
Abstract
In recent decades, stem cell therapy has been introduced as a novel therapeutic approach for patients suffering from bone disorders. Recently, menstrual blood has been identified as an easily accessible and recycled stem cell source. However, the osteogenic differentiation capacity of menstrual blood-derived stem cells (MenSCs) compared with other adult stem cells remained unsolved. The aim of this study was to investigate the osteogenic differentiation capacity of MenSCs compared to bone marrow-derived mesenchymal stem cells (BMSCs) in the presence of human platelet releasate (HPR). Our results showed that MenSCs were strongly positive for mesenchymal and negative for hematopoietic stem cell markers in a similar manner to BMSCs. In contrary to BMSCs, MenSCs exhibited marked expression of OCT-4 and a significantly higher proliferative capacity. Mineralization, as judged by alizarin red staining, was more pronounced in differentiated BMSCs than in differentiated MenSCs in an osteogenic medium fortified with fetal bovine serum (FBS). However, FBS substitution with HPR in a differentiation medium resulted in typical impact on intensity of MenSC mineralization. The results of semiquantitative reverse transcription–polymerase chain reaction showed comparable levels of parathyroid hormone receptor and osteocalcin transcripts in both types of differentiated stem cells in an HPR medium supplemented with osteogenic inducers. However, the upregulation level of alkaline phosphatase was relatively lower in differentiated MenSCs than that in differentiated BMSCs. We concluded that despite lower osteogenic differentiation capacity of MenSCs compared to BMSCs, substitution of FBS with HPR could equalize the osteogenic differentiation of MenSCs. Therefore, by taking advantage of osteogenic driving potential of HPR, MenSCs could be introduced as an apt and safe alternative to BMSCs for bone tissue-engineering purposes.
Introduction
Adult bone marrow-derived mesenchymal stem cells (BMSCs) are considered the gold standard stem cell source in bone regeneration, because they present no ethical problems of embryonic stem cells (ESCs), have bone generation ability in vitro and in vivo,2,3 and also exhibit immunosuppressive capabilities.4,5 However, problems such as less availability, invasive methods for sample collection, and lower proliferation capacity compared with ESCs limit BMSCs' applicability for research and clinical use.
Several studies have reported that menstrual blood (MB) contains a unique population of cells with properties similar to adult stem cells.6–8 Apparent evidence to support this assumption is high regenerative ability of human endometrium characterized by cyclic processes of cellular proliferation, differentiation, and shedding. 9 It was proposed that MB, in addition to the cells shed from the endometrium, contains circulating bone marrow mesenchymal stem cells, which contribute to endometrial regeneration. 10
MB-derived stem cells (MenSCs) display a long-term self-renewal capacity and much higher growth ability compared with umbilical cord-derived mesenchymal stem cells. In addition, the risk of karyotypic abnormalities in these cells is minimal.6–8,11 These characteristics as well as the ease of access and the possibility of cyclic sample collection make MB an appropriate stem cell resource for bone tissue engineering and regenerative medicine.
Recent studies present satisfying evidence about great potential of MenSCs to cardiomyogenesis and neurogenesis in vitro and in vivo.12–14 However, only little information is available about osteogenic differentiation potential of MenSCs.6,7 The study of osteogenic differentiation potential of MenSCs in comparison to BMSCs, as the well-established stem cell source in bone regeneration, will help researchers to realize whether these cells could find a place for future stem cell therapy of bone disease.
On the other hand, the main challenge is to introduce a robust protocol for osteoinduction of MenSCs. Therefore, finding a proper stimulus to trigger osteogenic differentiation of MenSCs may be promising for clinical therapy of bone diseases. Taking into consideration the platelet secretory factors, human platelet releasate (HPR) has been introduced as a suitable source of growth factors for appropriate expansion and differentiation of BMSCs.15–20 In addition, with regard to high cost and safety problems with fetal bovine serum (FBS) as a common supplement in stem cell culture, substitution of FBS with human-derived alternatives during human stem cell manipulation would be advantageous.
In this study, the differentiation capacity of MenSCs into an osteolineage has been compared to that of BMSCs. Furthermore, the effect of replacing FBS with HPR on osteogenic differentiation potential of MenSCs has been addressed.
Materials and Methods
Preparation of HPR
Five units of platelet-rich plasma (PRP) was provided from the Iranian Blood Transfusion Organization. HPR was prepared according to our protocol described previously. 17 In brief, PRP was centrifuged at 3000 g, 20°C for 10 min, to obtain platelet pellet. The platelet concentrates were pooled and incubated at room temperature for 30 min on a rotating platform to eliminate platelet aggregates. After cell counting using Sysmex K-1000 (Kobe), platelet density was adjusted to final concentration of 109/mL using phosphate-buffered saline (PBS). Next, 1 mL of prepared thrombin was added to 4 mL of platelet suspension and incubated for 1 h at room temperature to facilitate release of growth factors. The platelet releasate was centrifuged at 4000 g for 5 min to eliminate platelet membrane fragments. The supernatant was filtered through a 0.22-μm filter, divided into aliquots, and frozen at −20°C for future use.
The concentrations of epidermal growth factor (EGF), platelet-derived growth factor-AA (PDGF-AA), basic fibroblast growth factor (b-FGF), transforming growth factor-β (TGF- β), and tumor necrosis factor-α (TNF-α) in HPR were determined using enzyme-linked immunosorbent assay (ELISA) kits (R&D Systems), according to the manufacturers' instructions.
Isolation and culture of MenSCs and BMSCs
Donors for MB were selected from healthy volunteers at the age ranging between 22 and 30 years. All donors signed informed the consent form approved by the medical ethics committee of Avicenna Research Institute. Five milliliters of MB was collected using a sterile Diva cup (Diva International) in the second day of menstrual cycle. Collected MB was decanted into the tubes containing 2.5 μg/mL fungizone (Gibco), 100 μg/mL streptomycin, 100 U/mL penicillin, and 0.5 mM EDTA in PBS. Stem cells from MB were isolated as described earlier. 6 In brief, mononuclear cells were separated using Ficoll-Hypaque (GE-Healthcare) and washed. The cell pellet was suspended in complete Dulbecco's modified Eagle's medium-F12 (DMEM-F12) (Sigma-Aldrich) and cultured in polystyrene 75-cm2 tissue culture flasks. The flasks were maintained at 37°C in a humidified 5% CO2 incubator. After 1-to-2-day incubation, nonadherent cells were washed away and left behind the adherent cell population. The culture medium was refreshed every 3–4 days. When the cells reached 70% confluency, they were passaged using Trypsin/EDTA (Gibco). BMSCs were isolated from bone marrow specimens aspirated from donors aged from 19 to 32 years at the Bone Marrow Transplantation Center, Shariati Hospital, Tehran, Iran. Samples were obtained after getting signed informed consent according to guidelines of the Medical Ethics Committee, Ministry of Health, Iran. BMSCs were isolated according to the protocol we described before.17,21 All experiments were performed with cells at passage 2–4 from three to six donors.
Immunophenotyping of MenSCs and BMSCs
Aliquots of 105 cells/100 μL were incubated separately with fluorescein isothiocyanate (FITC)-conjugated mouse anti-human CD34 (clone 581/CD34; BD Pharmingen), CD38 (clone HIT2; BD Pharmingen), and CD45 (clone HI30; BD Pharmingen) or phycoerythrin (PE)-conjugated mouse anti human CD29 (clone 04-MAR; BD Pharmingen), CD73 (clone AD2; BD Pharmingen), CD44 (clone 515; BD Pharmingen), CD133 (clone TMP4; eBioscience), CD10 (clone HI10a; BD Pharmingen), CD146 (clone P1H12; BD Pharmingen), and CD105 (clone 43A3; BioLegend) for 40 min at 4°C. Cell surface staining of STRO1 was performed by an indirect method using sequential incubation with unconjugated mouse anti-human STRO1 (clone STRO-1; R&D Systems) and FITC-conjugated sheep anti-mouse polyclonal antibody (Aviccena Research Institute). To assess OCT-4 expression, cells were first fixed in 4% paraformaldehyde for 10 min. After being washed, cells were permeablized with 0.1% saponin and washed. Then, cells were treated with rabbit anti-human OCT-4 polyclonal antibody (Abcam) for 40 min followed by 30 min incubation with FITC-conjugated goat anti-rabbit Ig (Sigma). Afterward, all cell suspensions were fixed in 1% formaldehyde solution and examined using a flow cytometer (Partec) in reference to appropriate isotype controls (clone MOPC-21; BD Pharmingen).
Cell proliferation assay
MTT reduction test was employed to assess the proliferative capability of MenSCs in comparison with that of BMSCs as described elsewhere. 22 Briefly, cells were seeded at a concentration of 1.5×103 cells/well in 24-well plates, and their proliferative capacity was analyzed during the 10-day culture period. Optical densities were measured at 570 nm using an ELISA reader (Labsystem Multiskan), with background subtraction at 670 nm.
Osteogenic differentiation protocol
The osteogenic differentiation capacity of two experimental groups of cultured MenSCs or BMSCs in the presence of HPR or FBS was determined. For this purpose, both cell types were plated in six-well plates at a density of 3×103 cells/well. For osteogenic differentiation, cells at 70% confluency were cultured in 15% (v/v) FBS- or HPR-supplemented DMEM-F12 fortified with osteogenic agents, including 0.1 μM dexamethasone (DEX), 10 μM β-glycerophosphate (β-GP), and 50 μM ascorbic acid (Sigma-Aldrich). 23 The cells cultured in the medium supplemented with 15% FBS or HPR without osteogenic inducers served as control cells.
Cultures were continued for 14 days, and the media were changed twice a week. After 2 weeks, osteogenic properties were assessed in differentiated cells by calcium accumulation staining and mRNA expression of osteoblast-specific genes compared with those in control cells.
Alizarin red staining
Alizarin red staining was used to evaluate calcium accumulation in mineralized cells. Cultured cells were washed with PBS and fixed in 10% neutral buffered formalin for 20 min. Cells were then stained with alizarin red (Sigma-Aldrich) for 20 min. During this time, cells were checked microscopically for orange–red color development. As soon as the color appeared, cells were washed in deionized water to remove excess dye and then checked with an invert microscope (Olympus CKX41). Proportion of the positive surface area on 10 microscopic fields was determined separately for each cell population and averaged.
Semiquantitative reverse transcription–polymerase chain reaction analysis
Semiquantitative reverse transcription–polymerase chain reaction (RT-PCR) was performed to assess expression of a set of osteocyte-related genes, including bone alkaline phosphatase (ALP), osteocalcin (OCN), and parathormone receptor (PTHR). Human MG63 osteoblast cell line (National Cell Bank, Pasteur Institute) was used as a positive control. Briefly, total RNA was isolated from 106 cells by a standard RNA extraction protocol using RNA-bee (Biosite). Reverse transcription was performed using 2 μg purified RNA, 200 U/μL M-MuLV Reverse Transcriptase (Fermentase), 20 pM N6 Random-Hexamer (Cybergene), 5×RT buffer, and 20 pM dNTP Mix (Roche) in a thermocycler (Eppendorf) at 42°C for 60 min. Then, 1 μL of cDNA was admixed with 12.5 μL reaction master mix (Amplicon) and 1 μL of each primer (Table 1). After initial denaturation at 94°C for 3 min, PCR amplification was continued at 94°C for 30 s, annealing temperature (see Table 1) for 30 s, and 72°C for 30 s for total cycles of 35, and a final extension was performed at 72°C for 7 min. Each PCR was performed under linear conditions with β-actin used as an internal standard. The amplified DNA fragments were electrophoresed on a 1.5% agarose gel and visualized by an ultraviolet transilluminator (Uvitec-USA). For semiquantitative determination, gel images were analyzed using AlphaEase software (Genetic Technologies, Inc.). Values for all genes were normalized to that of the corresponding β-actin.
ALP, alkaline phosphatase, OCN, osteocalcin; PTHR, parathormone receptor.
Analysis of human leukocyte antigen-I and human leukocyte antigen-II expression
The expression of human leukocyte antigen (HLA)-I and HLA-II on osteoblast differentiated MenSCs and BMSCs was examined by flow cytometry using PE-conjugated mouse anti-human HLA-I (A, B, and C) and FITC-conjugated mouse anti-human HLA-DR monoclonal antibodies (clone G46-2.6 and G46-6; BD Pharmingen, respectively). The expression level in both cell populations was compared to that of undifferentiated cells.
Statistical analysis
All measurements were performed in triplicate. Data were expressed as a median with range, and results were analyzed using the nonparametric Mann–Whitney U-test. For all statistical analysis, SPSS 13 software was used, and a p-value<0.05 was considered significant.
Results
Concentration of growth factors in HPR
Mean levels of b-FGF and TNF-α in HPR were generally low. In contrast, HPR contained relatively high levels of EGF, PDGF-AA, and TGF-β reaching to 450±85 pg/mL, 1000±105 pg/mL, and 9000±182 pg/mL, respectively.
Characterization of isolated MenSCs
Morphological characteristics of MenSCs were compared with BMSCs at different time intervals postretrieval. Phase-contrast microscopy examination showed that MenSC morphology was more dependent upon cell density compared with BMSCs throughout the cell culture period. After the plating of 500 MenSCs/cm2, initial colony-forming units comprised of small and flat cells, whereas cells in BMSCs colonies had a fibroblastic shape with larger size and long cell protrusions at both poles. As the cells approached confluency, both cell types exhibited spindle-shaped, fibroblastic morphology and a homogeneous cell population with a characteristic nonhematopoietic phenotype (Fig. 1A). Also, MenSCs proliferated at a substantially faster rate than the control mesenchymal cell population derived from bone marrow (mean doubling time of 20.5 h and 40.1 h for MenSCs and BMSCs, respectively). The proliferation rate of MenSCs was approximately twofold greater than that belong to BMSCs as judged by the MTT assay (Fig. 1B).

Morphology and proliferation potential of menstrual blood-derived stem cells (MenSCs) in comparison to bone marrow-derived mesenchymal stem cells (BMSCs).
Flow cytometric analysis of expanded cells showed that MenSCs typically express surface antigens associated with mesenchymal stem cells such as CD44, CD29, CD73, CD105, and CD146, but they failed to express CD34, CD38, CD133, and CD45 (Fig. 2). MenSCs did not express STRO1 in a similar manner to BMSCs. However, marked expression of OCT-4 distinguished them from BMSCs.
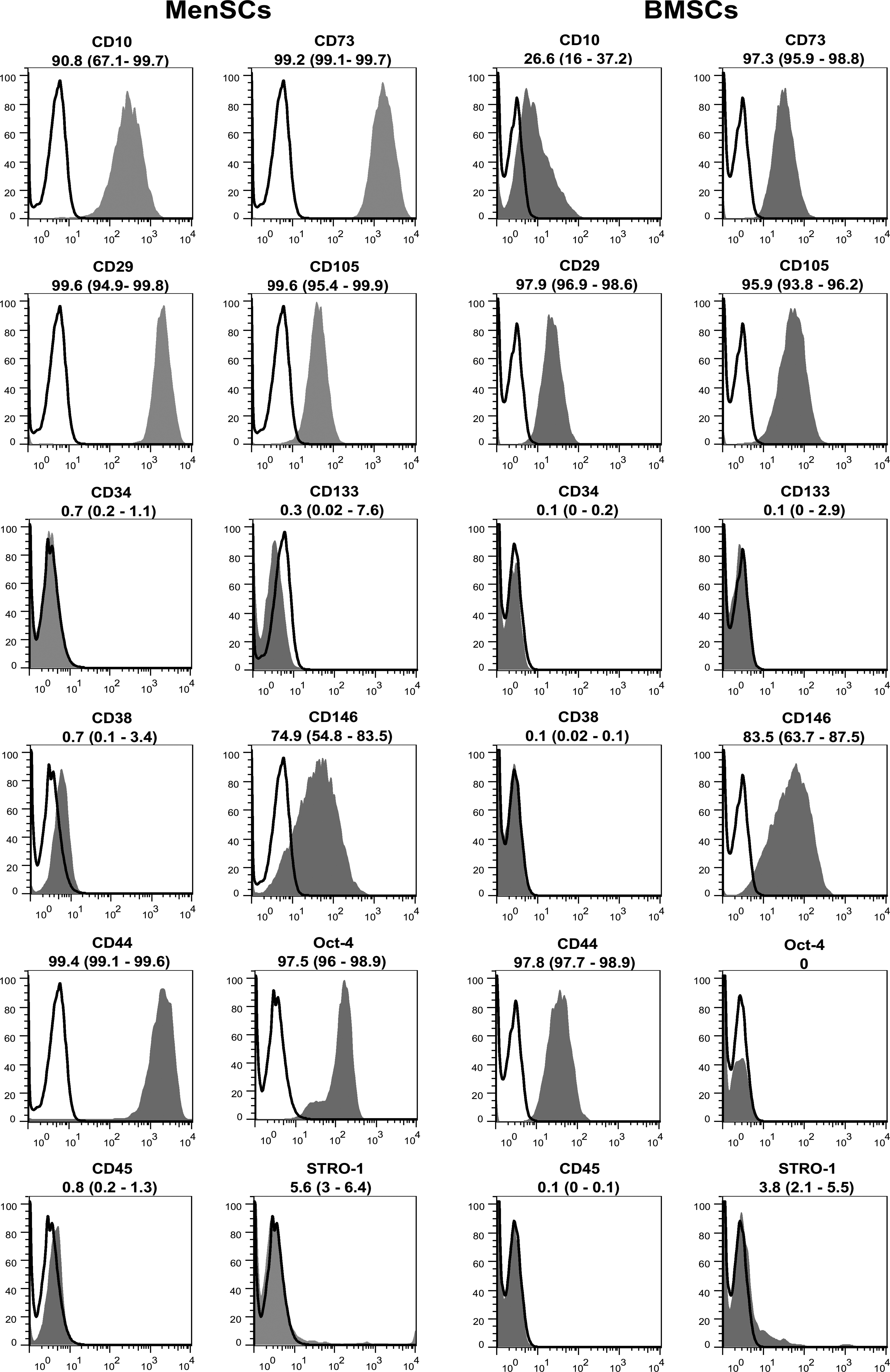
Immunophenotyping of MenSCs and BMSCs by flow cytometery. Representative histograms for CD markers are demonstrated (gray). The respective isotype control is shown as black line. The results are presented as median (range) of three to five independent experiments.
Evaluation of osteogenesis of MenSCs and BMSCs
Mineralization is a hallmark of end-stage osteoblastic differentiation, which, in the case of BMSCs, usually takes place in the presence of an FBS-supplemented medium fortified with osteogenic inducers comprising DEX, β-GP, and ascorbic acid. 23 In the first step, we investigated whether this medium is potent enough to induce mineralization of MenSCs. As shown in Figure 3A, in the presence of FBS and osteogenic inducers, both MenSCs and BMSCs reached the mineralization step. However, the mineralization potential of MenSCs was much lower than that of BMSCs (MenSCs: 5%–10%, BMSCs: 60%–70%). Expectedly, omission of osteogenic inducers resulted in no mineralization in both stem cell types.
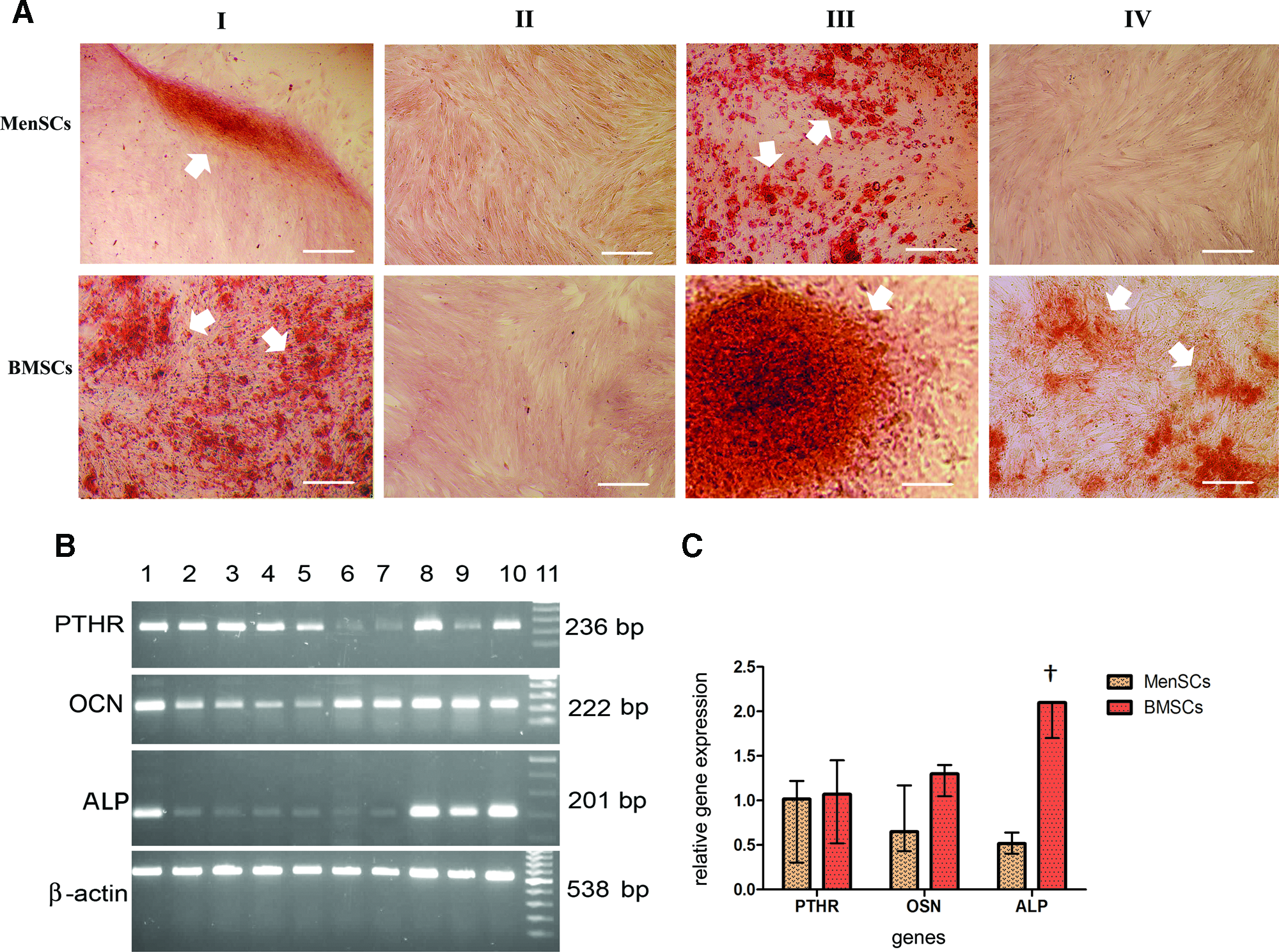
Cytochemical and molecular evaluation of MenSCs and BMSCs differentiated into osteoblasts. The accumulation of calcium deposits (arrowheads) in differentiated cells in the presence of conventional osteogenic medium fortified with fetal bovine serum (FBS)
The influence of FBS substitution with HPR on the osteogenic differentiation of MenSCs compared to BMSCs was investigated thereafter. Interestingly, substitution of FBS with HPR in the presence of osteogenic factors had a strong effect on the formation of nodule-like structures with calcium deposits in differentiated MenSCs (positive cells: 70%–80%). The positive effect of HPR on BMSC differentiation potential was appreciable either with or without osteogenic agents. However, osteogenic promoting efficiency of HPR in the presence of osteogenic inducers was more pronounced compared to when they were omitted (85%–95% and 15%–20%, respectively). Although in all culture conditions the level of calcium deposits in BMSCs was greater than that in MenSCs, it is noticeable that the effect of FBS substitution with HPR in mineralization of MenSCs was more pronounced compared to that of BMSCs.
Based on the results obtained by alizarin red staining, the expression pattern of osteogenic genes in BMSCs and MenSCs differentiated in the presence of HPR and osteogenic inducers was then investigated in reference to untreated cells (FBS-driven cells) using semiquantitative RT-PCR (Fig. 3B, 3C). There was a significant interindividual variation in the expression pattern of OCN and PTHR (Fig. 3B). However, the median upregulation level of these genes was not significantly different between MenSCs and BMSCs (PTHR: p<0.7, OCN: p<0.055) (Fig. 3C). In the case of ALP expression, a clear and significant difference between differentiated MenSCs and BMSCs was noted. The upregulation level of ALP in differentiated MenSCs was significantly lower compared to that of BMSCs (p<0.001) (Fig. 3C).
Expression of HLA antigens
Flow cytometric analysis showed that MenSCs expressed high levels of HLA-I, but failed to express HLA-II. As shown in Figure 4, osteogenic differentiation had no significant effect on their level of HLA expression. BMSCs express HLA-II neither before differentiation nor after osteogenic differentiation. A small percentage of BMSCs (4%–6%) expressed HLA-I, which did not change after differentiation.
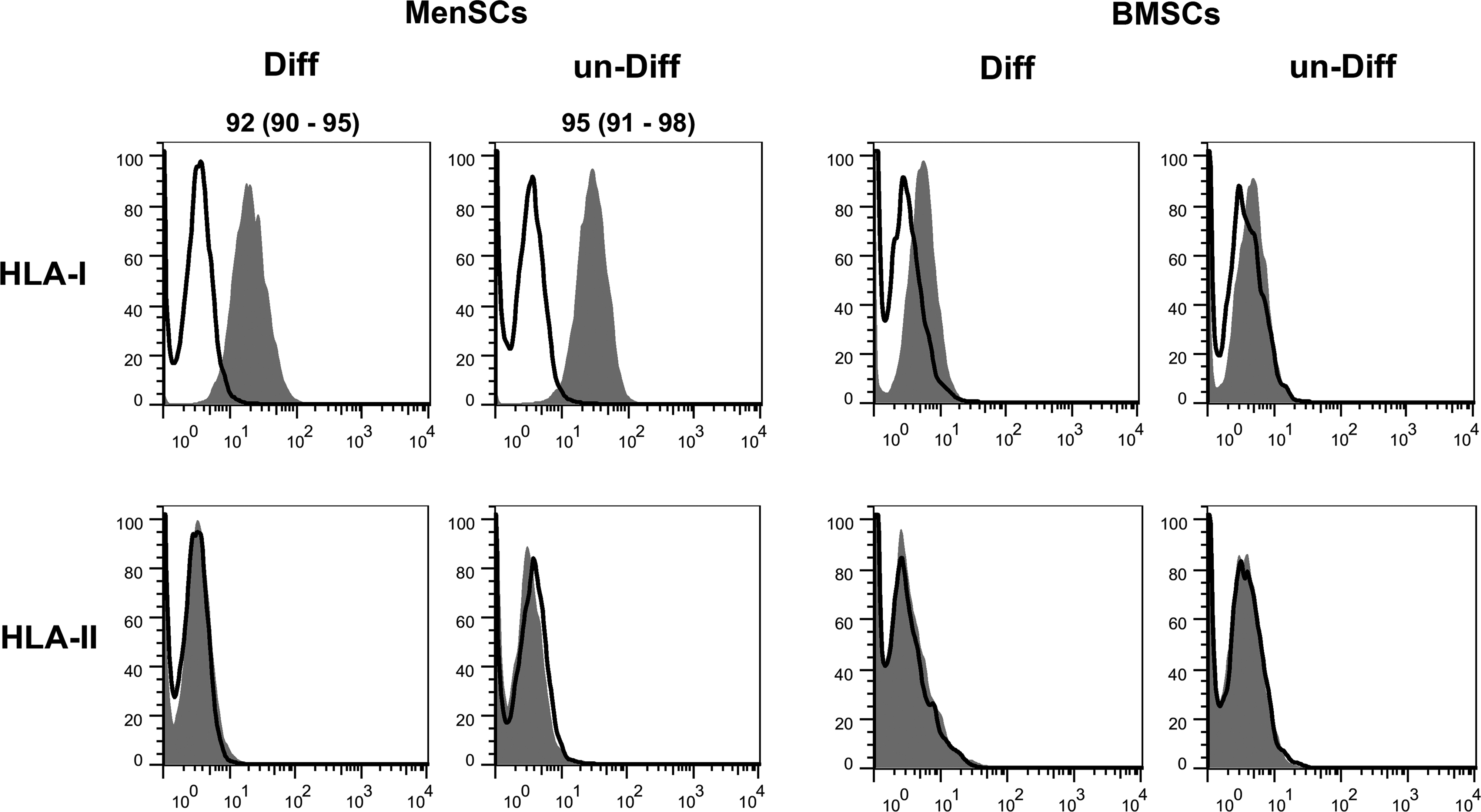
Human leukocyte antigen (HLA)-I and HLA-II expression by differentiated MenSCs and BMSCs. Expression of HLA-I and HLA-II in the cells was analyzed by flow cytometry (white background, isotype control; gray background, specific antibody) prior (un-Diff) or after (Diff) osteogenic differentiation. The results are presented as median (range) of three independent experiments. The expression values>3% are indicated.
Discussion
In few recent years, such problems with well-known stem cell sources as low availability, painful access, or limited proliferative ability have impelled scientists to take advantages of MenSCs in cell therapy and regenerative medicine. 24 The satisfying results of recent animal studies demonstrating improvement of some diseases such as cardiac disorders, 12 Duchenne muscular dystrophy, 25 and nervous system disorders 26 hold promise for feasibility of MenSC-based therapy of human diseases. However, relatively few data are available on the MenSC potential to generate bone tissue, particularly compared with other known stem cells like BMSCs. In this study, considerable differences were beheld between MenSCs and BMSCs regarding the proliferative capacity, immunophenotype, and osteogenic differentiation potential. We also demonstrated that substitution of FBS with HPR has a great impact on differentiation potential of MenSCs into the osteocyte lineage.
There are limited data available on the entity of MenSCs. These cells showed morphological variations throughout the cell culture period. It seemed that their morphology was influenced by cell density, and cell–cell interaction had a significant effect on morphology of MenSCs. To further characterize the nature of these cells, we evaluated expression of both mesenchymal and ESC markers. In a similar way to the CD146+ PDGF-Rβ+ subpopulation of endometrial stromal cells,27–29 MenSCs appeared to possess some markers of mesenchymal stem cells such as CD29, CD44, CD73, and CD105. Interestingly, a large percentage of MenSCs, similar to BMSCs, expressed CD146. This finding attests perception of others suggested that the CD146+ PDGF-Rβ+ subpopulation of endometrial stromal cells is shed in MB. 27 Regardless of these findings, MenSCs cannot be simply classified as mesenchymal stem cells due to much higher growth capacity than BMSCs and expression of ESC marker, OCT-4, that is not common in mesenchymal stem cells. In this manner, it is reasonable to assume that these cells represent a population of stem cells with a unique proliferation and differentiation ability.
Here, we showed that MenSCs could be committed to the osteogenic lineage, although with a relatively lower capacity compared to BMSCs. There are inconsistent reports about differentiation potential of endometrial stem cells into osteogenic lineages. While the experiments of Dimitrov et al. to induce osteogenic differentiation of the cultured CD29+CD73+CD90+ endometrial stem cells in a well-known osteogenic medium were not successful, 30 others presented primitive results implying positive osteodifferentiation of MenSCs or the CD146+PDGF-Rβ+ subpopualtion of endometrial cells.7,28 Although this discrepancy can be attributed to donor differences, sample source, sampling day, or stem cell isolation method, a comparative study between these cells and other known stem cells, as we showed here, is required to get robust decision on the osteogenic differentiation capacity of MenSCs.
Recently, the use of HPR for expansion and differentiation of stem cells has been suggested as a promising FBS substitute.15–20 Based on the present study, although MenSCs' commitment into the osteolineage in a well-known osteogenic medium was much lower than that of BMSCs, and FBS replacement with HPR in a differentiation medium presented a typical impact on intensity of MenSC mineralization. It is notable that the effect of FBS substitution by HPR on MenSCs was more pronounced compared to that of BMSCs. The promoting effect of HPR on osteogenic differentiation was more highlighted when some calcium deposits were visible in BMSCs in the absence of osteogenic inducers.
This differentiation-promoting effect may be due to the high concentration of a cocktail of natural growth factors present in HPR. According to our results, HPR contains remarkable concentration of TGF-β and PDGF and very low levels of b-FGF and TNF-α.
Concentration of PDGFs in a platelet lysate has also been investigated by several other laboratories. Because of different preparation methods, reported levels of growth factors by others could not be directly compared with our results. However, platelet lysates obtained by the other groups also contained high concentrations of TGF-β and PDGF in contrary to low levels of b-FGF or TNF-α.15,31–33 More importantly, TGF-β as the major constituent of HPR plays a key role in early osteogenic differentiation, but is not potent per se to induce terminal osteogenic differentiation as well.34–36 Therefore, osteogenic inducer-independent action of HPR on differentiation of BMSCs toward an osteogenic lineage, as we showed here, implies that osteogenic potential of HPR may due in part, but not restricted to the distinct presence of TGF-β.
With the pronounced effect of HPR and osteogenic inducers cocktail on osteogenic differentiation, we next corroborated the cytochemical observations with the RT-PCR analysis of osteogenic-specific markers such as OCN, PTHR, and ALP. 37 Our results clearly showed that PTHR and OCN transcripts were significantly upregulated in differentiated MenSCs compared with the baseline condition. It is noticeable that there was no considerable difference between MenSCs and BMSCs in this aspect. However, the upregulation level of ALP in differentiated MenSCs was significantly less than that of BMSCs. Considering the key role of ALP in bone mineralization, 38 we can attribute different mineralization levels in differentiated MenSCs and BMSCs to the different expression pattern of ALP in these stem cell populations.
One major potential clinical application of MenSCs is autologous or allogeneic cell transplantation with the aim of bone tissue regeneration. The immune system plays an important role in the success of any allogeneic stem cell transplantation, but the most important for transplants is the HLA. In consistent with other reports, 7 we showed typical expression of HLA-I contrary with no expression of HLA-II by MenSCs. Although in line with our results, many researchers reported the absence of HLA-II on BMSCs, and there is a general inconsistency in the case of HLA-I. It seems that the level of HLA-I expression on BMSCs depends on donor and cell culture conditions.39,40 In this study, we showed that osteogenic differentiation in presence of HPR was not inherently immunogenic and did not change the levels of HLA-I and HLA-II expression on both cells.
Taken together, MenSCs are a unique cell population with higher proliferation rate and lower osteogenic differentiation ability compared to BMSCs. So, to gain broader insight into the differences of MenSCs and BMSCs, more studies on cellular signaling pathways involved in propagation and differentiation process of MenSCs are required. On the other hand, considering the efficiency of HPR in osteoblast differentiation of MenSCs, the discussion on the substantial effect of FBS substitution by HPR during propagation and differentiation of stem cells has now been encouraged.
In conclusion, although lower osteogenic differentiation capacity of MenSCs compared to BMSCs could be viewed as a limiting factor for bone regenerative medicine, FBS substitution by HPR in the culture medium of MenSCs not only substantially improves this property, but also represents a major novel step toward safe and applied stem cell therapy of bone diseases.
Footnotes
Acknowledgments
This work was supported by a research grant from the Iranian Council of Stem Cell Technology and Tehran University of Medical Sciences. The authors would like to thank Dr. Kourosh Kamali for assistance in statistical analysis.
Disclosure Statement
No competing financial interests exist.