
Abstract
Human mesenchymal stem cells (hMSCs) can be derived from various adult and fetal tissues. However, the quality of tissues for the isolation of adult and fetal hMSCs is donor dependent with a nonreproducible yield. In addition, tissue engineering and cell therapy require large-scale production of a pure population of lineage-restricted stem cells that can be easily induced to differentiate into a specific cell type. Therefore, human embryonic stem cells (hESCs) can provide an alternative, plentiful source for generation of reproducible hMSCs. We have developed efficient differentiation protocols for derivation of hMSCs from hESCs, including coculture with murine OP9 stromal cells and feeder layer-free system. Our protocols have resulted in the generation of up to 49% of hMSCs, which expressed CD105, CD90, CD29, and CD44. The hMSCs exhibited high adipogenic, chondrocytic, and osteogenic differentiation in vitro. The latter correlated with osteocalcin secretion and vascular endothelial growth factor (VEGF) production by the differentiating hMSCs. hMSC-derived osteoblasts further differentiated and formed ectopic bone in vivo, and induced the formation of blood vessels in Matrigel implants. Our protocol enables generation of a purified population of hESC-derived MSCs, with the potential of differentiating into several mesodermal lineages, and particularly into vasculogenesis-inducing osteoblasts, which can contribute to the development of bone repair protocols.
Introduction
In the developing skeleton, the endochondral cartilage template is replaced by highly vascularized tissue. 21 The formation and development of an active microvasculature are essential for bone remodeling and fracture healing.22,23 Recent studies have suggested that vascular endothelial growth factor (VEGF), a potent angiogenic stimulator, may play a role in bone formation. VEGF is a subfamily of homodimeric proteins referred to as VEGF-A, B, C, and E, and placenta growth factor.24,25 VEGF-A exists in five different isoforms. VEGF165, the most abundant of these isoforms, is soluble and can bind to the extracellular matrix. 26 Maes et al. and Street et al. showed that administration of VEGF stimulates, and its antagonist FLT-1 suppresses, bone repair.27,28 VEGF has also been shown to demonstrate a beneficial effect on bone healing elicited by bone morphogenic protein 4.29,30 Further, the absence of VEGF isoforms in genetic models indicates a completely disrupted vascular pattern associated with impaired differentiation of hypertrophic chondrocytes and osteoblasts, suggesting that VEGF not only mediates bone vascularization but also affects the differentiation of progenitors to hypertrophic chondrocytes and osteoblasts. 31
The current study implements considerable improvements in two protocols for derivation of hESC-MSCs: coculture with murine OP9 and feeder layer-free systems. The result is highly efficient and reproducible directed differentiation of hMSCs. After CD73-based sorting, a pure population of hMSCs was established in long-term cultures. Two lines of hESC-MSCs showed mesenchymal morphology, extended in vitro expandability, and robust multilineage potential, giving rise to chondrocytes, adipocytes, and osteoblasts in vitro. The hESC-MSCs also secreted osteocalcin and VEGF during osteoinduction. We further demonstrated the capability of the hESC-MSCs to differentiate into ectopic bone in vivo without the use of an osteoconductive carrier, and to induce endothelial cell sprouting in Collagen gels as well as invasion of blood vessels into hMSC-derived bone-like implants.
Materials and Methods
Cell culture and fluorescence-activated cell sorting
The undifferentiated hESC lines I-4 (P68-71) and H9.2 (P54) used in this study were derived from the preimplantation embryo as described previously.32,33 The blastocysts that were produced by in vitro fertilization for clinical purposes were donated by individuals after informed consent and after the approval of the Rambam Medical Center Helsinki Committee and the Israeli Ministry of Health. The hESCs were cultured on mitotically inactivated mouse embryonic fibroblasts and maintained under growth conditions and passaging techniques as previously described.
34
In brief, hESCs were cultured with a medium consisting of 80% Dulbecco's modified Eagle's medium F12 (DMEM-F12; Biological Industries), 1 mM
Cell expansion and growth curves
Population-doubling time (PDT) was calculated by the formula t/PD; t is the number of hours in culture. PD was calculated by the formula ln(Xf/X0)/ln2, where X0 is the number of seeded cells, and Xf is the number of cells at confluence before splitting. The growth curve was established by using the XTT kit (Biological Industries) according to the manufacturer's instructions.
Flow cytometry analyses
hESC-MSCs were harvested using 0.25% trypsin (Biological Industries), and after neutralization, resuspended in a staining buffer, phosphate-buffer saline (PBS) +1% FBS. About 1.5×105 cells were incubated with each of the following monoclonal conjugated antibodies: CD73, CD45 (IgG1–PE; BD Pharmingen), CD105, CD29, CD90, CD31 (IgG1–PE; eBioscience), CD44 (IgG2b-PE; eBioscience), and CD34 (IgG1–PE; IQ Products) and acquired using FACSCalibur. Nonspecific fluorescence was determined by incubation of similar cells with matched isotype control antibodies. Samples were analyzed using an FACSCalibur flow cytometer with CellQuest Acquisition software (Becton Dickinson Immunocytometry Systems).
Adipogenic differentiation
hESC-MSCs were seeded at a density of 20,000 cells/cm2, and grown in a DMEM F-12 medium containing 10% FBS and 1 mM
Osteogenic differentiation
hESC-MSCs were seeded at a density of 2000–3000 cells/cm2 and grown in an osteogenic medium (OM): GMEM BHK-21 (Gibco BRL) medium supplemented with 10% FBS, 1 mM sodium pyruvate (Gibco BRL), 1% nonessential amino acids, 50 μg/mL
Chondrogenic differentiation
For chondrogenic differentiation, 2×105 hESC-MSCs were centrifuged at 300 g for 5 min in 15-mL polypropylene falcon tubes to form a cell pellet. The cells were grown for 9 weeks in the DMEM (Gibco BRL), supplemented with 10−7 M dexamethasone 1% ITS, 50 μg/mL
Myogenic differentiation
For myogenic differentiation, hMSCs (2000 cells/cm2) were cultured for 2 weeks with an α-MEM containing 20% serum. The cells were then tested for expression of NCAM/CD56, a marker known to be expressed on skeletal myoblasts, by FACS (CD56; eBioscience).
Reverse transcription polymerase chain reaction
Total RNAs were isolated using TRIzol/Tri-reagent (Invitrogen), and reverse transcribed by the iScript™ cDNA synthesis kit (BIO-RAD). Reverse transcription polymerase chain reaction (RT-PCR) was performed by the DreamTaq™ Green Master Mix (Fermentas). Primer sequences and product sizes are listed in Supplementary Table S1 (Supplementary Data are available online at www.liebertpub.com/tea).
Concentrations of VEGF165 in hMSC-conditioned medium
hESC-MSCs were cultivated with an hMSC growth medium (see the section Cell culture and FACS) or with an OM (see the section Osteogenic differentiation) for 3 weeks. To measure the secretion of VEGF165, the medium was replaced at the indicated time, and the cell number determined. Human VEGF165 in a conditioned medium was detected by an enzyme-linked immunosorbent assay (ELISA) kit (PeproTech) according to the manufacturer's instructions.
Quantification of osteocalcin in hMSC-conditioned medium
Osteocalcin was quantified in a serum free-OM, according to the manufacturer's instructions, using the human osteocalcin instant ELISA kit (eBioscience). The assay was performed on culture days 7, 14, and 21.
Spheroid sprouting assay
The assay was performed as described elsewhere. 35 In brief, human umbilical vein endothelial cell (HUVEC) spheroids were generated overnight in a hanging-drop culture consisting of 400 cells in 80% endothelial cell medium (ScienCell) and 20% methylcellulose (Sigma). Spheroids were embedded in collagen (collagen extract of rat tails) and stimulated with 5 ng/mL bFGF (R&D systems) or with an osteogenic conditioned medium of hESC-MSCs in the presence or absence of 10 μM SU6668 inhibitor (Tocris).
Proliferation assay
HUVECs (1×104 cells/well) were seeded in 24-well plates. The cells were incubated with 5 ng/mL bFGF or with a conditioned medium of hESC-MSCs in the presence or absence of 10 μM SU6668 inhibitor (Tocris). The cells were counted 3 and 5 days after seeding.
Osteogenic Matrigel implants
The formation of ectopic osteogenic structures in vivo was evaluated using a xenograft model of Matrigel implants. hESC-MSCs (1.5×106) undergoing osteoblast differentiation in vitro for 3, 7, or 14 days and noninduced hESC-MSCs were mixed with 250 μL Matrigel on ice and injected subcutaneously to immune-deficient NOD/SCID 8–12-week-old mice. All implants were removed after 2 weeks and fixed in 4% PFA. Empty Matrigel implants served as control. For bone maturation, the hESC-MSCs (1.5×106) underwent osteoblast differentiation in vitro for 14 days; the cells were mixed with 250 μL Matrigel on ice and injected subcutaneously to immune-deficient NOD/SCID 8–12-week-old mice. The implants were removed after 1, 2, or 4 weeks, fixed in 4% PFA, and stained with anti-osteocalcin. Placenta-derived MSCs were isolated as previously described 36 and served as a positive control. All procedures were approved by the committee for the Use and Care of Animals of the Technion-Israel Institute of Technology.
Teratoma formation
hESCs and hESC-MSC lines I-4 and H9.2 were injected into the rear leg muscle of 4-week-old, male SCID/beige mice (Harlan). The number of cells per injection ranged from 2.5×106 to 5.0×106. Ten weeks or more after the injection, the resulting teratomas were examined histologically.
Histology immunofluorescence and immunohistochemistry
Paraffin-embedded sections of the implants were stained with H&E and Alizarin red for evaluation of calcium deposit content. For fluorescence microscopy, the following uncoupled anti-human primary antibodies were used: alkaline phosphatase (ALP; BD Pharmingen; 1:100), rabbit-anti human MHC class I (Epitomics; 1:100), mouse-anti human osteocalcin (Abcam; 1:200), and goat anti-human osteopontin (1:50). The secondary conjugated antibodies included Alexa-488-conjugated donkey anti-goat (1:100; Invitrogen), goat anti-mouse Cy-3 (1:100; Jackson), and donkey anti-rabbit Cy-3 (1:100; Jackson). The nucleus was labeled with DAPI (4′,6-diamidino-2-phenylindole, dihydrochloride; Molecular Probes; 1:1000) for 5 min at room temperature.
Fluorescence in situ hybridization
Aquarius probes (human chromosome 1-red, human DAPI antifade; Cytocell) were used for whole-chromosome painting, according to the manufacturer's instructions.
Microvessel density analysis
Microvessels were quantified by evaluation of 10 randomly selected fields of H&E-stained sections obtained from various parts of the implants. Microvessels were identified as lumenal structures containing red blood cells. Microvessel density was reported as the average number of red blood cell-containing microvessels from the fields analyzed, and expressed as the number of lumens/mm2.
Statistical analysis
Statistical significance (p<0.05; p<0.01) was determined using analyses of variances (one-way analysis of variances), followed by Tukey's post hoc testing.
Karyotype
Karyotype was performed as described elsewhere. 37
Results
Isolation of hESC-MSCs grown in coculture with OP9 stromal cells or on Matrigel-coated plates
We achieved mesenchymal differentiation by plating undifferentiated hESC lines I-4 and H9.2 at a high density of 30,000 cells/cm2 on a monolayer of murine OP9 stromal cell–coculture system, or alternatively on Matrigel-coated plates as a feeder-free system. At day 35, the hESC-MSCs were harvested and sorted by FACS using the surface antigen CD73, which is known to be expressed in hMSCs. CD73-positive cells (CD73+) comprised 26±7% of the differentiated hESCs grown on OP9 cells (Fig. 1A, upper panel). The specificity of the CD73 antibody for detection of hMSCs was to the human protein only; cross-reactivity with the murine OP9 feeder was not shown (Fig. 1B). We also used a feeder-free system based on gelatin and Matrigel matrices to induce mesenchymal differentiation similar to that in the coculture system. Undifferentiated hESCs (lines I-4 and H9.2) were seeded on gelatin- or Matrigel-coated plates. Cells that were seeded on gelatin detached from the plates during the culture period, significantly reducing the number of cells available for CD73-based sorting. This phenomenon did not occur on Matrigel-coated plates. The feeder-free system yielded twice the percentage of CD73+ derivatives (49±12%) as the coculture system (Fig. 1A, lower panel). When hESCs were spontaneously differentiated in 3D culture to form EBs, only 4% of the cells were detected as CD73+ in 14-day-old EBs (Supplementary Fig. S1). Therefore, the feeder-free system seems to be the most efficient means of differentiating hMSCs (Fig. 1C). The procedure for isolating CD73+ cells appears reproducible, since a total of 10 independent experiments, four in the coculture system and six in the feeder-free system, yielded similar populations of cells. Sorted hESC-MSCs were further efficiently expanded (up to 108 cell) in long-term culture for 20 passages, with normal karyotype retained (Supplementary Fig. S2). hESC-MSCs from the different systems exhibited different growth potential. hESC-MSCs from the feeder-free system had PDT of about 60 h in the first 4 weeks, whereas cells from the coculture system had PDT of 80 h (Fig. 1D, E). In addition, freeze and thaw cycles demonstrated good recovery of at least 90%.

Isolation of hESC-MSCs.
Characteristics of hESC-MSCs
Sorted hESC-MSCs, from both coculture and feeder-free systems, exhibited heterogeneous morphology, depending on the cell density, ranging from fibroblastic shape to rectangular cells in confluent culture. The hESC-MSCs from both systems were analyzed by flow cytometry for the expression of a comprehensive set of markers known to be expressed in hMSCs. hESC-MSCs coexpressed CD105, CD29, CD44, and CD90 (Fig. 2A), and were devoid of the hematopoietic markers CD45 and CD34, as well as the endothelial/monocytic cell surface marker CD31/PECAM. Immunofluorescence staining and RT-PCR analyses demonstrated that long-term cultured CD73+ cells did not express the pluripotency markers Oct3/4 and Nanog, whereas their parental source H9.2 and I-4 hESC lines were positively stained for Oct3/4, Nanog, and TRA1-60, and also expressed Oct3/4 and Nanog, as demonstrated by RT-PCR analyses (Fig. 2B, C). Immunofluorescence staining and RT-PCR analyses revealed that similar to native tissue-derived hMSCs, 36 long-term cultured CD73+ cells expressed nestin, but did not express brachyury (Fig. 2B, C).

Characterization of hESC-MSCs.
Differentiation of hESC-MSCs into adipocytes and chondrocytes in vitro
Adipogenic differentiation, first detected 5 days after culture initiation by the appearance of lipid vesicles, was completed within 3 weeks. The majority of the resultant cells were of the multilocular type, with polygonal morphology, and containing a large number of lipid droplets of various sizes in the cytoplasm. As differentiation progressed, the lipid-rich vacuoles within the adipocytes increased in size and became brighter. Adipogenic differentiation was highly efficient; after 3 weeks of induction, more than 95% of cells displayed Oil Red O-positive lipid droplets within the adipocytes (Fig. 2D). In addition, RT-PCR analyses revealed that upon adipogenic induction, hESC-MSCs expressed the early adipogenic transcription factor peroxisome proliferator-activated receptor gamma, as well as adipoprotein 2 (AP2) (Fig. 2D). Upon chondrogenic induction, mature lacunae appeared in the center of all pellets, and a perichondrium, a sheath of dense connective tissue surrounding the cartilage, was also detected. Positive Alcian blue staining of cartilage-specific glycosaminoglycans appeared throughout the cell section, with particular enhancement in the marginal area (Fig. 2E). Differentiated cells expressed aggrecan, Sox 9, and collagen type II, as demonstrated by RT-PCR analyses (Fig. 2E). hESC-MSCs had limited myogenic potential. After only a few of the trails, a limited number of cells expressing CD56/NCAM were observed. Sorting for these CD56-positive cells had failed. In addition, expression of MyoD was not observed with immunostaining (data not shown).
Differentiation of hESC-MSCs into osteoblasts is accompanied by secretion of osteocalcin
Osteogenic differentiation was detected by brown deposits first appearing 5 days after culture initiation. Robust osteogenic differentiation was detected at 2 weeks of culture, and remained stable until day 28, as demonstrated by an increased level of mineralized matrix stained positive by Alizarin red (Fig. 3A and Supplementary Fig. S3A). In addition, immunostaining of ALP was only identified in hESC-MSCs grown with an OM, and not in cells grown in the control medium in the absence of osteogenic inducers (Fig. 3B and Supplementary Fig. S3B). RT-PCR analyses revealed that hESC-MSCs expressed osteoblast-related genes, including osteocalcin, osteonectin, osteopontin, and collagen type I (Fig. 3C). Osteopontin expression was observed exclusively in hESC-MSCs grown in the OM. However, osteocalcin, osteonectin, and collagen type I were observed in hESC-MSCs grown either in an hMSC growth medium or in a control medium (without osteogenic inducers) as well (Fig. 3C). Yet, significant increases in osteonectin and collagen type I expression were detected in hESC-MSCs grown in the OM. Densitometry values demonstrated a twofold increase in osteonectin expression by hESC-MSCs grown in the OM as compared to cells grown in the control medium (without osteogenic inducers) (Supplementary Fig. S4). Levels of osteocalcin secretion from hESC-MSCs and placenta-derived MSCs that served as a positive control in OM were detected in vitro by ELISA. Osteocalcin levels increased during osteogenesis in both hESC-MSCs and placenta-derived MSCs. Levels of osteocalcin secreted by the placenta-derived MSCs were higher at each time point than the level secreted by the hESC-MSCs (Fig. 3D).
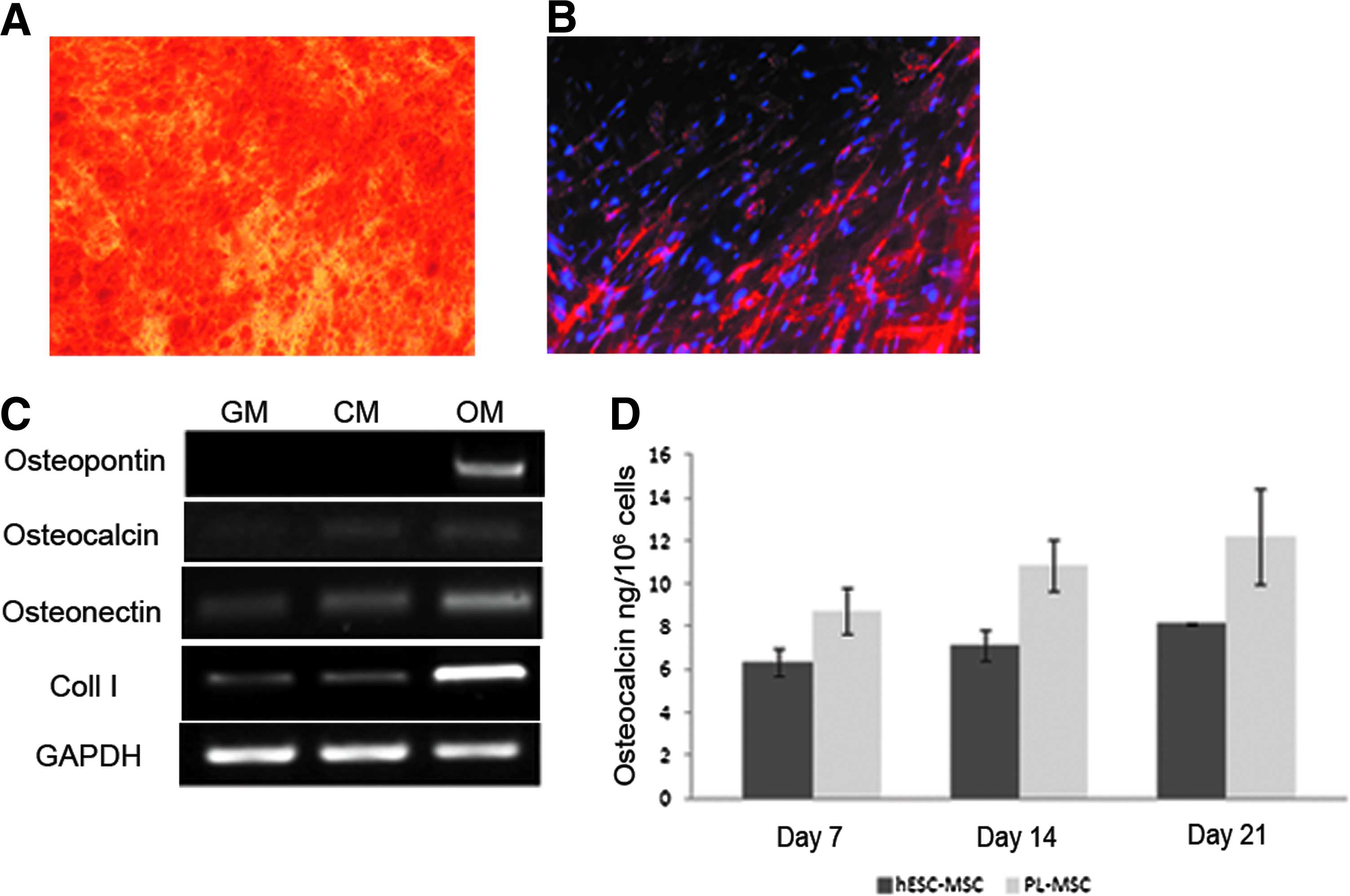
Osteogenesis in vitro and osteocalcin secretion.
hESC-MSCs form ectopic vascularized bone in vivo
The ability of the cells to produce a mineralized matrix in vitro, which although indicates osteogenesis, may nevertheless not predict the capacity of the cells to develop bone tissue in vivo. Therefore, the hESC-MSCs were tested for their capacity to survive and to produce human bone tissue after ectopic subcutaneous transplantation in NOD/SCID mice without the use of an osteoconductive carrier or scaffold. The hESC-MSCs were subjected to osteogenic induction of 3, 7, or 14 days in vitro. The injected cells remained within a small well-limited area at the injection site, and the developed tissue showed clearly defined boundaries toward the murine tissue (Fig. 4A), while no teratoma formation was observed (Fig. 4B). As we have previously demonstrated, the parental lines of the hESC-MSCs (I-4 and H9.2) did form teratoma after intramuscular injection into SCID/beige mice. 38 After 2 weeks, the comprehensive area of a newly formed mineralized bone matrix was observed in the central region of all implants. The calcified matrix was identified by positive Alizarin red staining of the engineered tissue and relevant controls (Fig. 4A, inset). Furthermore, an extensive expression of osteopontin and osteocalcin was detected by immunofluorescence labeling in sectioned implants (Fig. 4C, D). To verify the human cell identity in 2-week-old bone-like Matrigel implants, implanted cells were positively labeled with antibodies that specifically recognize human MHC class I (Fig. 4C). In addition, human cells were identified by red fluorescence staining of chromosome 1 in the cell nuclei within the newly formed tissue, by in situ hybridization using the whole-chromosome painting technique (Fig. 4C, inset). Empty Matrigel was implanted as well, and did not induce bone-like tissue formation or the formation of blood vessels (Fig. 4E). Vascularization of engineered tissue is critical for cell survival and remodeling at initial stages of implant integration to the host tissue. In line with this demand, an extensive network of blood vessels (266±86 lumens/mm2) was formed when the hESC-MSCs of the feeder-free system were subjected to osteogenic induction for 7 days in vitro (Fig. 4F, G). When the cells were exposed to osteogenic induction for 3 days, the number of blood vessels was reduced significantly to 38±16 lumens/mm2 (Fig. 4G). All blood vessels contained non-nucleated erythrocytes, indicating that the blood vessels within the implant anatomized the murine circulation (Fig. 4F). Cells from the coculture system contained significantly less blood vessels (89±35 lumens/mm2, p<0.01) when the hESC-MSCs were subjected to osteogenic induction for 7 days in vitro; when exposed to osteogenic induction for 3 days, the implants did not contain any blood vessels (Fig. 4G).

Ectopic bone formation in vivo. hESC-MSCs were subjected in vitro to osteogenic differentiation for 3, 7, and 14 days
We analyzed VEGF165 concentrations in a conditioned medium of hESC-MSCs 7, 14, and 21 days after osteogenic induction in vitro by ELISA. VEGF165 concentrations were found to be higher in cells grown in an hMSC osteogenic growth medium than in those grown in the control medium (w/o osteogenic inducers). VEGF165 concentrations increased during osteogenesis (Fig. 5A). These findings correlate with the observed mineralization appearance and indicate that VEGF165 is maximally secreted at the late phase of osteogenic differentiation, when the matrix is mineralized (Fig. 5B). We further tested whether hESC-MSCs are involved in the regulation of angiogenesis using an in vitro endothelial cell-sprouting assay, wherein spheroids of HUVECs were incubated with either the HUVEC medium in the presence or absence of bFGF, or the conditioned medium of osteogenic differentiated hESC-MSCs in the presence or absence of 10 μM of the broad inhibitor SU6668 (ATP-competitive platelet-derived growth factor receptor [PDGFR], VEGF, and fibroblast growth factor receptor [FGFR] inhibitor). In the presence of bFGF or in the osteogenic hESC-MSC-conditioned medium, the spheroids invaded the gel and formed elongated capillary-like structures 24 h after stimulation (Fig. 5C). In the presence of SU6668, the number and length of sprouts were reduced significantly by ∼30% and 55%, respectively (p<0.01, Fig. 5D[i, ii]). In addition, the osteogenic conditioned medium of hESC-MSCs induced proliferation of HUVECs, whereas the presence of the inhibitor reduced proliferation (Fig. 5E).

VEGF secretion, spheroid sprouting assay, and proliferation.
The kinetics of bone maturation was assessed by immunofluorescence staining of osteocalcin. Implants of hESC-MSCs, after osteogenic induction of 14 days in vitro, were removed after 1, 2, or 4 weeks in vivo. Placenta-derived MSCs served as positive control. After 1 week in vivo, low expression of osteocalcin was detected in sectioned implants, both in hESC-MSCs and placenta-derived MSCs (Fig. 6A, B). However, after 2 weeks in vivo, a significant increase in the expression of osteocalcin was observed. The expression of osteocalcin remained high thereafter, with no additional increase in either hESC-MSCs or placenta-derived MSCs (Fig. 6A, B). Altogether, these findings demonstrate that the kinetics of osteogenic maturation of hESC-MSCs is similar to that of their native tissue MSC counterparts, further highlighting their equivalent MSC characteristics.
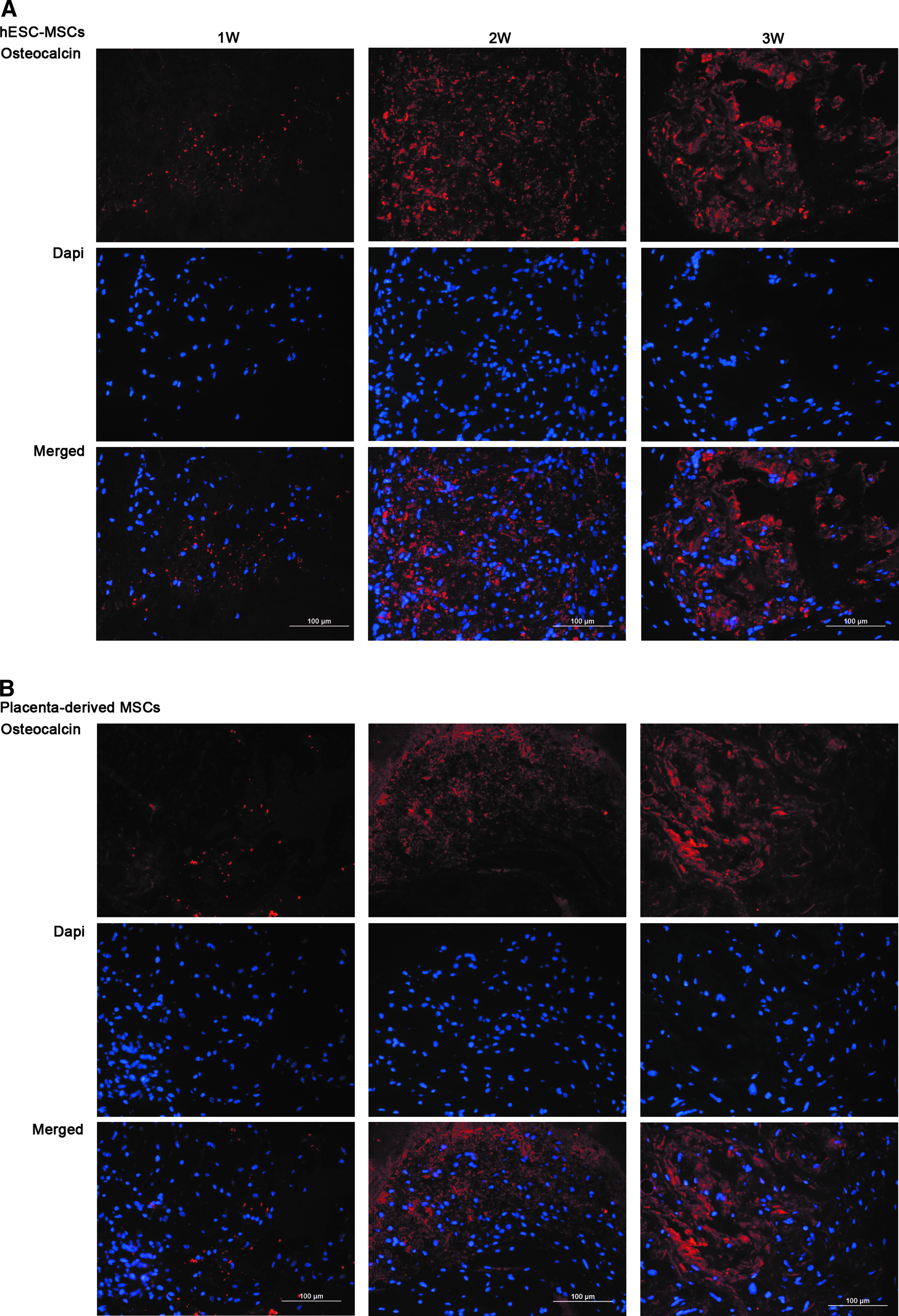
Kinetics of bone maturation–osteocalcin expression. hESC-MSCs were subjected in vitro to osteogenic differentiation for 14 days and then implanted subcutaneously into NOD/SCID mice using Matrigel. Implants were harvested 1, 2, or 4 weeks after transplantation. Placenta-derived MSCs served as positive control.
Discussion
In this study, we displayed two simple methods, based on cell sorting, to derive from hESCs a pure population of hMSCs (hESC-MSCs) with high osteogenic potential. The hESC-MSCs exhibited characteristics of hMSCs, including expression of known surface markers and the capability of differentiating into adipocytes, chondrocytes, and osteoblasts in vitro. In addition, the osteogenic differentiated hESC-MSCs secreted osteocalcin and VEGF, induced capillary sprout formation in collagen gels, and formed an ectopic vascular bone when implanted subcutaneously into immunodeficient mice. Previous protocols for the establishment of hESC-MSCs include coculturing with OP9 stromal cells,10,11 isolation based on manual/selective selection, consecutive passaging,12–15 or cell sorting.16,17 However, in some of the protocols, only a small proportion of the cells differentiated into hMSCs, thereby requiring further enrichment of the target population. In others, the population obtained was heterogeneous. In contrast, both systems that were used for derivation of hMSCs from hESCs in the present study, namely feeder free and coculture, with OP9 stromal cells, are easy to perform, do not require expensive growth factors, and exhibit a high degree of reproducibility and homogeneity in the resultant cells. Nevertheless, the feeder-free system demonstrated a greater efficiency in inducing mesenchymal differentiation. Moreover, since differentiation through EBs results in a heterogeneous cell population, and coculture using cells from a different species can lead to contamination from animal products, the feeder-free system is highly relevant to the development of techniques for clinical applications.
The differentiation potential of hESC-MSCs toward the osteogenic lineage was demonstrated by secretion of a mineralized matrix and by the expression of osteoblast-associated genes in vitro. The observation that the hESC-MSCs did not express the pluripotent genes Oct3/4 and Nanog implies the derivation of the osteoblasts from a restricted cell lineage that lacks undifferentiated cells.
In the few studies that have demonstrated the ability of hESC-MSCs to form bone in vivo, the cells were mixed and implanted with an osteoconductive scaffold, which further stimulates osteogenic differentiation of implanted hMSC post-transplantation.9,14,18–20 The hESC-MSCs generated in the current study formed new vascular bone-like tissue when implanted in vivo, without the need of any osteo-inducing scaffold. The implants exhibited bone-like morphology and mineralized matrix. The cells also secreted noncollagenous bone proteins such as osteopontin and osteocalcin. Osteocalcin secretion by the differentiated hESC-MSCs was detected in vitro as well.
The possibility of teratoma formation has been a major obstacle to the clinical use of hESCs. However, hESC-MSCs, unlike their parental line, did not induce teratoma formation, indicating phenotypic stability and highlighting their safety. Moreover, detection of human cells in the newly formed bone matrix, both by antibodies that specifically recognize human MHC class I and by in situ hybridization, confirmed the fate of the transplanted human cells as the bone-forming cells, which are able to further survive and remodel after transplantation. However, their capacity to regenerate impaired bone should be further examined in bone repair models. VEGF and bFGF, potent angiogenic stimulators, have been shown to play important roles in stimulating the restoration of blood flow at the site of a fracture, thus promoting the bone repair process. 31 In accordance with a previous study, 31 we have demonstrated that in the process of osteogenic differentiation, hESC-MSCs first secrete low levels of VEGF, and subsequently increased levels during terminal differentiation. Mineralization appearance increased during osteogenesis as well indicates that VEGF165 is maximally secreted at the late phase of osteogenic differentiation, when the matrix is mineralized. The correlation between secreted levels of VEGF and the degree of matrix mineralization suggests a direct role of VEGF-A in the mineralization process.
All blood vessels observed in the implants and adjacent to the newly formed bone contained un-nucleated erythrocytes, indicating that the vessels that invaded the implant anatomized the murine circulation, similarly to another Matrigel implant model, in which human bone marrow-derived MSCs were shown to secrete VEGF and to promote remodeling of newly formed human blood vessels in the implant. 39 In addition, we also observed reduction in the number of blood vessels similarly to this study, in which the durability of blood vessels as examined in vivo was found to decrease over time. 39 These findings suggest different kinetics of VEGF and other angiogenic factors in vivo over culture conditions.
The capability of MSCs to recruit murine blood vessels into the implant can be attributed to the secretion of angiogenic factors, including VEGF, bFGF, and platelet-derived growth factor, as was previously reported.36,39,40 In agreement, we have demonstrated in vitro VEGF secretion by hESC-MSCs, and capillary sprout formation originating from HUVEC spheroids that were stimulated with an osteogenic differentiated hESC-MSC-conditioned medium. The cell sprouting was significantly reduced by the use of ATP-competitive PDGFR, VEGF, and FGFR inhibitor, implying that implanted osteogenic hESC-MSCs participate in regulation of neovascularization in the newly formed bone via secretion of paracrine angiogenic factors.
To apply differentiated hESCs in clinically relevant scenarios, a large number of cells are required. The high expansion rate of hESC-MSCs and the fact that freeze and thaw cycles do not affect cell features enable high survival rates. In addition, it has been shown that hESC-MSCs may be more immunoprivileged than adult hMSCs, which is another advantage of their clinical application.13,16 The generation of induced pluripotent stem cells (iPSCs) from adult somatic cells by reprogramming is a significant scientific breakthrough. iPSCs hold a great promise, particularly because they provide an alternative source of functional MSCs for tissue repair, without raising the ethical and immunological issues integral to the use of hESCs. Graft rejection, another problem concerning hESCs, can be overcome with the use of iPSC-derived MSCs for autologous transplantations. Nevertheless, there are several challenges that need to be overcome before iPSCs and their derivatives can be used clinically, such as the low efficiency of iPSC generation and the safety of viral integration of transgenes.
In conclusion, the cell culture protocol presented herein affords a high yield of pure populations of hESC-MSCs with a high osteogenic capability. The osteoblasts derived from these hESC-MSCs were able to form a new mineralized and vascularized bone matrix in vivo. Once the functionality of these derived cells will be thoroughly examined in a bone repair model, the potential of the described method as a source of osteoblasts for bone tissue engineering may be realized.
Footnotes
Acknowledgments
This work was performed at the Berlin Family Laboratory for Stem Cell and Tissue Regeneration Research at the Sohnis and Forman Families Center for Stem Cell and Tissue Regeneration Research. We thank Dr. Shoham Shivtiel for assistance with in vivo experiments. This study was partially supported by the Technion R&D Foundation. J.I.-E. holds the Sylvia and Stanley Shirvan Chair in Cell and Tissue Regeneration Research at the Technion-Israel Institute of Technology.
Disclosure Statement
No competing financial interests exist.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.