL cells from deciduous teeth also have a higher proliferation rate. However, they used cells isolated via enzymatic dissociation, and a small population of PDL cells obtained by immunomagnetic cell sorting (CD105+, CD34−, and CD45− cells). The characteristics of cells isolated by enzyme-digested methods differ from those isolated by the outgrowth method.28,29 This difference in experimental conditions may be an explanation for the disharmony between the obtained results; further investigations are needed to clarify this issue.
CFU-F assays have been used to evaluate self-renewal ability, which is a characteristic of mesenchymal stem cells. 30 In accordance with some previous studies, but not with others, we found that the pPDLSCs exhibited a greater number of CFU-Fs than BMMSCs.26,31,32 Although the dPDLSCs also exhibited a greater number of CFU-Fs than did BMMSCs, it is not possible to compare the findings of the present study to those of previous studies because this is the first study to perform CFU-F assays of dPDLSCs.
The present study revealed that pPDLSCs and dPDLSCs represented mesenchymal stem-cell surface markers such as Stro-1, CD146, CD105, and CD90,4,33–35 but not CD31, which is an endothelial cell marker, 36 and CD45, which is a hematopoietic cell marker known as common leukocyte antigen.37,38 Therefore, the pPDLSCs and dPDLSCs in this study appeared to have arisen from the perivascular area, where Stro-1 and CD146 antigen reacted strongly.8,39 In addition, compared with BMMSCs, pPDLSCs and dPDLSCs expressed more periodontium-related genes (e.g., CP23, identified as a cementoblast marker and regulator of the biomineralization of cementum40,41 and collagen XII, identified as a PDL marker and found mainly in tissues subject to high tensile stresses such as tendons and PDLs).42–45 This finding suggested that these stem cells indeed originated from PDL tissues and had previously differentiated into the cell-forming periodontium.
We found the adipogenic differentiation potentials to be lower for the pPDLSCs and dPDLSCs than for the BMMSCs, which is consistent with the findings of previous studies.31,32,46 The comparison of pPDLSCs and dPDLSCs by Silverio et al. 9 revealed that the adipogenic differentiation of pPDLSCs was superior to that of dPDLSCs. This finding does not concur with ours. Furthermore, while Silverio et al. 9 reported that the osteogenic differentiation potential of dPDLSCs was inferior to that of pPDLSCs, we did not. Whether different experimental conditions are responsible for this disagreement requires further investigation.
It has been reported that PDLSC transplants are capable of producing tissues that resemble cementum/PDL complex.4,20,47 In the present study, both pPDLSCs and dPDLSCs were able to make cementum-like and adjacent PDL-like tissues, but not the lamellar pattern of hard tissue observed in the BMMSC transplants. In addition, pPDLSC and dPDLSC transplants expressed more cementum/PDL-related genes rather than BSP, osteocalcin, and osteopontin genes; the latter are involved in the mineralization of both cementum and bone.42,48 It was recently reported that stem cells from the PDL adjacent to the alveolar bone could make more structural bone-like tissue than those from PDL attached to the root. 49 Therefore, it is reasonable that stem cells obtained from the PDL attached to extracted teeth might be capable of making cementum/PDL-like tissue rather than bone-like tissue, as shown previously and in our own studies. The pPDLSC transplants made more typical cementum/PDL complex and expressed relatively more cementum/PDL-related genes than did the dPDLSC transplants (i.e., CP23 and collagen XII). In addition, it was reported that cementoblasts of deciduous teeth could express more osteopontin and BSP—which were thought to be associated with odontoclast adhesion and subsequent root resorption—than could those of permanent teeth.50–53 Although dPDLSC transplants expressed similar levels of BSP and a slightly higher level of osteopontin genes relative to pPDLSC transplants in the present study, the newly formed hard tissue in the dPDLSC transplants might have been easily resorbed as cementum of deciduous teeth. 54
The finding that cells that originate from permanent teeth make more typical tissues resembling the original tissues in vivo than those of deciduous teeth has been reported previously for studies of dental pulp.1,5,55 The DPSCs formed vascularized pulp-like tissue surrounded by dentin-like tissues,1,55 but SHED were unable to regenerate a complete dentin/pulp-like complex, 5 and instead formed bone-like tissues that stained negatively for antidentin sialoprotein and antidentin sialophosphoprotein antibodies.5,56 It seems that the cells that produce bone-like tissues originated from both murine cells and transplanted SHED, as shown by their reaction to the anti-human-specific mitochondria antibody and the expressions of both mouse mRNAs and human mRNAs.5,56 Transplanted human PDLSCs were revealed to differentiate into cementoblasts and PDL fibroblasts4,20,47; however, it is not clear that the dPDLSCs in the present study could differentiate into osteoblasts and induce recipient murine cells to differentiate into bone-forming cells, as the SHED could, because we did not observe the absence of the cementum-specific protein (CP23) and the presence of mouse mRNA expression within the lacunar cells. Further investigations are needed to clarify this issue.
Conclusions
The proliferation rate, expressions of stem-cell markers, and in vitro differentiation into adipogenic and cementogenic/osteogenic lineages were similar in dPDLSCs and pPDLSCs. The pPDLSC transplants made more typical cementum/PDL complex-like tissues than did the dPDLSC transplants in vivo, which suggests that pPDLSCs are better candidates for use in periodontal regeneration.
Footnotes
Acknowledgments
We thank the division of Anatomy and Developmental Biology, Department of Oral Biology, the Yonsei University for providing the research space and equipment. We also gratefully acknowledge Professor Hwal Suh (Department of Medical Engineering, the Yonsei University, Korea) for kindly providing the bone marrow-derived mesenchymal stem cells. This study was supported by grants of the Korea Healthcare Technology R&D Project, the Ministry for Health, Welfare, and Family Affairs, the Republic of Korea (no. A100061) and of the Basic Science Research Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Education, Science and Technology (no. 2011-0022160).
Disclosure Statement
No competing financial interests exist.