
Abstract
Extensive efforts have been made to prepare osteoconductive collagen gels for the regeneration of normal bone and the pathological examination of diseased bone; however, collagen gels are often plagued by limited controllability of their rigidity and mineral deposition. This study reports a simple but efficient strategy that tunes the mechanical properties of, and apatite formation in, collagen gels by incorporating hydrolyzable poly(lactic-co-glycolic acid) (PLGA) microparticles within the gels. The PLGA microparticles are associated with the collagen fibrils and increased both the gel's elasticity and rigidity while minimally influencing its permeability. As compared with pure collagen gels, the PLGA microparticle-filled collagen gels, termed PLGA-Col hydrogels, significantly enhanced the deposition of apatite-like minerals within the gels when incubated in simulated body fluid or encapsulated with mesenchymal stem cells (MSCs) undergoing osteogenic differentiation. Finally, PLGA-Col hydrogels mineralized by differentiated MSCs led to an enhanced formation of bone-like tissues within the hydrogels. Overall, the PLGA-Col hydrogel system developed in this study will serve to improve the quality of osteoconductive matrices for both fundamental and clinical studies that are relevant to bone repair, regeneration, and pathogenesis.
Introduction
Collagen gel systems, which are chemically and structurally similar to natural extracellular matrices, have been extensively studied for the induction of mineralization, because of their advantageous properties that support cellular proliferation and osteogenic differentiation in a three-dimensional (3D) matrix.6–11 Nonetheless, collagen gels often encounter limited mineral deposition because of an absence of chemical cues for promoting apatite formation. 12 In addition, collagen gels encapsulating calcium-secreting cells tend to shrink significantly due to a lack of matrix rigidity and, thus, an increased susceptibility to the contractions exerted by cells. 13
Several strategies have been developed to improve either the mechanical rigidity or the mineral deposition of collagen gels. First, collagen molecules are often cross-linked with various reactive chemicals, including glutaraldehyde, succinic anhydride, and acyl azide, to improve mechanical rigidity and stability13,14; however, many of these cross-linking agents have cytotoxic properties that are detrimental to cellular viability, and do not allow in situ cell encapsulations. These approaches, therefore, commonly require additional processing for the purification and formation of interconnected micropores through which cells are incorporated. There are certain approaches that cross-link collagen molecules with enzymes such as lysyl oxidase via glycation, but these approaches do not provide broad controllability of mechanical rigidity as compared with synthetic cross-linking molecules.15,16 Separately, collagen molecules are often combined with anionically charged polymers, such as glycosaminoglycans and poly(acrylic acid), as they are reported to facilitate calcium deposition within a matrix; however, the incorporation of these anionic molecules within collagen gels requires the use of cytotoxic chemical linkers or additional processing steps, which prevent in situ cell encapsulation.17–19 In addition, these charged molecules do not contribute toward the enhancement of the gel's mechanical rigidity.
In this study, we hypothesized that the incorporation of anionically charged microparticles in a collagen gel would allow us to improve both the mechanical rigidity and apatite formation of the collagen gels incubated in conditions that promote mineralization. To examine this hypothesis, we used poly(lactic-co-glycolic acid) (PLGA) microspheres as model particles that act as a filler to bridge interconnected collagen fibrils and present charged carboxylic groups in physiological conditions via hydrolysis.20,21 The PLGA microparticles' role in the mechanics of the collagen gels was examined by measuring the gels' elastic moduli. The permeability of the PLGA microparticle-filled collagen gels was also evaluated by measuring the diffusion coefficients of dextran molecules within the gel matrices, in order to ensure that the particles minimally affect gel permeability and subsequent cell viability. In parallel, the effects of the PLGA microparticles on mineralization were examined by incubating cell-free hydrogel matrices in SBF, and separately activating the calcium secretion of the mesenchymal stem cells (MSCs) encapsulated in the hydrogels via osteogenic differentiation. Finally, MSC-encapsulating hydrogels incubated in osteogenic differentiation media were further implanted on chicken chorioallantoic membranes (CAMs) to evaluate the effects of the matrices' osteoconductivity on bone-like tissue formation.
Materials and Methods
Microparticle preparation
PLGA (50/50 lactic acid/glycolic acid) microparticles were synthesized using a double emulsion/evaporation process. First, a water-in-oil emulsion was created by adding water to dichloromethane (DCM; Fisher Scientific) containing 100 mg/mL PLGA, followed by sonication for 20 s (Sonic Dismembrator; Fisher Scientific). The mixture was immediately vortexed for 20 s in a 1% poly(vinyl alcohol) (PVA; Sigma-Aldrich) aqueous solution, creating an oil-in-water emulsion. Next, the mixture was added to a 0.1% PVA aqueous solution and vigorously stirred for 3 h, allowing the DCM to evaporate. The resulting microparticles were repeatedly centrifuged and resuspended in deionized water to remove residual PVA. The microparticles were then sterilized by resuspending them in a mixture of ethanol and water with a volumetric ratio of 70 to 30, respectively. Finally, the microparticle solution was freeze dried (FreeZone6; Labconco) to attain a powder.
Preparation of collagen hydrogels encapsulating PLGA microparticles (PLGA-Col hydrogels)
PLGA-Col hydrogels were prepared through an in situ physical association of collagen molecules premixed with PLGA microparticles. First, sterile solutions of 1 M sodium hydroxide (Sigma-Aldrich) and reconstitution buffer, consisting of 0.2 M sodium carbonate (Sigma-Aldrich), 0.2 M 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (Sigma-Aldrich), and 0.4 M sodium hydroxide, were prepared. Next, Dulbecco's modified Eagle's medium (DMEM; Gibco) containing 0, 1, or 10 mg of PLGA microparticles were mixed with 0.5 mL of collagen solution. The collagen concentration in the solution was kept constant at 3 mg/mL collagen I (PureCol; Advanced BioMatrix). Then, reconstitution buffer and 1 M sodium hydroxide solutions were added to the pregel mixtures, resulting in PLGA microparticle concentrations of 0, 1, and 10 mg/mL. Finally, volumes of 100 μL of the mixtures were poured into 96-well plates, covering the entire surface area of each well. The mixtures were then incubated at 37°C for 30 min to complete the gel formation. All procedures were performed under sterile conditions.
Rheological characterization of hydrogels
The viscoelastic properties of the pure collagen gels and the PLGA-Col hydrogels were characterized by measuring the elastic and loss moduli of the hydrogels subject to a low-amplitude oscillatory shear deformation at varied frequencies. The samples were prepared within the space between the walls of a rheometer's cup and bob (Bohlin; CD-50). Then, the samples were oscillated at a stress of 1.1 Pa to deform the gel in the linear viscoelastic region. The frequency was varied from 1 to 10 Hz. The storage moduli, loss moduli, and dynamic viscosities were recorded at various frequencies.
Analysis of molecular diffusivity within the hydrogels using fluorescence recovery after photobleaching
Molecular diffusivity within the hydrogels was evaluated using fluorescence recovery after photobleaching (FRAP). Gels were prepared using the same procedures as just described, except that fluorescein isothiocyanate-dextran (FITC-dex, MW 40,000; Sigma) was incorporated into the gels. FRAP experiments were performed using a Ziess LSM 710 confocal microscope equipped with a ZEN FRAP module. A small area of 0.01 mm2 was exposed to a high-intensity light pulse, which photobleached the FITC-dex emission at 525 nm. The subsequent recovery of emissions intensity of FITC-dex was monitored over an extended period of time. Molecular diffusivities in three to four different hydrogels were examined per condition. Six to eight different areas were examined per sample.
Analysis of biomineralization of hydrogels induced by modified SBF
Collagen gels encapsulating varied concentrations of PLGA microparticles were incubated in modified SBF (mSBF) to induce mineralization within the gels. First, a sterile mSBF solution consisting of 141 mM sodium chloride (Sigma-Aldrich), 0.4 mM potassium chloride (Sigma-Aldrich), 0.5 mM magnesium sulfate (Fisher Scientific), 1 mM magnesium chloride (Fisher Scientific), 4.2 mM sodium bicarbonate (Sigma-Aldrich), 5 mM calcium chloride (Sigma-Aldrich), and 2 mM potassium phosphate monobasic monohydrate (EMD Chemicals) was prepared. PLGA-Col hydrogels prepared with PLGA concentrations of 0, 1, and 10 mg/mL were incubated in mSBF at 37°C for 2 weeks, while exchanging the solution with fresh mSBF every 2 days. Throughout the duration of the experiment, changes in the transparency of the gels were monitored through images captured using an S6E stereomicroscope (Leica) linked with a D-Lux E Camera (Leica).
The overall concentrations of calcium and phosphate formed within the gels were determined using colorimetric calcium assays. Briefly, the hydrogels were enzymatically degraded using 1000 U/mL of type I collagenase (Sigma-Aldrich) at 37°C for 30 min. The calcium and phosphate concentrations of the resulting collagen solutions were determined using a Quanti Chrom™ Calcium Assay kit (Bio Assay Systems) and a Malachite Green Phosphate Assay kit (Bio Assay Systems), following the procedures described in the manufacturer's instructions. Briefly, the degraded collagen solutions were mixed with a phenolsulphonephthalein dye that forms a stable blue-colored complex with free calcium in solution, and, in parallel, mixed with a malachite green solution that forms a stable green-colored complex with free phosphate in solution. The intensity of the colors was measured at 612 nm and 650 nm for calcium and phosphate, respectively, using a spectrophotometer (Bioteck Synergy HT). In addition, standard samples with known concentrations of calcium and phosphate were prepared to construct two separate calibration curves of their absorbance values.
Analysis of biomineralization induced by osteogenic differentiation of MSCs
Mouse MSCs (D1, ATCC) between passages 21 and 25 were encapsulated in PLGA-Col hydrogels prepared with PLGA concentrations of 0 or 10 mg/mL. The cell concentration was kept constant at a density of 1 million/mL. Briefly, concentrated cell suspensions were mixed with pregel solutions consisting of collagen and PLGA microparticles. Then, the pH of the pregel solutions was increased to 7, in order to activate gel formation. The mixtures were further incubated at 37°C for 30 min, before adding DMEM supplemented with 10% fetal bovine serum (FBS; Gibco) and 1% penicillin/streptomycin (P/S; Gibco). The resulting MSC-encapsulating gels were incubated in osteogenic differentiation media: DMEM supplemented with 10% FBS, 1% P/S, 1 μM dexamethasone (Sigma-Aldrich), 50 μg/mL L-ascorbic acid (Sigma-Aldrich), and 10 mM β-glycerol phosphate (Fluka). Osteogenic differentiation media were exchanged every 2 days with fresh media over 1 week.
Analysis of biominerals using scanning electron microscopy equipped with energy dispersive X-ray spectroscopy
Cross-sections of hydrogels incubated in mSBF and MSC-encapsulating hydrogels incubated in osteogenic differentiation media were examined using scanning electron microscopy–energy dispersive X-ray spectroscopy (SEM-EDS). The collagen gels encapsulated with MSCs were fixed with a 10% neutral buffered formalin solution (NBF, 4% formaldehyde in phosphate-buffered saline; Sigma-Aldrich). All samples were flash frozen using liquid nitrogen, fractured, and lyophilized for imaging. Cross-sections of samples were coated with palladium and imaged at an electron intensity of 25 kV (JOEL 6060LV SEM). EDS (Oxford Instruments ISIS EDS System) was used to examine the chemical compositions of the minerals deposited within the cross-sections. Control samples, which were not exposed to conditions that induce biomineralization, were also examined. Mineral formation in the MSC-encapsulating hydrogels over a 10 day period was analyzed by imaging freeze-fractured cross-sections using SEM. In addition, freeze-fractured cross-sections of PLGA-Col hydrogel samples incubated in DMEM were also imaged using SEM imaging over 2 weeks to examine the degradation of the PLGA microspheres.
X-ray diffraction analysis
The minerals formed within the hydrogels were further characterized with X-ray diffraction (XRD). For XRD analysis, the hydrogels were washed in deionized water for 1 h to remove excess sodium chloride, and, subsequently, lyophilized. Each lyophilized hydrogel was mounted on a flat zero background sample holder. XRD patterns were collected on a Bruker D5005 (Bruker Germany), using CuKα radiation (λ=1.5406 Å) at 40 kV and 40 mA. The 2θ region of 25°–40° was analyzed at a scanning rate of 1°/min. The reference pattern of apatite was reconstructed from known structural data. 22
Hydrogel implantation on CAM
Fertilized chicken eggs (Hy-Line W-36) were obtained from the University of Illinois Poultry Farm. Initially, all the eggs were incubated for 7 days, placed horizontally inside an incubator at 37°C and 65% humidity. After the initial incubation period, a hole with a diameter of 2 cm was made at the top of the egg shells by carefully puncturing and removing shell fragments. The holes were covered with tape (Scotch) and incubated overnight to ensure survival after initial shell opening. Next, collagen gels were placed on the CAMs of individual embryos. Before implantation, MSC-encapsulating collagen and PLGA-Col hydrogels were incubated in osteogenic differentiation media for 1 week. The cell density in the gels was kept constant at 1 million/mL. For the PLGA-Col hydrogels, the PLGA microparticle concentration was kept constant at 10 mg/mL. The openings were then covered with tape, and the eggs were incubated at 37°C for 7 days. All procedures were performed under sterile conditions.
Histological analysis
After incubation for 7 days, the CAMs were fixed using 10% NBF and excised using suture scissors. The explants were then embedded in paraffin, sectioned, and stained with hematoxylin and eosin (H&E) or an antibody to collagen I, following standard histological procedures. In parallel, samples were cryo-sectioned and stained with von Kossa reagents following standard histological procedures. In addition, calcium deposition within the MSC-encapsulating collagen and PLGA-Col hydrogels was examined over a 10 day period by cryo-sectioning the hydrogels and staining the cross-sections with von Kossa reagents.
Analysis of hydrogel rigidity
The mechanical rigidity of the collagen and PLGA-Col hydrogels was examined by measuring the compressive elastic moduli of the MSC-encapsulating hydrogels after incubation in osteogenic differentiation media and those which were first incubated in osteogenic differentiation media and subsequently implanted on CAMs. The hydrogels implanted on CAMs were collected by excising the CAMs over which the gels were placed. The compressive elastic moduli of the hydrogels were measured using a mechanical tester (Insight; MTS System) by compressing the hydrogels at a rate of 1 mm/min. The elastic moduli were calculated from the linear slope of the stress versus the strain curve for the first 10% strain. Additionally, the shear moduli were calculated from the linear slope of the stress versus–(v–v−2) for the first 10% strain (Γ), where v=1- (Γ).
Statistical analysis
Student's t-tests were used to determine significant differences between groups, with p<0.05.
Results
Preparation and characterization of hydrogels
The PLGA microparticle-filled collagen hydrogels, termed PLGA-Col hydrogels, were prepared by mixing varying concentrations of premade PLGA microparticles with a collagen solution and subsequently activating physical associations of collagen fibrils. The diameters of the spherical PLGA microparticles, prepared using a double emulsion process, ranged from 1 to 30 μm (Supplementary Fig. S1; Supplementary Data are available online at www.liebertpub.com/tea). The PLGA microparticles, prepared in a powder form via lyophilization, were readily dispersed in pregelled collagen solutions. The subsequent change in pH of the collagen solution activated the physical association between collagen fibrils to form a hydrogel (Fig. 1a–c). The PLGA microspheres did not interrupt the formation of collagen fibrils at a concentration of 10 mg/mL when compared with the pure collagen gels. Rather, the PLGA microspheres associated and interlinked with the collagen fibers, according to microscopic images captured by SEM (Fig. 1c). Further increasing the PLGA concentration greater than 30 mg/mL inhibited the gel formation. The PLGA microparticles encapsulated in collagen gels gradually degraded in DMEM over 2 weeks (Supplementary Fig. S2).

Characterizations of microstructure, stiffness, and permeability of collagen and poly(lactic-co-glycolic acid) (PLGA)-Col hydrogels.
The PLGA microparticles incorporated in the collagen gels at concentrations between 0 and 10 mg/mL significantly changed the viscoelastic properties of the gels while minimally affecting gel permeability. According to low-amplitude oscillatory shear measurements, the pure collagen gels behaved similar to a liquid, characterized by a linear dependency of the storage moduli on the frequency and an independency of the dynamic viscosity on the frequency (Fig. 1d and Supplementary Fig. S3). In contrast, the hydrogels encapsulating 10 mg/mL PLGA microparticles presented a solid-like elastic behavior represented by an independency of storage moduli on the frequency and a steeper dependency of the dynamic viscosity on the frequency (Fig. 1d and Supplementary Fig. S2). In addition, the difference between the loss and elastic modulus at a low frequency of 1 Hz was smaller for the PLGA-Col hydrogels. Therefore, the phase angle of the PLGA-Col hydrogels, quantified through the inverse tangent of the ratio between loss moduli and storage moduli, was smaller than the pure collagen hydrogels—the phase angle of the PLGA-Col hydrogels at 1 Hz was 17°, while that of the pure collagen hydrogels was 26°. In addition, the storage moduli of the PLGA-Col hydrogels measured at 1 Hz was five-fold higher than the pure collagen gels—an increase from 4.0±2.5 to 21.3±1.2 Pa.
These changes in gel rigidity minimally affected the gel's permeability, which was evaluated by measuring the diffusivity of fluorescently labeled dextran. According to FRAP measurements, there was a statistically insignificant difference between conditions for the fluorescence recovery in the photo-bleached region of the hydrogels (Fig. 1e). The fluorescence recovery was fit to a double exponent relation (Eq. 1),
where I is the FITC-dex emission intensity, IE is the plateau intensity, I1 is the first mobile fraction constant, I2 is the second mobile fraction constant, t1 is the first half-life time constant, t2 is the second half-life time constant, and t is the time. There was a minimal difference in t1 and t2 values between the pure collagen gel and the PLGA-Col hydrogel (Table 1).
The PLGA concentration in the PLGA-Col hydrogels was kept constant at 10 mg/mL.
PLGA, poly(lactic-co-glycolic acid).
Biomineralization of hydrogels
The PLGA-Col hydrogels with a PLGA particle concentration of 10 mg/mL exhibited a larger mineralization within their 3D matrices compared with the pure collagen gels, when they were incubated in mSBF for 2 weeks. The transparent collagen gels encapsulating 10 mg/mL PLGA microparticles became opaque and white over the 2 week-incubation period in mSBF, while the pure collagen gels or PLGA-Col hydrogels encapsulating 1 mg/mL PLGA microparticles remained transparent (Fig. 2a). The concentration of calcium deposited within the PLGA-Col hydrogels encapsulating 10 mg/mL PLGA microparticles was 1.5 times higher than the pure collagen gels or PLGA-Col hydrogels encapsulating 1 mg/mL PLGA microparticles (Fig. 2b). In contrast, there were no significant differences in the phosphate concentrations among these three conditions. The PLGA-Col hydrogels incubated in DMEM did not present any change in transparency with minimal mineral depositions (Results not shown here).
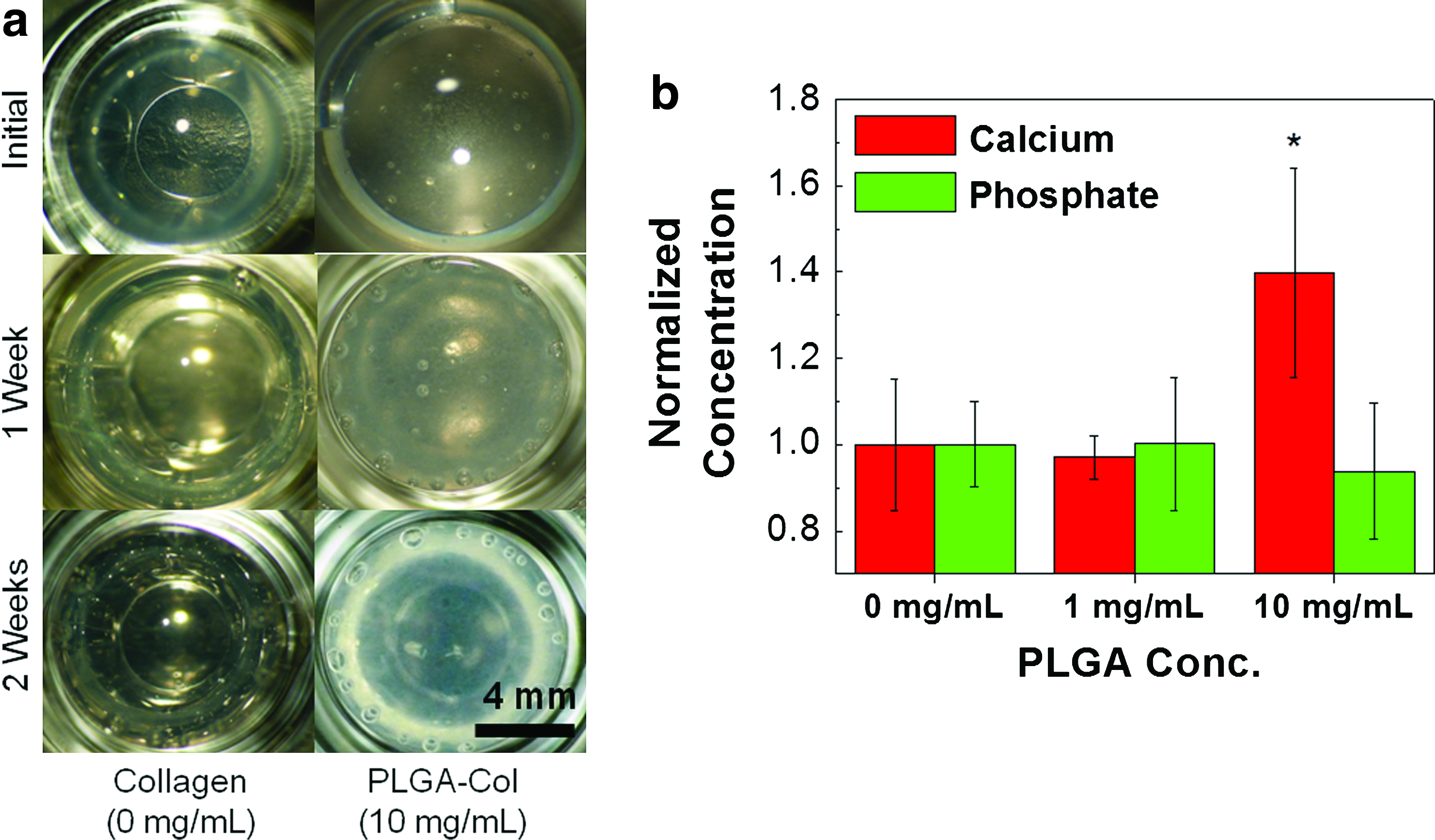
Characterization of hydrogels mineralized with modified simulated body fluid (mSBF).
According to scanning electron microscopic images, the pure collagen gels incubated in mSBF presented limited deposition of minerals within the gels (Fig. 3a). In contrast, the PLGA-Col hydrogels encapsulating 10 mg/mL PLGA microparticles displayed larger sizes and densities of minerals, which were mostly localized on the surfaces of the PLGA microparticles (Fig. 3b). Some parts of the matrix had continuous mineral plates due to the interconnections between minerals on the microparticles. A chemical analysis of the minerals using SEM equipped with EDS displayed a larger ratio of calcium to phosphate (Ca/P) for the PLGA-Col hydrogels than for the pure collagen gels (Fig. 3c). The Ca/P ratio attained with the PLGA-Col hydrogel was ∼2.0, while the Ca/P ratio with the pure collagen gel was about 1.0. The Ca/P ratio attained for the PLGA-Col hydrogels was also closer to that of apatite minerals in bone (∼1.66) than the Ca/P ratio of the pure collagen hydrogels.

Mineral analysis for hydrogels incubated in mSBF
XRD was used to analyze the structures of the minerals formed in the collagen and PLGA-Col hydrogels during the biomineralization process. The minerals formed within the PLGA-Col hydrogels exhibited diffraction peaks with stronger intensities, the most notable one being at a 2θ of 32° (Fig. 3d), compared with minerals formed within the pure collagen hydrogels. In addition, the minor peaks at 2θs of 26°, 28°, and 29° were more defined for the PLGA-Col hydrogels. These diffraction peaks were closely related to the reference pattern of pure hydroxyapatite, reconstructed from known structural data. 23 Note that the broad peak characteristics and slightly shifted peak positions for the mineralized PLGA-Col hydrogels were similar to those observed for bone samples. These results are attributed to the small size of the crystallites and various crystal imperfections.
Mineralization of MSC-encapsulating hydrogels
The PLGA microparticles also enhanced the mineralization of MSC-encapsulating collagen gels, which was driven by mineral ions secreted from differentiated osteoblasts. In this study, MSC-encapsulating gels were cultured in media supplemented with osteogenic differentiation-inducing factors, including L-ascorbic acid, β-glycerophosphate, and dexamethasone. After 1 week of incubation, the PLGA-Col hydrogels encapsulating 10 mg/mL PLGA microparticles became more opaque than the pure collagen gels (Fig. 4a, b). Accordingly, both gel conditions became more rigid than the original gels, but the elastic modulus of the PLGA-Col hydrogels was almost twice larger than that of the pure collagen gels (Fig. 4c). Hydrogel cross-sections stained with von Kossa reagents showed a larger increase in the calcium concentration of the PLGA-Col hydrogels than that of the pure collagen gels 5 days after incubation (Fig. 4d, e). The difference in the calcium concentration between the conditions became more significant on the 10th day of incubation.
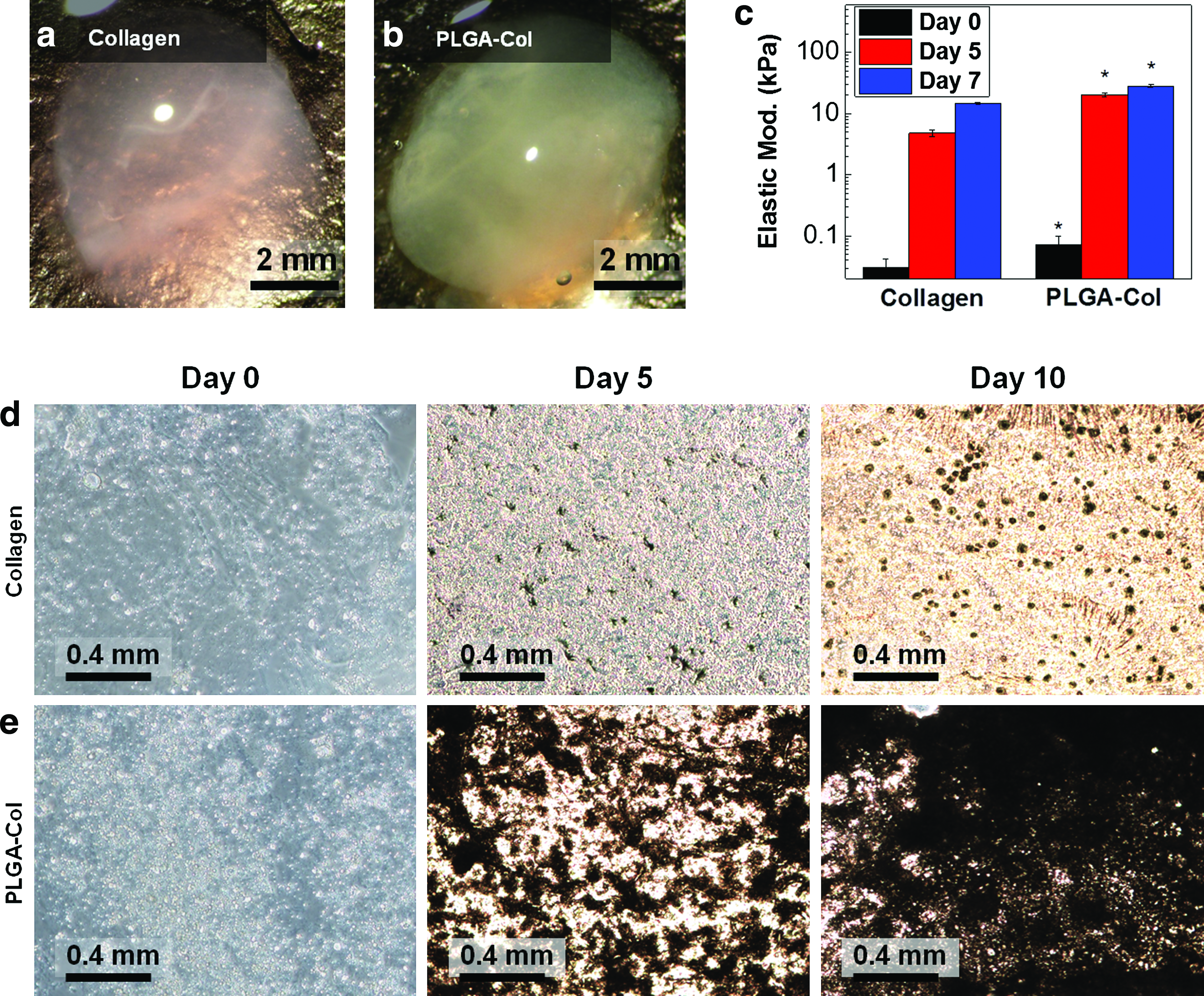
Characterization of hydrogel properties mineralized by the osteogenic differentiation of mesenchymal stem cells (MSCs) encapsulated within the gels.
Microscopic images acquired through SEM analysis also showed more active mineral deposition in the PLGA-Col hydrogels compared with the pure collagen gels, even after 5 days of incubation in the osteogenic differentiation media (Fig. 5a, b). In the PLGA-Col hydrogels, the minerals initially formed on the PLGA microparticle surfaces that were interconnected to form layered plates, similar to those attained by incubating PLGA-Col hydrogels in mSBF (Fig. 5b). According to the spectroscopic analysis using SEM coupled with EDS, the minerals formed in the PLGA-Col hydrogels consisted of calcium and phosphate, while the minerals sparsely distributed in the pure collagen gels were mostly calcium (Fig. 5c). The Ca/P ratio of minerals localized on PLGA microparticles was ∼1.6, which is comparable to apatite in a bone matrix (i.e., Ca/P ∼1.66). 22 In contrast, the Ca/P ratio of minerals formed within the pure collagen gel was 25.7.
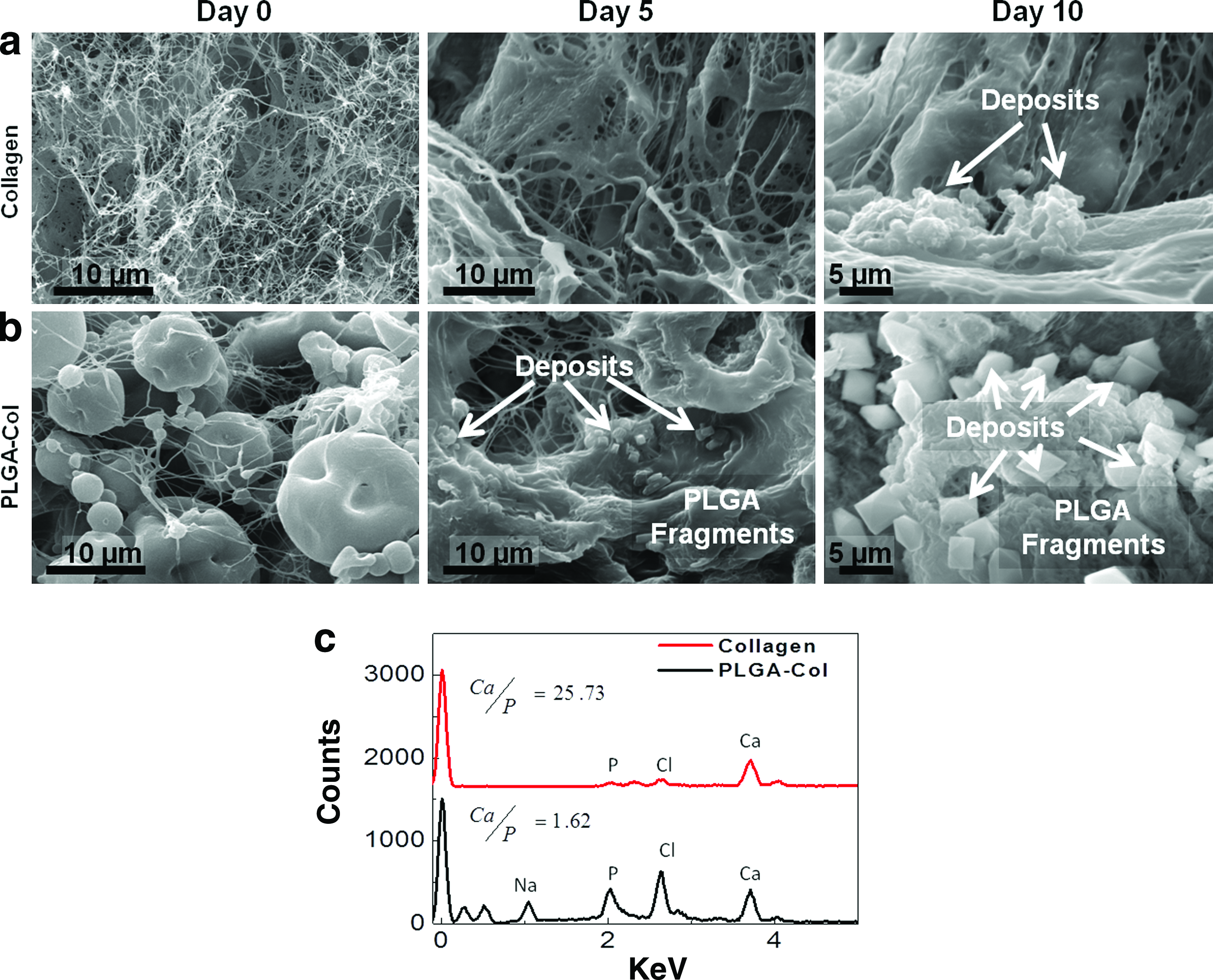
Analysis of minerals formed within MSC-encapsulating hydrogels.
In vivo bone-like tissue formation (osteoid) within MSC-encapsulating PLGA-Col hydrogels
After the incubation of MSC-encapsulating PLGA-Col hydrogels in osteogenic differentiation media over 1 week, the mineralized gels were implanted on chicken CAMs to test the osteogenic potential of the mineralized matrices. CAMs have been previously used as in vivo models for examining bone formation because of the strong osteogenic environment of the developing embryo.24–27 The subsequent implantation of MSC-encapsulating hydrogels on CAMs over 1 more week further increased their opaqueness along with the formation of vascular networks around the implants (Fig. 6a). No significant host immune response was observed at the implant site. The volume of the collagen hydrogel explants was qualitatively smaller than the PLGA-Col hydrogel explants, likely because of cell-exerted volumetric shrinkage (Fig. 6a). Additionally, both collagen and PLGA-Col hydrogels exhibited increased mechanical rigidity throughout the mineralization process; however, PLGA-Col hydrogel explants exhibited a significantly larger increase in mechanical rigidity compared with the collagen gel explants (Supplementary Fig. S4).
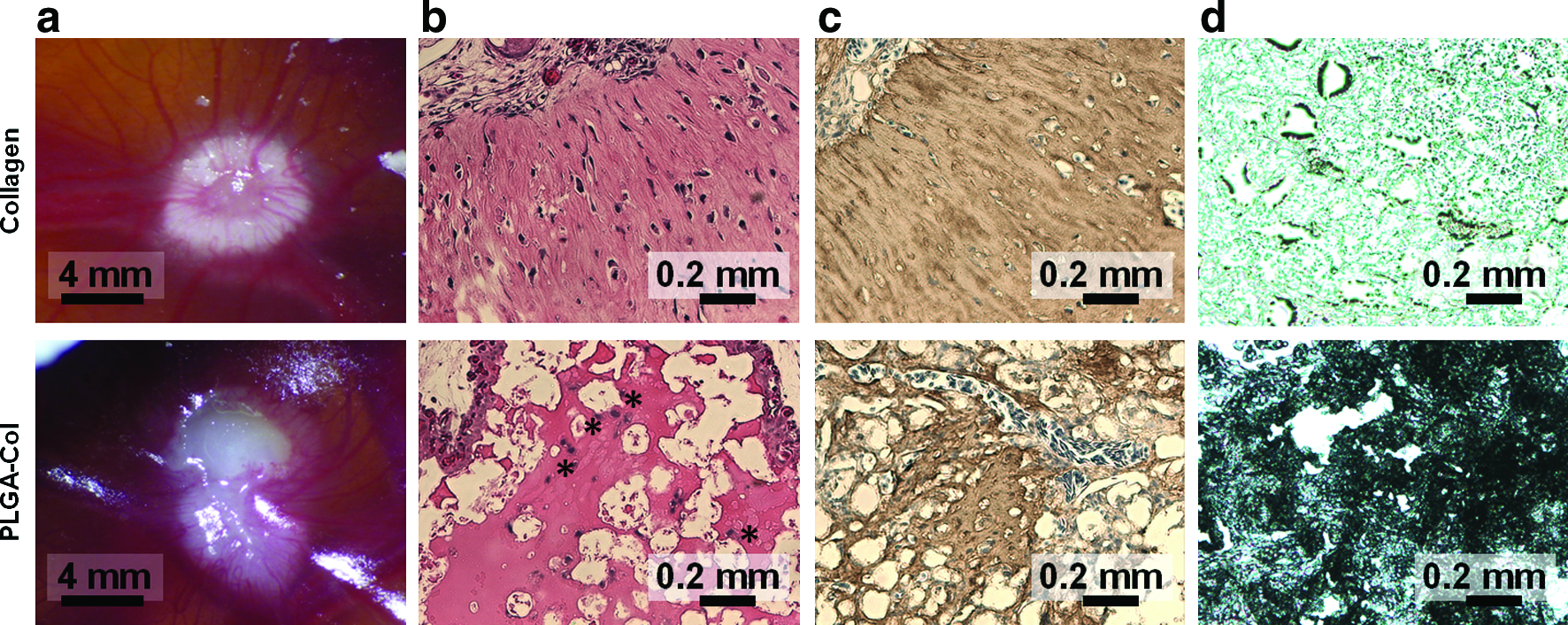
Histological analysis of MSC-encapsulating hydrogels, which were first incubated in osteogenic differentiation media over 1 week and subsequently implanted on chorioallantoic membranes (CAMs) over 1 week.
According to excised CAM cross-sections stained with H&E, the pure collagen gels generated a connective tissue with textures similar to fibrous tissue (Fig. 6b). In contrast, the PLGA-Col hydrogels displayed cells with large volumes of cytoplasm, which represents osteoblasts differentiated from MSCs, as well as a highly eosinophilic matrix in the spaces between the PLGA microparticles, which represents the active ossification site of the transplanted osteoblasts (Fig. 6b). In addition, the PLGA-Col hydrogels presented larger amounts of type I collagen and calcium than the pure collagen gels, as characterized with the cross-sections of explants stained with antibodies to type I collagen and von Kossa reagents, respectively (Fig. 6c, d).
Discussion
The results of this study demonstrate that PLGA microparticles incorporated within collagen gels enhance the gels' mechanical rigidity and capacity for mineralization. The PLGA microparticles increased both the elasticity and stiffness of the collagen hydrogels, while minimally affecting the molecular diffusivity. In addition, the degradation of the PLGA microparticles in the collagen hydrogels facilitated the deposition of apatite-like minerals in both cell-free hydrogels incubated in mSBF and MSC-encapsulating hydrogels incubated in osteogenic differentiation media, resulting in a significant increase in the hydrogels' rigidity. 28 Finally, as demonstrated with in vivo CAM assays, the MSC-encapsulating PLGA-Col hydrogels with enhanced deposition of hydroxyapatite-like minerals promoted bone-like tissue formation.
The PLGA microparticles incorporated in the collagen gels increased their stiffness and elasticity, because of the particles' capability to associate with interconnected collagen fibers. As exhibited through SEM imaging, the hydrophobic surface of the PLGA microparticles facilitated the adsorption of collagen molecules and subsequently generated a microstructure similar to a particle-filled composite. Therefore, we propose that these microparticles increase the resistance of the interconnected collagen network to deformation and also increase the material's capability to store deformation energy within the network, demonstrated by the independence of the storage moduli from frequency and the decrease of the phase angle at a low frequency. In addition, PLGA-Col hydrogels can be incorporated into micro-sized pores in a rigid and biodegradable polymeric scaffold to maintain its structural integrity in skeletal tissue defects subjected to large mechanical pressures on the megapascal scale.
Another interesting feature of the PLGA-Col hydrogels was the increase in gel stiffness and elasticity with a minimal change in gel permeability, characterized through molecular diffusivity. It is common to increase the gel's stiffness by increasing the number of intermolecular cross-links, but this conventional approach is often plagued by a decrease in gel permeability.29,30 This inverse dependency between gel permeability and stiffness is disadvantageous for its use as a cell transplantation device implanted in a load-bearing zone. In contrast, the PLGA microparticles did not significantly decrease the space between interconnected collagen fibers, as compared with the pure collagen gels. Therefore, our approach to incorporate micro-sized reinforcing fillers in the gel may be more advantageous for cell encapsulations in hydrogels with controlled stiffnesses.
In addition, the enhanced mineralization of the PLGA-Col hydrogels is attributed to negatively charged carboxylate groups that result from the hydrolytic degradation of the microparticles. We suggest that these carboxylates attract the calcium in mSBF or that secreted by differentiated MSCs, and facilitate calcium deposition within the collagen gels (Fig. 7). 31 Significant calcium deposits on the PLGA particle surfaces account for the larger calcium concentration within the PLGA-Col hydrogels compared with the pure collagen gels, as characterized by a calcium assay, SEM images, EDS-based chemical analysis, and XRD-based structural analysis.
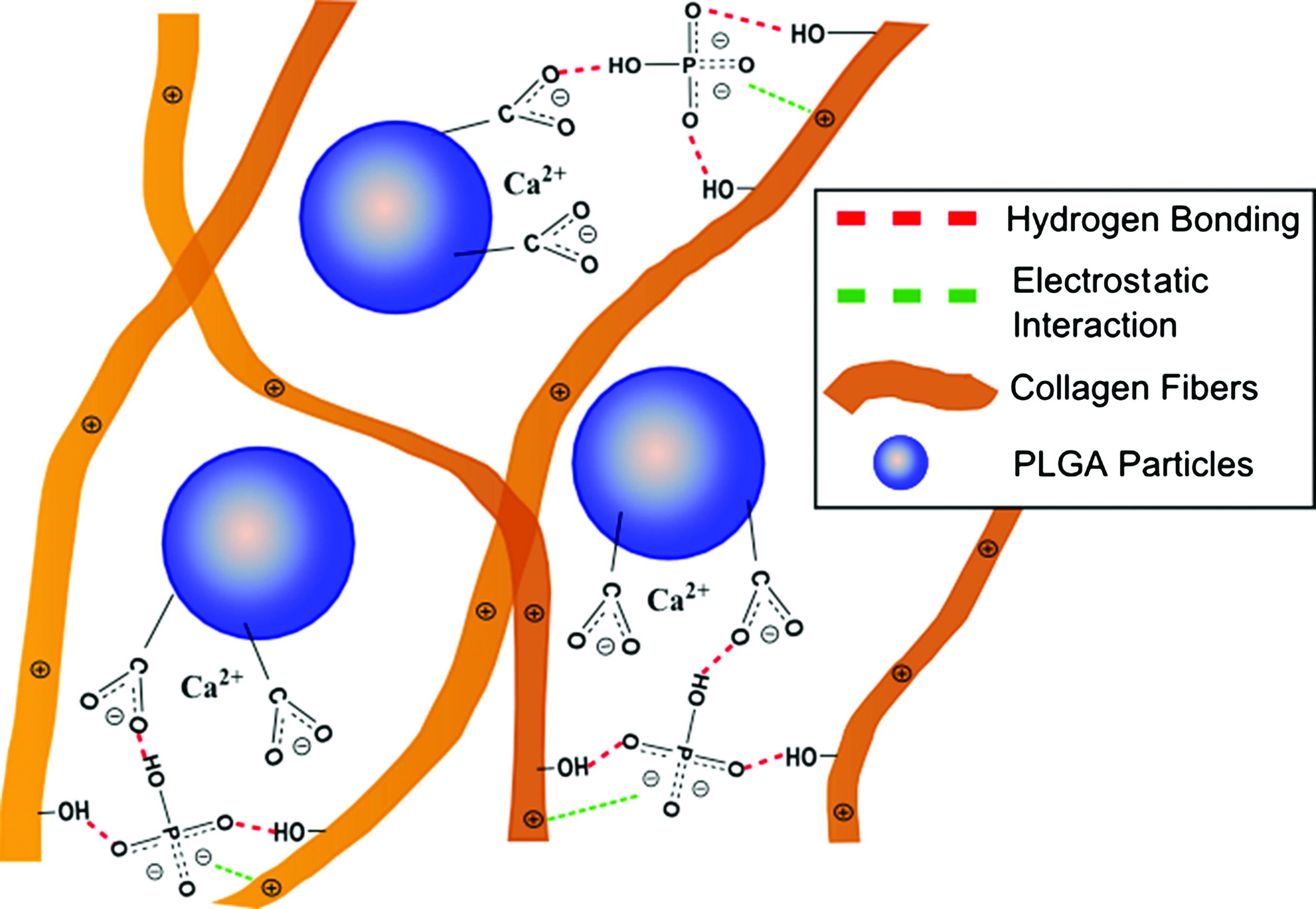
Schematic depicting the enhanced mineralization of PLGA-Col hydrogels. Cell-secreted calcium was electrostatically bound to hydrolyzed PLGA microparticle surfaces, and the calcium readily associates with phosphates, also electrostatically bound to collagen fibrils. The immobilizations of calcium and phosphates facilitate the formation of apatite within the PLGA-Col gels, as compared with pure collagen gels. Color images available online at www.liebertpub.com/tea
Furthermore, it is suggested that the collagen fibers attached to the particle surfaces also play an important role in the formation of apatite minerals in the gel's matrix because of their capacity to bind with phosphate (Fig. 7). It is likely that these phosphates bound to the matrix have reduced entropies compared with free phosphates in solution, and subsequently react with calcium in a thermodynamically favorable manner. This orchestrated activity between the microparticles and fibers is addressed by the limited apatite formation on PLGA microparticles either freely suspended in mSBF or encapsulated in neutral polymer matrices such as poly(ethylene glycol). 32 The minimal difference in phosphate concentrations between the pure collagen and PLGA-Col hydrogels also addresses the preferential binding of phosphates to collagen fibers. Therefore, we interpret that the enhanced mineralization and structural rigidity of the PLGA-Col hydrogels, as compared with the pure collagen gels, significantly contributes to the formation of bone-like tissue within the hydrogels implanted in vivo.
Conclusion
Overall, this study presents a simple but efficient way of facilitating the 3D mineralization of gels without significant volumetric shrinkage by controlling both the mechanics and mineralization capabilities of a collagen gel using PLGA microparticles. The enhanced formation of apatite minerals within the PLGA-Col hydrogels is attributed to the gradual degradation of PLGA microparticles to form anionic carboxylates and their subsequent association with phosphate ions bound to collagen fibers. The PLGA microparticles used in this study can be further modified by encapsulating various soluble factors so that the particles will act as multi-functional tools which release the soluble factors in a sustained manner, while controlling gel mechanics and mineralization. We, therefore, propose that the results of this study will be highly useful for controlling 3D mineralization processes and ultimately contributing to fundamental and clinical studies of bone repair, regeneration, and pathogenesis.
Footnotes
Acknowledgments
This work was supported by the National Science Foundation (CAREER: DMR-0847253) (H.J.K.), the National Institute of Health (1R25CA154015A) (R.D.), and the Korean Research Foundation (KRF-2008-331-D00117) (I.K.).
Disclosure Statement
No competing financial interests exist.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.