
Abstract
Mesenchymal stem cells (MSCs) from adult exhibit self-renewal and multilineage differentiation capacities, making the MSCs promising candidates for cell therapy and tissue engineering. Although bone marrow (BM) is the most universal source of MSCs, other tissues may also contain MSCs. Peripheral blood (PB), in particular, arises as the most attractive source of MSCs due to easy accessibility and noninvasive procedure. However, it is not certain that PB-MSCs have the equal biological characteristics to those of BM-MSCs. The purpose of this study was to compare the biological characteristics between BM-MSCs and PB-MSCs. We adopted granulocyte colony-stimulating factor combined with CXCR4 antagonist AMD3100 to stimulate MSCs to release into blood circulation of the rats. PB-MSCs were obtained from mobilized PB and expanded in long-term culture. BM-MSCs were isolated from the femur and tibia medullary canal of the same rats by density gradient centrifugation. After cell expansion in vitro, cell surface markers and multipotentiality analysis were performed to identify MSCs. Apoptosis resistance to H2O2-induced apoptosis, proliferation kinetics, cellular senescence, and karyotype analysis were measured to compare the biological characteristics of PB-MSCs and BM-MSCs. PB-MSCs with the typical adherent fibroblast-like morphology were similar to that of BM-MSCs. Both PB-MSCs and BM-MSCs were positive for CD44 and CD90, and negative for CD34 and CD45. They both exhibited trilineage differentiation potential and expressed lineage-specific genes. Although the BM-MSCs showed stronger osteogenic and adipogenic differentiation, PB-MSCs displayed a more chondrogenic capacity. Further, BM-MSCs have greater proliferation ability. Apoptosis resistance and cellular senescence were similar in MSCs derived from both sources. The results of our study demonstrate that PB-MSCs have similar biological characteristics to those of BM-MSCs despite certain minor differences, suggesting PB as a possible alternative source for MSCs.
Introduction
Currently, it has been well accepted that MSCs are mainly derived from bone marrow (BM). However, the MSCs from BM have the problems of marrow harvesting, the limited number of cells, and the tendency to senescence with aging. Therefore, it is necessary to develop other sources to obtain MSCs. At present, in addition to BM, MSCs could be yielded from almost all the tissues of the body, such as adipose, periosteum, synovial membrane, skin, cord blood, umbilical cord, amniotic fluid, cartilage, and even menstrual blood.6–11
MSCs derived from blood have been confirmed and successfully isolated and expanded in cord blood12,13 or fetal blood,14,15 although with a low number and frequency. Compared with cord blood or fetal blood, MSCs from peripheral blood (PB) of adult have no ethical and legal controversy and have a more clinical significance for achieving autologous transplantation. 16 MSCs derived from PB became a promising candidate for the above purposes because of its minimally invasive harvesting, easy accessibility, and the availability of autologous transplantation in the clinical application.
The main problem remained is that PB-MSCs have a very low colony formation under physical or nonmobilized conditions in adult. The existence of MSCs in PB has always been controversial, and it is still uncertain that whether the stem cells could be isolated from PB or not. Limited successes have been reported on the isolation of MSCs from circulation blood. Even with the administration of mobilizing agents, such as granulocyte colony-stimulating factor (GCSF), the PB-MSCs have very limited expanding potential and become senescent 20–25 days after isolating. 17 Some researchers reported that GCSF combined with CXCR4 antagonist AMD3100, which was also known as a new and rapid mobilization agent, could lead to a massive release of hematopoietic stem cells (HSCs). 18 Could MSCs be mobilized from BM or other relative tissues into circulation in the same way as HSC mobilization? If so, whether the adherent cells from mobilized circulation have the same biological characteristics as MSCs derived from BM? We hypothesized that PB-MSCs could be isolated and expanded in long-term culture from mobilized circulation with GCSF plus AMD3100. The adherent spindle cells were also identified as MSCs by morphological and functional properties. In this study, the biological characteristics of MSCs derived from BM and PB were compared for similarities and differences in the following seven respects: (1) morphology, (2) immunophenotypic characterization, (3) proliferation capacity, (4) differentiation ability, (5) antiapoptotic ability, (6) cellular senescence, and (7) karyotyping analysis.
Materials and Methods
Mobilization, harvest, and culture of MSCs from PB and BM
The experimental procedures were approved by the Animal Ethics Committee of Peking University Health Science Center. Sprague Dawley rats, about 200 g, aged 8–12 weeks, were used for this study. Rats were first administered with GCSF (100 μg/kg; Qilu Pharmaceutical Co. Ltd.) by intraperitoneal injection once daily for 5 days; on the 6th day, CXCR4 antagonist AMD3100 (5 mg/kg i.p.; Sigma) was administrated. One hour later, 4–5 mL of blood was sterilely harvested with tube containing 300 U/mL heparin through the abdominal aorta under anesthesia.
The blood samples were diluted with 1:1 phosphate-buffered saline (PBS). Mononuclear cells (MNCs) were isolated with density gradient centrifugation (Lymphoprep Ficoll 1.077 g/mL; Dingguo Biotechnology), following the supplier's instructions. After centrifugation, cells at the gradient interface were collected, washed with PBS, and passed through a 100-mm mesh filter, and resuspended in complete culture medium (minimum essential medium alpha medium with 10% fetal bovine serum [FBS], 2.2 g NaHCO3, 100 U/mL penicillin, 100 μg/mL streptomycin, 25 ng/mL amphotericin B, and 2 mM L-glutamine) at the density of 2.5×106 cells/mL, incubated at 37°C with 5% humidified CO2 incubator. After 3 days, the nonadherent cells were removed and fresh medium was replaced. The medium was changed every 3–4 days. Approximately after 10 days, the cells were harvested by 0.25% trypsin/0.1% EDTA for passage when reaching 90% confluence, and then replicated. The cells were subcultured onward in the same manner up to passage 10 (P10) at the later passage.
The lower limbs of the same rats were collected, and then sterilized in 70% ethanol for 5 min. Under sterile conditions, the muscle and connective tissue were cleaned from both the tibia and the femur by scraping the diaphysis of the bone, and then the BM-MSCs were harvested by flushing with PBS supplemented with 1% penicillin/streptomycin. The cell suspension was filtered through a 100-mm filter mesh to remove any bone spicules or muscle and cell clumps. MNCs were isolated with density gradient centrifugation, and the isolation and culture of cells were the same as PB-MSCs.
Surface marker identification
FACS analysis of surface markers was performed on cultured MSCs at P3. Cells aliquots of P3 (1×106 cells) were trypsinized with 0.25% trypsin/EDTA; washed twice; suspended in 500 μL PBS; stained with fluorescein isothiocyanate (FITC)-, Alexa FLUOR488-, PE-Cy5-, and PE-Cy5-conjugated monoclonal antibodies against rat CD44, CD90 (Thy-1), CD34, and CD45 (Biolegend); and incubated on ice for 30 min. For CD105, collected cells were incubated with anti-CD105 primary Ab (Santa Cruz) for 60 min, and then labeled with FITC-conjugated rabbit anti-goat secondary Ab for 30 min. A homologous isotype control IgG was included in each experiment to eliminate nonspecific staining. Cells were then fixed for 30 min in flow buffer fixative (1% paraformaldehyde, 2% FBS, and 0.1% sodium azide). At least 10,000 cells were collected for further analysis on an FACS Vantage cytometer using Cell Quest software (BD).
Proliferation analysis
The cultured P3 MSCs were used for cell proliferation assay with Cell Counting Kit-8 (CCK8) proliferation assay kit (Dojindo), following manual's instructions. One hundred microliters of cell suspension (2000 cells/well) was dispensed in a 96-well plate. After incubated for 7 days, 10 μL of the CCK8 solution was added to each well of the plate, and incubated for 2 h; the absorbance at 450 nm was measured using a microplate reader. The growth curve of MSCs from PB and BM was depicted by OD value of each group at various times.
In vitro differentiation
To evaluate the multilineage capacity of PB-MSCs and BM-MSCs, cells were differentiated toward the adipogenic, osteogenic, and chondrogenic lineages by using lineage-specific induction factors. For osteogenesis, the cultured P3 MSCs were grown to 80% confluence and then induced with osteogenic medium (OM) containing dexamethasone, β-glycerophosphate, and ascorbate. After 3 weeks, alkaline phosphatase (ALP) activity and Alizarin Red staining were used to evaluate osteogenic matrix production. To test for adipogenesis, when MSCs were grown to 80% confluence, the cells were treated with adipogenic medium (AM) with 0.5 μM 1-methyl-3 isobutylxanthine, 1 μM dexamethasone, 10 μg/mL insulin, and 100 μM indomethacin for 3 weeks. To visualize lipid droplets, the cells were fixed with 4% formalin and stained with Oil Red O. For chondrogenic differentiation, P3 MSCs were cultured in a pelleted micromass culture consisting of serum-free medium with chondrogenic medium (CM): 100× ITS (Sigma), 1 mM pyruvate, 0.17 mM ascorbate, 0.1 μM dexamethasone, 0.35 mM proline, and 10 ng/mL TGFβ3. After 3 weeks, HE staining, Toluidine blue metachromasia, and immunohistochemical staining for type II collagen (COL II) were carried out to evaluate the chondrogenic differentiation and the matrix production.
Quantitative real-time polymerase chain reaction
The expression of specific markers for extracellular matrix and transcription factors was assessed by real-time polymerase chain reaction (PCR) at 21 days after induction for quantitating differentiation capability of MSCs from PB and BM. Total RNA from samples was extracted using RNAVzol reagent (Vigrous) according to the manufacturer's instructions. Concentration of each RNA sample was measured using UV spectrophotometer. The integrity of RNA samples was also assessed by agarose gel electrophoresis. Two micrograms of total RNA was subjected to a reverse transcription using the Superscript First Strand Synthesis System (Invitrogen). Quantitative real-time PCR was performed with 15 μL of reaction detection system consisting of 7.5 μL 2× SYBR Green PCR Master Mix (Toyobo, Japan), 1 μL of cDNA product, 1 μL of each primer, and 5.5 μL of nuclease-free water. Primer sets were designed using Oligo 6 primer analysis software, as followed (5′–3′): Runx2: forward, AACCCACGAATGCACTATCCA, reverse, CTTCCATCAGCGTCAACACCA; OPN: forward, CTTCCAAGCAACTCCAATG, reverse, CTCAGTCCGTAAGCCAAG; ALP: forward, GACTGTGGTTACTGCTGAT, reverse, GCCTGGTAGTTGTTGTGA; Col I: forward, GAAGACTCGATCCTCCATAG, reverse, ATGCTGAATCTAGAGAGGAG; peroxisome proliferator activated receptor γ (PPARγ): forward, TGGAGCCTAAGTTTGAGTTTGC, reverse, TGACAATCTGCCTGAGGTCTG; lipoprotein lipase (LPL): forward, GAGATTTCTCTGTATGGCACA, reverse, CTGCAGATGAGAAACTTTCTC; SOX9: forward, AGGAAGCTGGCAGACCAGTA, reverse, ACGAAGGGTCTCTTCTCGCT; GAG: forward, CCACTGGAGAGGACTGCGTAG, reverse, GGTCTGTGCAAGTGATTCGAG; COL II: forward, CACCGCTAACGTCCAGATGAC, reverse, GGAAGGCGTGAGGTCTTCTGT; GAPDH: forward, ATGGGGAAGGTGAAGGTCG, reverse, TAAAAGCAGCCCTGGTGACC. All the PCRs were performed under following conditions: 2 min at 50°C, 10 min at 95°C, and 40 cycles of 15 s at 95°C and 1 min at 60°C by employing ABI Prism 7000 Sequence Detection System software. The specificity of amplicons was verified by melting curve analysis. Levels of mRNA were normalized against GAPDH using the comparative cycle threshold (Ct) method.
H2O2-induced apoptosis
H2O2-induced apoptosis model was performed to compare the ability of antiapoptosis of MSCs from BM and PB at P3. Apoptosis was induced by H2O2 at different concentrations and was measured with Annexin V-FITC/PI apoptosis detection kit (Biosea Biotechnology Co. Ltd.) by FACS analysis and TUNEL staining using in situ cell death detection kit (Roche) under laser scanning confocal microscope. 19 Cells were treated with different concentrations of H2O2 after 1 h; they were harvested, washed, and suspended in binding buffer containing Annexin V-FITC for 15 min and PI for 5 min at appropriate concentrations according to the instructions, and then analyzed by FACScan FCM. The membrane phosphatidylserine residues of apoptotic cells would be labeled with Annexin V. The percentage of Annexin V–positive cells was used as a measurement of apoptosis. Apoptotic cells were visualized by the TUNEL according to the manufacturer's instructions.
Cellular senescence
Both of the cultured cells at P3 and P10 were prepared for senescence-associated-β-gal (SA-β-gal) staining to determine cellar senescence. SA-β-gal activity was performed to evaluate the long-term culture MSCs from BM and PB, as previously described. 20 In brief, cells were washed, fixed in 2% formaldehyde/0.2% glutaraldehyde, and incubated overnight at 37°C with freshly prepared β-gal staining solution (1 mg/mL X-gal, 40 mM citric acid/sodium phosphate [pH 6.0], 5 mM potassium ferrocyanide, 5 mM potassium ferricyanide, 150 mM NaCl, and 2 mM MgCl2). The slides were viewed under bright field at a magnification of ×100. Cellular senescence was determined by the detection of blue precipitate over the cells.
Karyotype analysis
Karyotyping of MSCs was performed at early and later passages to check whether the cells were diploid in long-term culture. BM-MSCs and PB-MSCs at P3, P5, and P10 cultured near confluence were subjected to metaphase mitotic arrest using 100 μg/mL colchicine overnight at 37°C incubator. Cultures were trypsinized, suspended for hypotonic shock in 0.075 M hypotonic potassium for 15 min at 37°C, and fixed with 3:1 methanol/acetic acid fixative. Metaphase preparations were made by dropping the fixed cell suspension onto precleaned slides and stained to visualize G-banding using 2% trypsin and Giemsa stain. At least 20 metaphase spreads were analyzed from each MSC preparation. Images were acquired using Nikon microscope.
Statistical analysis
All data were calculated as mean±standard deviation. The differences between two groups were analyzed by using the Student's t-test or ANOVA by SPSS16.0. p-Value<0.05 was considered statistically significant.
Results
Isolation and culture of MSCs from mobilized PB and BM
Adherent cells were obtained from mobilized PB by administration of GCSF plus AMD3100. The MNCs were isolated from PB and BM with lymphocyte separation medium and density gradient centrifugation. A few sparse adherent spindle-shaped cells appeared (Fig. 1A, F) after 3–5 days. The primary cultures became 50%–70% confluent after 7–9 days, and showed a typical plastic adherence and a typical fibroblast-like morphology under phase-contrast microscope (Fig. 1B, G). The morphology of PB-MSCs was similar to BM-MSCs. The culture was nearly confluent and treated with 0.25% trypsin/EDTA for passage after 10 days. The growth of PB-MSCs was slower ∼2 days than that of BM-MSCs before subculture. A purified population of MSCs from both sources could be obtained at P3 after the initiation of culture, showing a homogeneous performance. It took about 3–4 days for each passage until P10. With the gradual passage, MSCs at the late passage exhibited larger shape, cells of the typical spindle shape decreased, grew slower with a prolonged passage time, and an increase of the granular debris in the culture medium and cytoplasm (Fig. 1C–E, H–J).
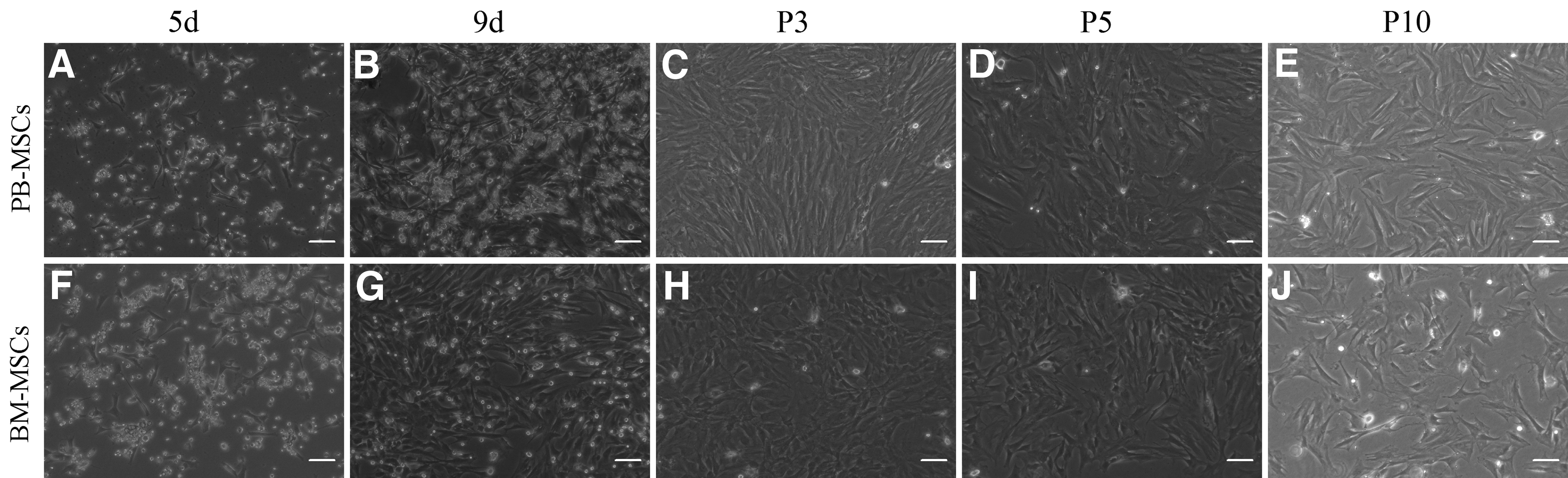
Morphologic change of adherent cells from mobilized peripheral blood–mesenchymal stem cells (PB-MSCs) and bone marrow (BM)–MSCs at early and later passages. A few sparse spindle-shaped adherent cells after initial plating derived from PB
Phenotypic characteristics of MSCs
For further characteristics of PB-MSCs, we chose CD44, CD90, CD105, CD34, and CD45 for identifying surface markers of MSCs. PB-MSCs at P3 had a high expression of the typical MSC marker proteins CD44 (96.36±5.87) and CD90 (98.87±0.28), a low expression of CD105 (20.86±0.54), but a negative expression of hematopoietic progenitor cell marker CD34 (1.09±0.49) and leukocyte common antigen CD45 (1.27±0.57). MSCs from BM were also positive for CD44 (99.13±1.14), CD90 (98.21±0.32), and CD105 (22.92±3.87), and negative for CD34 (1.13±0.69) and CD45 (0.95±0.13) (Fig. 2A). There was no significant difference between the MSCs derived from two different sources (Fig. 2B).
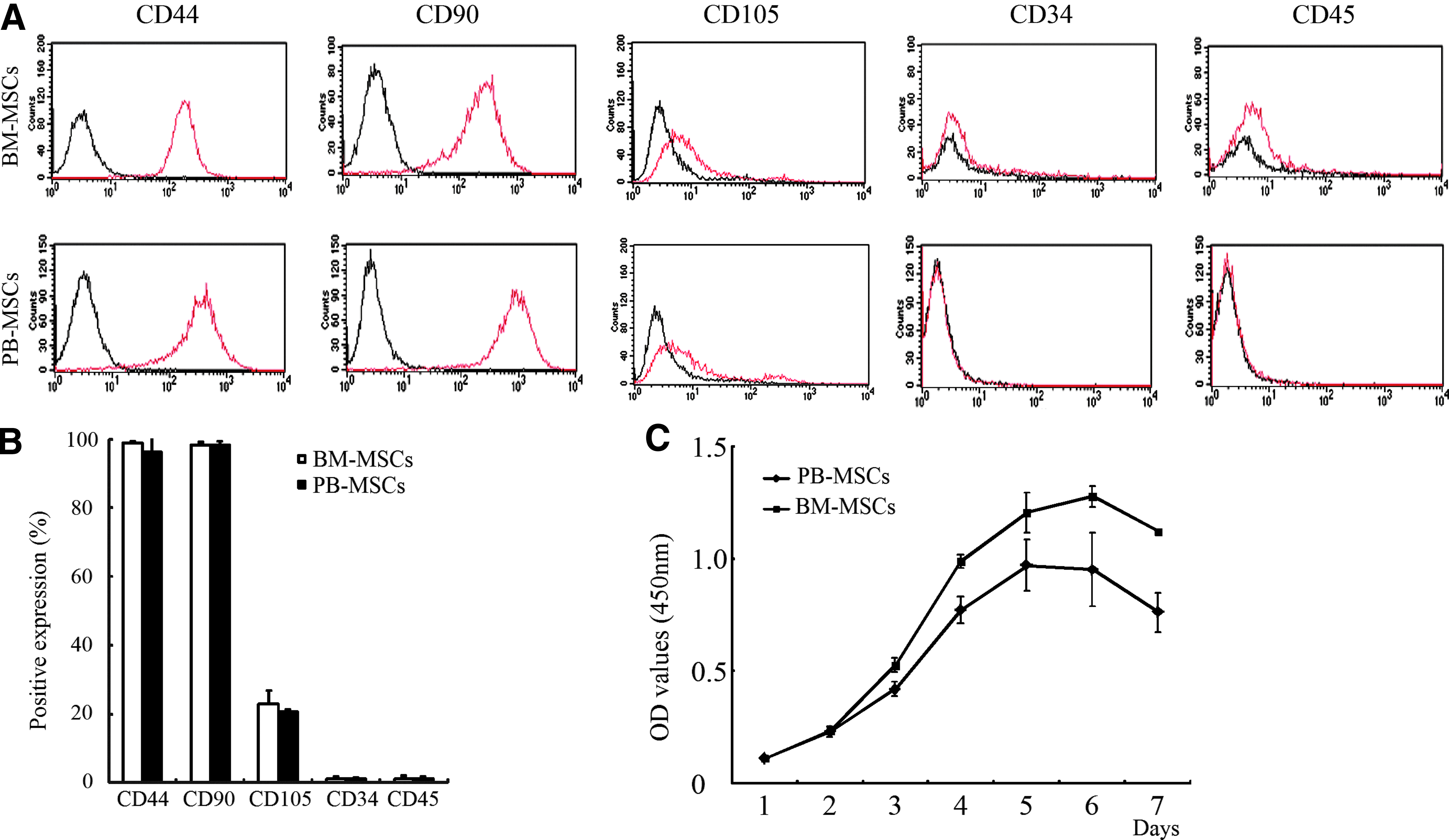
Comparison of immunophenotypic characteristics of MSCs at P3 derived from PB and BM as analyzed by FCM. BM-MSCs and PB-MSCs were both positive for CD44 and CD90, relatively low for CD105, and negative for CD34 and CD45
Growth characteristics of MSCs from PB and BM
The growth curve of MSCs from PB and BM was depicted by CCK8 assay, which both showing typical “S-shaped curve” (Fig. 2C). After seeding for 1–2 days, both MSCs were in lag phase and then entered into exponential phase between 2 and 5 days in BM-MSC group in contrast to 2–6 days in PB-MSC group, and finally reached a plateau or stationary phase. Significant differences were observed between two groups after 2 days, demonstrating that BM-MSCs possessed a stronger proliferation activity than PB-MSCs.
Multilineage differentiation potential
MSCs from the two sources possessed the trilineage differentiation capability; however, the difference of the differentiation intensity existed in respective lineage.
For osteogenesis, after cultured in OM for ∼1 week, MSCs underwent a change in cellular morphology: from spindle shape to a more polygonal appearance accompanied with a gradual aggregation growth of the cell and an increase in ALP activity (Fig. 3A3, A4). Formation of mineralized calcium nodules was observed by Alizarin Red staining; the area of positive staining was larger in BM-MSCs than that in PB-MSCs, showing that the amount of matrix mineralization of BM-MSCs is more than that of PB-MSCs (Fig. 3A1, A2). PCR illustrated that osteogenic-specific markers Runx2, OPN, ALP, and COL I of BM-MSCs were stronger than those of PB-MSCs (Fig. 3B–E). Those significant differences suggested that BM-MSCs have more osteogenic potential than PB-MSCs.

In vitro trilineage differentiation potential of rat MSCs derived from BM and mobilized PB at P3. Osteogenesis was evaluated by Alizarin Red (A1, A2) and alkaline phosphatase (ALP) activity staining (A3, A4) of MSCs from two sources 21 days after induction
Adipogenic differentiation was demonstrated by accumulation of cytoplasmic lipid vacuoles with Oil Red O staining in the MSCs derived from both sources (Fig. 3F1, F2). The expression of adipogenic-specific genes, such as PPARγ and LPL (Fig. 3G, H), was stronger in BM-MSCs than those in PB-MSCs, showing a more adipogenic potential of BM-MSCs compared with PB-MSCs.
The expression of cartilage-specific matrix GAG and COL II was determined by HE (Fig. 3I1, I2), Toluidine blue (Fig. 3I3, I4), and immunohistochemical staining (Fig. 3I5, I6) to assess chondrogenesis. On the further test for chondrogenesis, PB-MSCs have stronger chondrogenic differentiation according to the expression of cartilage-specific genes, such as SOX9, COL II, and GAG (Fig. 3J–L).
Ability of antiapoptosis induced by H2O2
MSCs at P3 were treated with H2O2 of various concentrations and examined by Annexin V-FITC/PI and TUNEL assay. With the increasing concentration of H2O2 the proportion of apoptotic cells increased, which showed that the apoptosis of rat PB-MSCs and BM-MSCs induced by H2O2 was dose dependent. Apoptosis was detected at concentrations as low as 0.2 mM of H2O2 in BM-MSC group while no positive cells was detected with TUNEL assay after 1 h of treatment with H2O2 (Fig. 4A). Dose-dependent increase in apoptosis was observed up to 0.8 mM of H2O2. At 0.4 mM of H2O2 concentration or greater, a large portion of the cells detached from the culture plates and most of the cells became necrotic. At H2O2 concentrations of 0.8 mM, the percentage of Annexin V–positive cells increased rapidly. Dose-dependent increase in apoptosis was also observed by Annexin V-FITC/PI FACS analysis, which was consistent with the results of TUNEL assay. There were no statistical differences at various H2O2 concentrations between the MSCs from two sources (Fig. 4B).

TUNEL
Cellular senescence
With the passage in long-term culture in vitro, MSCs gradually lead to replicative senescence and showed some typical morphological changes: cells became much larger with irregular and flat shape, and nuclei became more circumscribed in phase-contrast microscopy. The cytoplasm began to be granular with many inclusions and aggradations, and the cell debris increased (Fig. 1). At pH 6, β-gal activity was associated with senescence in vitro. One theory suggests that β-gal activity could be associated with the RAS pathway and with lysosomal dysfunction. The result of our study is consistent with the reports of Wagner. 21 β-Gal-positive cells increased with passages (Fig. 5A). No significant difference was observed in PB-MSCs and BM-MSCs (Fig. 5B).

Cellular senescence examined by β-gal staining. β-Gal-positive cells increased at P10
Karyotype analysis
G-band karyotype of MSCs at early and later passages was examined to assess their genetic stability in in vitro long-term culture. Chromosomal analysis was performed on BM-MSCs and PB-MSCs at P3, P5, and P10. No chromosomal abnormality was observed up to P10. The karyotype images of BM-MSCs and PB-MSCs at P10 are shown in Figure 6, indicating normal chromosomes of rat (2n=42).
Karyotype analysis of BM-MSCs
Discussion
BM is the most common source of MSCs, but obtaining BM is an invasive operation for patients. Besides, the limited number, transformation, or tendency of senescence of BM-MSCs in in vitro expansion make them difficult for clinical application. And they are likely to lose the differentiation ability and proliferation potential while aging. Therefore, the search for alternative sources of MSCs is of significant value. Compared with BM-MSCs, PB-MSCs are more easy to obtain with minimally invasive procedures, and also benefit for autologous MSC transplantation in clinical application. These advantages make PB-MSCs an excellent candidate for tissue regeneration.
It is well accepted that PB has essentially replaced BM as the main source of HSCs for autologous and allogeneic transplantation, with the advantages of a less-invasive collection method, faster engraftment, enhanced immune reconstitution, shorter hospitalization, and reduced cost. 22 Whether MSCs are available in PB remains controversial. Many studies have shown that under certain pathological conditions, MSCs in PB could be significantly increased, such as injury, inflammation, cancer, dysmetabolism, autoimmune diseases, and so on.23–27 There is growing evidence that MSCs could enter into the peripheral circulation under the right circumstances. We assume that MSCs could be mobilized into PB in the similar way as the mobilized HSCs. In this study, adherent fibroblast-like cells were successfully isolated with GCSF and AMD3100 in rat. Then, the cells were identified as MSCs by the means of adherent ability, morphology, surface markers, and differentiation potential according to the standard of the International Society for Cellular Therapy. 28 BM is mainly comprised of two types of stem cells: HSCs and non-HSCs (mainly MSCs). Stem cells resided in BM niche rely on the interaction between the ligand on BM microenvironment and the receptor on cell surface. 29 Emerging evidence indicates that stem cells from BM migrate to peripheral circulation in respond to tissue injury, also hematopoietic stem and progenitor cells are themselves mobile and are able to repopulate ectopic niches and contribute more directly to inflammatory responses in the periphery. 30 It was well known that HSCs could mobilize into PB with the administration of cytokines, such as GCSF. But recent evidence indicates that HSCs are able to mobilize into the periphery at low frequency even during steady state.
MSCs are primarily resided in BM niche microenvironment, which controls the self-renewal and the fate of MSCs and benefits stem cell expansion and maintenance. Stem cell niche is located proximate to trabecular or cortical bone (the endosteal compartment) and three dimensionally intertwined with blood vessels (the perivascular compartment). 31 BM niche is a very complicated microenvironment for MSCs that includes osteoblasts, osteoclasts, stromal cells, extracellular proteins, and signaling molecules. 32 The regulation of remaining and releasing MSCs consists of a series of molecular events. The releasing regulation of MSCs—migration out of BM into PB—involves a complex interaction among chemokines, adhesion molecules, cytokines, proteolytic enzymes, hematopoietic cells, and stromal cells. The mechanism of MSC mobilization is to disrupt the interaction between an array of cell surface adhesion molecules (receptors) expressed on MSCs and their ligands expressed on the BM niche, which allows the egress of MSCs from BM. 33
Similar to HSCs in BM, SDF1/CXCR4 is an essential chemokine/receptor for MSC engraftment, recruitment, homing, and migration, which can control MSCs' release from the BM into the circulation. 34 GCSF is the most common drug to mobilize PB in clinical procedures. The mobilization mechanism is to promote HSCs to release from the BM into PB by blocking the SDF1/CXCR4 chemotactic interaction. 35 The efficacy of GCSF alone for mobilization of stem cellss for auto-HSC transplantation results in fewer CD34+cell yields. 36 Unfortunately, 5%–30% of patients do not respond to these agents. 37 Studies have shown that the CXCR4 inhibitor AMD3100 in conjunction with GCSF significantly increased HSC yield when compared with GCSF used alone. 38 The mechanism of GCSF-induced mobilization is to interrupt the connection of MSCs and their niche to stimulate MSC release into PB. Further, MSCs were also considered as stromal cells supporting HSCs. The mobilization of HSCs may lead to the lease of MSCs. Pitchford et al. 39 reported that MSC mobilization was detected by CXCR4 antagonist pretreated with VEGF via VEGFR2 in mice models, indicating that HSCs and MSCs have their own specific receptors. The mobilization of stem cells in BM involves a complex interplay between stromal cells, cytokines, chemokines, and adhesion molecules; though details of this regulatory process are poorly understood. The exact mechanism of MSC mobilization needs to be further explored. Further, culture method is another critical factor affecting the isolation of MSCs from mobilized PB. The methods include classical plastic adherence, erythrocyte lysis buffer, lymphocyte separation medium combined with density gradient centrifugation, immunomagnetic beads select, adding growth factors or some protein, and so on. In this study, we isolated PB-MSCs by using lymphocyte separation medium combined with density gradient centrifugation, which is associated with MSCs' own adhesion properties. In addition, more homogeneous and pure MSCs were obtained by passage (Fig. 1).
H2O2-induced apoptosis model in vitro simulates the microenvironment of ischemia-reperfusion injury that MSCs would encounter in vivo. The central role for oxidative stress in apoptosis is strongly supported by the ability of various cellular antioxidants to block apoptosis, and targeted therapies aiming at generating ROS might be proven effective. Apoptosis rates were detected at various H2O2 concentrations. The survival capacity of MSCs in host tissues in the conditions of ischemia or ischemic reperfusion is another important characteristic to be considered. The use of graft approach of MSCs is limited by the fact that most of the transplanted MSCs are readily lost, which is potentially triggered by the in vivo ischemic or ischemia-reperfusion environment. In our study, we investigated the antiapoptosis ability of these MSCs toward oxidative stress induced by hydrogen. ROS is a major regulator of the cell metabolism and mediators of apoptosis. However, oxidative stress occurs when the antioxidant capacity of the tissue is inadequate for removing ROS, such as in osteoarthritis or rheumatoid arthritis, all of the above would result in cartilage degradation. Studies on H2O2, which is one of the main ROS in cartilage tissues, have revealed its contribution to cartilage degradation by inducing chondrocyte apoptosis. 40
In this study, the biologic properties of MSCs from BM and PB were identified and compared. The data clearly demonstrated significant similarities between them, such as morphology, surface markers, trilineage differentiation ability, antiapoptotic capacity, senescence properties, and karyotype analysis. However, the distinct differences are also observed with regard to growth kinetics and differentiation potential. BM-MSCs showed stronger osteogenic and adipogenic potential while demonstrated weaker chondrogenic differentiation than PB-MSCs. Whereas, the growth kinetics of BM-MSCs confirmed greater in contrast to those of PB-MSCs.
It is generally believed that the mobilized PB-MSCs are mainly released from BM under the appropriate conditions, but why the differences exist in biological characteristics between BM-MSCs and PB-MSCs? In addition to BM, MSCs are present in almost all tissues of the body. Zhao et al. observed that MSCs derived from mobilized PB may come from other tissues besides BM, which also contains MSCs. 41 MSCs derived from various tissues display different cytokine expression profile and with the tissues' specific biological characteristics. MSCs derived from different tissues share very similar characteristics and minor differences probably due to their microenvironment of the origin. 42 Recently, Crisan and coworkers concluded that MSCs are a subpopulation of vessel-lining pericytes that may enter into circulation to relocate or retain vessel homeostasis by reacting to tissue damage or the stimulation of different cytokine.43,44 Still, there is no definite answer on the real origin of MSCs in PB. The stem cells in blood or a variety of tissues may be related with embryonic development and the model of differentiation, and the movement of stem cells between primary hematopoietic sites occurs naturally throughout life. 45
Limitations
Our main interest in this study was to isolate PB-MSCs of rats with a new combined administration of GCSF and AMD3100, and compare their biological characteristics at early passage with BM-MSCs. But we do not know the characteristics of PB-MSCs in long-term culture and their effectiveness of repairing damaged tissues in vivo, which need to be further studied.
Conclusions
In summary, PB-MSCs were successfully isolated from mobilized PB of rats, showing the typical adherent fibroblast-like morphology. Both PB-MSCs and BM-MSCs reveal the similar biological characteristics despite of some minor differences in proliferation and differentiation. The results of our study demonstrate that the PB-MSCs have the similar biological characteristics to BM-MSCs, and suggest that PB may have a promising potential for tissue regeneration as an alternative source for MSCs.
Footnotes
Acknowledgments
The authors thank Mrs. Ling Han and Hui Liu for excellent assistance with karyotype analysis. This study was supported by the National Natural Science Foundation of China (No. 30471783), the Key Doctoral Foundation of Chinese Education Commission (No. 20030001009), and the Beijing Natural Science Foundation (No. 7052046).
Disclosure Statement
The authors declared no potential conflicts of interest.