
Abstract
There is a rise in the popularity of arthroscopic procedures in orthopedics. However, the majority of cell-based bone tissue-engineered constructs (TECs) rely on solid preformed scaffolding materials, which require large incisions and extensive dissections for placement at the defect site. Thus, they are not suitable for minimally invasive techniques. The aim of this study was to develop a clinically relevant, easily moldable, bone TEC, amenable to minimally invasive techniques, using human mesenchymal stromal cells (hMSCs) and calcium phosphate microparticles in combination with an in situ forming platelet-rich plasma gel obtained from human platelets. Most conventional TECs rely on seeding and culturing single-cell suspensions of hMSCs on scaffolds. However, for generating TECs amenable to the minimally invasive approach, it was essential to aggregate the hMSCs in vitro before seeding them on the scaffolds as unaggregated MSCs did not generate any bone. Twenty four hours of in vitro aggregation was determined to be optimal for maintaining cell viability in vitro and bone formation in vivo. Moreover, no statistically significant difference was observed in the amount of bone formed when the TECs were implanted via an open approach or a minimally invasive route. TECs generated using MSCs from three different human donors generated new bone through the minimally invasive route in a reproducible manner, suggesting that these TECs could be a viable alternative to preformed scaffolds employed through an open surgery for treating bone defects.
Introduction
However, it is often difficult to fit a preformed scaffold into a defect with a complex geometric shape as can be the case following trauma or tumor resections. Further, preformed scaffolds are not amenable to arthroscopic techniques. An open operation is often required to place the graft at the defect site. Such open surgeries are often associated with significant manipulation of the soft tissues with consequent risk of infections and longer intraoperative times. 9 This is of special concern in cases of staged surgical interventions, where multiple invasive surgeries may be associated with increased patient morbidity and negative aesthetic effects. Thus, the development of tissue-engineered bone grafts, which can be implanted through minimally invasive procedures, can result in immediate clinical benefits, both in terms of convenience for the surgeon and faster recovery time for the patient. 10
Several injectable gels and cements, such as calcium phosphate cements (CPC), polymeric gels, agarose, hyaluronate, cellulose, fibrinogen-based gels, collagen, and pluronic acid have been suggested as injectable biomaterials for the purpose of bone tissue engineering.11–13 While the CPCs provide good biomechanical stability, the exothermic reactions associated with their setting process prove to be a deterrent in combining them with cells. One of the approaches reported to overcome this drawback involves the use of self-dissolving alginate beads, which serve as a protective capsule for the cells during setting of the injectable cements. Though an interesting approach, the results have not yet been verified in vivo.14,15 Agarose gels, on the other hand, are a suitable delivery system for cartilage tissue engineering, but do not support blood vessel ingrowth, thus making them inadequate for bone tissue engineering. The degradation products of pluronic acid are cytotoxic, while the curing of hyaluronic acid from a solution to a gel is difficult making it unsuitable for routine clinical use. Gels from synthetic polymers often result in inflammatory reactions. 12 Collagen-, alginate-, and fibrinogen-based gels are biocompatible, and thus suitable for use with cells. However, of these three in situ gel forming compounds, fibrinogen-based gels offer distinct advantages as it gels at body temperature, can be prepared from the patient's own plasma, and has proven biocompatibility and biodegradability.15–17 Trombi et al. further demonstrated in vitro that fibrin-based gels supported human MSC (hMSC) proliferation and differentiation and could be used as a delivery vehicle for hMSCs. 18
The use of fibrinogen as an osteoconductive material and a medium for compacting grafts has been well reported. 19 They have been used in conjunction with other biomaterials, such as coral or hydroxyapatite to heal bone defects.20,21 It has been demonstrated that fibrin glue can be used as a carrier for calvarial osteoprogenitor cells to repair critical-sized osseous facial defects 22 and for periosteal cells to induce heterotropic osteogenesis in rat experiments. 23 It has also been reported that fibrin glue, together with periosteum-derived osteoprogenitor cells, can heal segmental bone defects in rabbits. 9 An indirect evidence of the benefits of the osteoinducitivity of fibrin- and plasma-based products was provided by Rochet and colleagues who used blood clotted around ceramics and MSCs to develop a cohesive moldable TEC with osteogenic properties. 24
To test the possibility of employing fibrin-based gels for injectable purposes, Yamada et al. used a combination of fibrin gel with rat MSCs and beta tricalcium phosphate (β-TCP) scaffolds to generate bone at an ectopic site. 25 In a follow-up study, they replaced the fibrin gel with the platelet-rich plasma (PRP) gel (the PRP gel is a fibrin gel enriched with the growth factors released from platelets) with in vitro expanded canine MSCs for treating mandibular defects in dogs. 26 Recently, a study using hMSCs derived from adipose tissue was used to develop an injectable TEC for bone regeneration. 27 In all the above-mentioned studies, the PRP gel functioned as the scaffolding material as well as the delivery vehicle. To add further osteoconductive and osteoinductive properties to the resulting graft and to improve its mechanical stability, Mankani et al. used a fibrin-based gel in combination with a single-cell suspension of hMSCs and hydroxyapatite (HA)/tricalcium phosphate (TCP) particles. 10 This formulation when injected into ectopic and orthotopic sites in mice generated bone, thus providing a proof of principle on the possible use of ceramics and hMSCs with fibrinogen as an injectable formulation for generating bone in vivo. 10 However, the fibrin gel was of mouse origin and the HA/TCP particle size used in this study was in the range of 0.5–1 mm. Studies demonstrating the possibility of employing smaller particle sizes for bone tissue engineering applications have been reported.24,28 This can potentially facilitate the delivery of the TECs via a minimally invasive approach.
It is well known that the in vitro culture conditions of MSCs can influence their in vivo performance. In that context, it has recently been reported by multiple researchers that culturing MSCs as aggregates can improve their osteogenic and chondrogenic differentiation capacities.29–31 Within our laboratory, we observed that greater amounts of bone were obtained in vivo when a single-cell suspension of hMSCs conventionally used in bone TECs was replaced by hMSC aggregates (manuscript submitted).
The aim of the present study is to combine hMSC aggregates, microceramic particles, and the human platelet-derived PRP gel into a safe (due to the autologous origins of the cells and gel), relatively low cost, easily obtainable TEC, with an in vivo osteogenic potential that can be delivered at the defect site using minimally invasive techniques.
Materials and Methods
Cell culture
Bone marrow aspirates (5–20 mL) were obtained from healthy donors during hip replacement surgery with written informed consent. Alternatively, cryopreserved vials of hMSCs were purchased (Lonza group Ltd). When isolated from fresh marrow aspirates, the aspirates were resuspended using 20G needles and plated at a density of 5×105/cm2 and cultured in a basic medium supplemented with 1 ng/mL basic fibroblast growth factor (Instruchemie). This medium is, henceforth, referred to as proliferation medium. The basic medium was made of the a-minimal essential medium (Life Technologies), 10% fetal bovine serum (Cambrex), 0.2 mM ascorbic acid (Asap, Life Technologies), 2 mM L-glutamine (Life Technologies), 100 U/mL penicillin (Life Technologies), and 10 mg/mL streptomycin (Life Technologies). Cells were grown at 37°C in a humid atmosphere with 5% CO2. The medium was refreshed twice a week and on reaching near confluence, cells were trypsinized and cryopreserved till further use. At the start of the experiments, the cryovials were expanded further in the proliferation medium until numbers sufficient for experiments were obtained.
Generation of cell aggregates
Four hundred micrometer diameter wells with a depth of 200 μm were patterned on poly (dimethylsiloxane) (PDMS) stamps using etched silicon wafers (Fig. 1A) according to established protocols. 32 After sterilizing the PDMS stamps for 10 min in 70% ethanol, they were placed in the wells of a standard six-well plate and completely covered with 3% agarose solution (Ultra-pure agarose, Invitrogen). Upon solidification, the chips were demolded and the agarose templates were placed in a nontissue culture-treated 12-well plate. After wetting the chips with the medium, a concentrated suspension (1 mL) of 1.5 million cells was uniformly dispersed over the wells on each agarose chip. Agarose chips were then centrifuged briefly at 1500 rpm, to facilitate the cells to settle down on the chips. For culturing on the chips, the medium used (termed Exp. Medium) was serum free and composed of Dulbecco's modified Eagle's medium supplemented with 10−7M dexamethasone, 50 mg/mL Asap (Life Technologies), 40 mg/mL proline (Sigma-Aldrich), 100 mg/mL sodium pyruvate, and 50 mg/mL ITS 1 Premix (Becton-Dickinson). 1.5 mL of the Exp. Medium was added to each chip. Cells spontaneously compacted to form aggregates within 24 h. For the study comparing the viability of the cells after 1 day, 1 week, and 2 weeks of in vitro culture, the aggregates were left in the agarose chips for 1 day, 1 week, and 2 weeks, respectively. The medium was changed every alternate day during culture in the agarose chips.
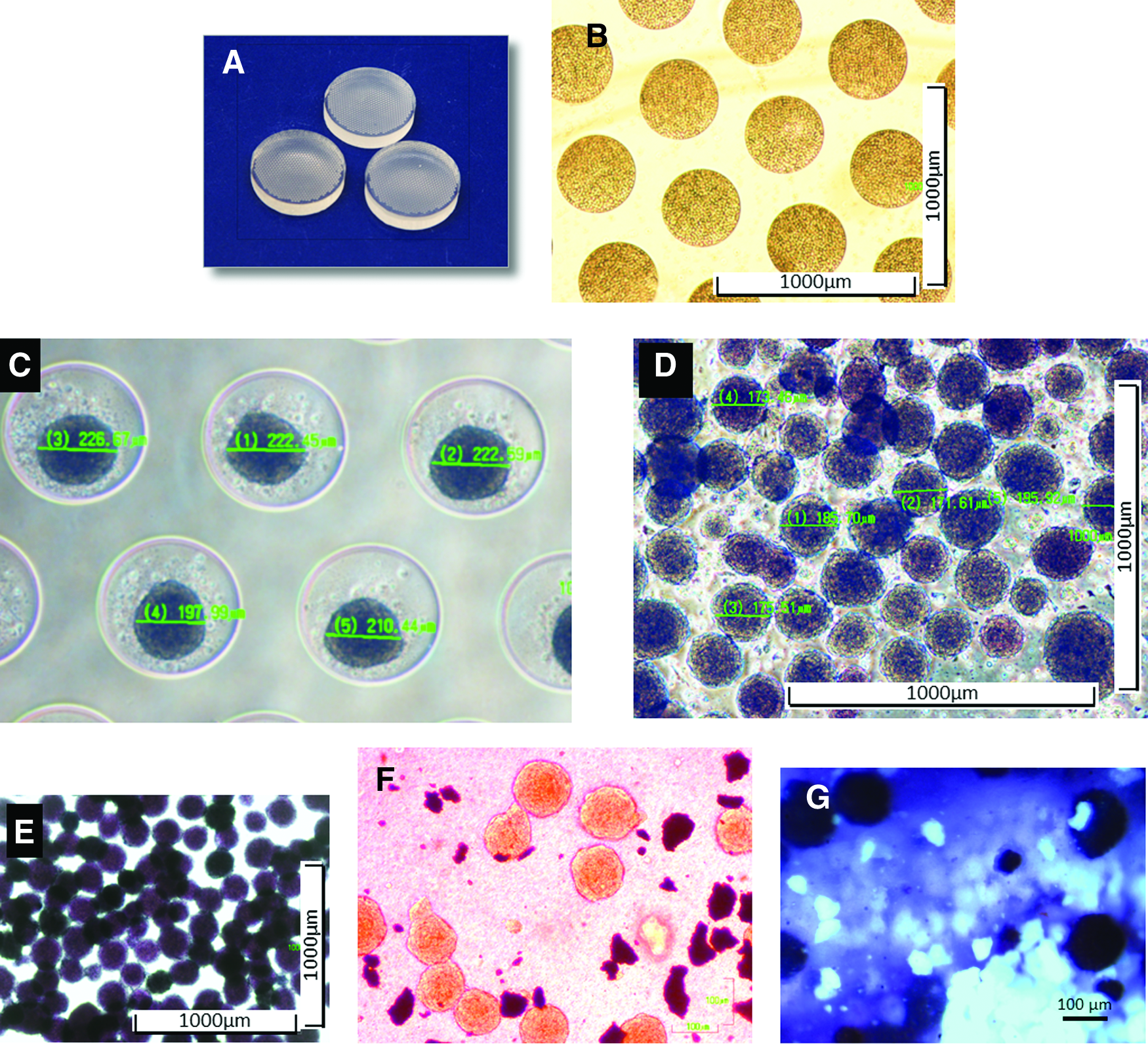
Characterization of cell aggregates.
PRP gel
After written informed consent, venous blood was withdrawn from healthy blood donors and passed through an apharesis unit at the Institut für Klinische Transfusionsmedizin. Platelets were isolated and immediately frozen at −80°C. At the time of the experiment, the bag was thawed at 37°C. This freeze–thaw cycle resulted in lysis of the platelets, with subsequent release of the growth factors and formation of the platelet lysate. 235 μL 1 M calcium chloride solution was added per 10 mL of platelet lysate and gently mixed on a shaker at 37°C degrees for approximately 10 min. This resulted in the separation of the platelet lysate into a gel-like component and a clear liquid. The clear liquid was the source of the thrombin component, which when added at a 1:1 ratio at 37°C to the rest of the platelet lysate (fibrin source), activated the clotting pathway, thereby resulting in the formation of a PRP gel in 10–12 s.
Calcium phosphate microceramics
Biphasic calcium phosphate (BCP) microceramics of sizes 53–63 μm and sintered at 1150°C were provided by Xpand Biotechnology BV. The particles contain 20%±5% beta-tricalcium phosphate and 80%±5% hydroxyapatite. The particles were autoclaved at 121°C for 30 min before use.
Generation of constructs for in vivo implantation
In the minimally invasive approach, the cell aggregates were flushed out of the microchips and immediately mixed with approximately 25 μg of BCP microceramics and fibrin and thrombin components (1:1 ratio) of the PRP gel just before implantation. The mix was then directly introduced into the defect through a small hole measuring approximately 1 mm and just enough to hold the tip of the pipette. The pipette tips were precooled, and the cell ceramic constructs were also kept on ice to slow the gelling time. The contents were very slowly introduced into the defect and the edges of the pocket were held up to contain the spreading in the subcutaneous space and to ensure that the contents did not leak out from the sides. The injected material gelled within 20–30 s after injection. The pocket was then closed using 5–0 vicryl sutures. As a control, we implanted in the same animals, with an open 2-mm incision, the construct generated after the cells were mixed with scaffolds, fibrin, and thrombin, and then left to gel at 37°C for 15 min before implantation.
Cell viability assays
The viability of the cell aggregates within the constructs generated using cell aggregates, microceramics, and platelet gel, and deposited using a pipette tip at the bottom of a six-well plate was checked using an MTT assay. The constructs were incubated with 1 mL Exp. Medium and 20 μL MTT solution (5 mg/mL; Gibco) per well for 2 h at 37°C in a 5% CO2 atmosphere incubator. Images were captured using a phase-contrast microscope and MATRIX Vision SRGB 32 Bit software.
The viability of cells in the aggregates cultured for 1 day, 1 week, and 2 weeks within the microwells of the agarose chips were checked using a live–dead assay (Invitrogen). The two components of the kit, ethidium homodimer 1 and calcein were mixed in a ratio of 4:1 and diluted in phosphate-buffered saline (PBS) as per the protocol recommended by the manufacturers. The constructs were incubated in the resulting mixture for 30 min in the dark. The fluorescence from the calcein dye was observed using a filter for the FITC 488 dye, while the fluorescence for ethidium homodimer was observed using filters for the Texas red dye. The images were captured using a color camera (Nikon FDX-35) and QCapture software.
Gene expression analysis
Cells aggregated in the microwells for 1 day, 1 week, or 2 weeks were washed briefly with PBS and lysed using the Trizol reagent (Invitrogen). The samples were stored for at least 12 h at −80°C for RNA isolation. After chloroform addition and phase separation by centrifugation, the aqueous phase containing the RNA was collected, mixed with equal volume of 75% ethanol, and loaded onto the RNA-binding column of a Nucleospin RNA II kit (Bioke). Subsequent steps were in accordance with the manufacturer's protocol. The RNA yields were determined by spectrophotometry using the Nanodrop1000 (ND-1000 spectrophotometer; Isogen Life Science). Subsequently, cDNA was synthesized using iScript (BioRad) according to the manufacturer's recommendations. 3 μL of undiluted cDNA was used for subsequent analysis. For quantitative polymerase chain reaction (qPCR), a master mix, containing distilled water, forward primer, reverse primer (Sigma Genosys), bovine serum albumin, and SYBR green I mix (Invitrogen), was prepared. Real-time qPCR was performed, for the osteogenic genes, on a MyIQ single color real-time PCR detection system (BioRad). Gene expression was normalized to the expression of Beta-2 microglobulin (B2M) gene. My IQ data were analyzed using iQtm5 optical system software (Biorad). Ct values were normalized to the B2M housekeeping gene and the comparative ΔCt method (Ct control−Ct sample) was used to calculate the fold inductions. Primer sequences are listed in Table 1.
Tl: transcript length.
Ta: annealing temperature.
In vivo studies
An immune-deficient mouse model was used for assessing the bone forming capacity of the hMSCs. TECs were introduced into their subcutaneous pockets either by injection or by an open approach. Ten immune-deficient male mice (Hsd-cpb: NMRI-nu) were used for each experiment. The mice were anesthetized by inhalation of isofluorane and carbon dioxide. For the open approach, a 2-mm incision was made on the dorsal aspect of each mouse, while for the injectable approach, a hole was made using an18-gauge needle, and then enlarged to approximately 1 mm to fit the pipette tip. The openings in either case were closed using a vicryl 5–0 suture. The constructs were left in the mice for 6 weeks, at the end of which the mice were euthanized using carbon monoxide and the samples were explanted. The size of the explanted samples in both cases averaged around 2 mm. All the experiments were approved by the local animal experimental committee.
Bone quantification
The explanted samples were fixed in 4% paraformaldehyde (Merck) and embedded in methacrylate for sectioning. Approximately 300 μm-thick, undecalcified sections were processed on a histological diamond saw (Leica saw microtome cutting system). At least four sections were cut per sample. Each section was stained with basic fuchsin and methylene blue to visualize new bone formation. The newly formed mineralized bone stains red with basic fuchsin, the unmineralized osteoid stains light pink, while all other cellular material stains blue with methylene blue. The ceramic material remains black and unstained by both the dyes. For quantitative histomorphometric analysis, three randomly selected slides were first scanned using a PathScan Enabler IV histology slide scanner. Mature and immature bone was manually pseudocolored green, while the scaffold material was pseudocolored red using Photoshop CS2 (Adobe Systems). The total scaffold area was defined as the area of interest. A custom-made Matlab script was used to measure the amount of bone within the area of interest. These values were then averaged and expressed as a percentage of the total scaffold area occupied by newly deposited bone in each condition.
Statistics
All in vitro experiments were performed in triplicate. Statistical analysis was performed using One-way Anova followed by the Tukey's multiple comparison test (p<0.05) when more than two groups were compared. When the statistical analysis was performed between two groups, a Student's paired t-test was used. As in the case of ANOVA, a p-value less than 0.05 was considered as significant.
Results
Characterization of cell aggregates
To generate cell aggregates, 1.5 million MSCs were dispersed uniformly over the microwells on each agarose chip. The agarose chip was then centrifuged briefly to permit the MSCs to settle within the chips. At this stage, the MSCs occupied the entire microwell, which measured 400 μm in diameter and 100 μm in depth (Fig. 1B). However, within 24 h, the cell aggregates condensed and on an average measured 220 μm in diameter (Fig. 1C). The average size of the aggregates after flushing was approximately 175 μm (Fig. 1D). To determine if the loss in size was associated with a decrease in cell viability, MTT assays were performed on the aggregates before and after flushing from the agarose chips. The results of the assay suggested that majority of the cells were viable even after flushing out of the chips (Fig. 1E). Next, cell aggregates, microceramic particles, and the PRP gel were combined within a pipette tip before being ejected out as a TEC. To investigate if the shear force experienced by the cell-ceramic-PRP construct, during passage through the pipette tips, alters the integrity of the cell aggregates, the ejected TECs were visualized under a light microscope. Cell aggregates were seen interspersed with the microceramic particles indicating that the majority of the aggregates remained intact even after passing through the pipette tips (Fig. 1F). MTT staining of the aggregates within the resulting TECs suggested that passage through the pipette tip did not affect their viability (Fig. 1G).
In vitro evaluation of the possibility to engineer TECs of different shapes
One of the major drawbacks of using preformed scaffolds is the difficulty associated with adapting it to complex geometric shapes as can be the case following trauma or tumor resections. In such cases, the surgeon needs to either fabricate ex vivo complicated scaffold geometries or carve the defect site. To test the possibility of adapting our injectable TEC into defects of varying shapes and sizes, cell aggregates were combined with microceramics and the PRP gel and injected into moulds having rectangular, cross, and triangular shapes. The PRP gel guided the cells and ceramics to fit the contours of the different morphologies before gelling, thereby resulting in TECs with varying shapes and sizes (Fig. 2). Our results thus suggest that our PRP gel-based injectable TEC can be used to engineer in vivo bone of different sizes and shapes.
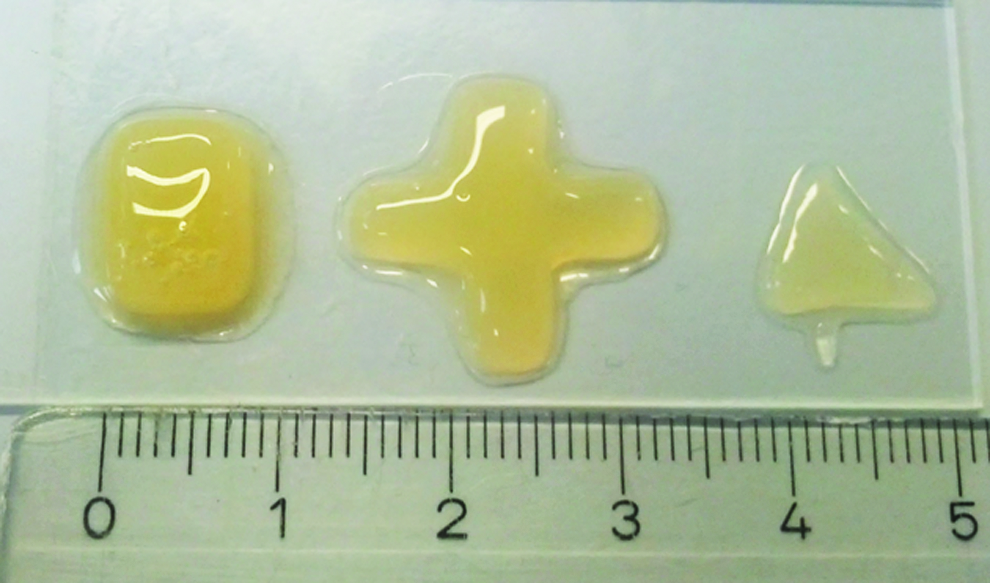
In vitro evaluation of the possibility to engineer TECs of different shapes. TECs generated by combining PRP gel, cell aggregates, and microceramic particles, were then placed within rectangular, cross-shaped, and triangular moulds. The TECs adapted to the shape of the mould. This suggested that the TECs developed with our system could be used to engineer bone of different shapes and sizes. Color images available online at www.liebertpub.com/tea
Optimization of the in vitro culture time of cell aggregates
To maximize cell–cell interactions in vitro, we investigated the possibility of a longer in vitro culture time of the cell aggregates on the agarose chips by assessing its influence on cell viability and osteogenic gene expression. MSCs were seeded and cultured on the agarose chips for 1 day, 1 week, and 2 weeks. In all cases, the medium was refreshed every alternate day. Cell viability was performed using a live/dead staining kit. We observed a steady increase in red fluorescence (suggestive of dead cells) with an increase in the in vitro culture duration. This suggested that cell viability is compromised upon extended culture (Fig. 3A–C). On qPCR analysis of the expression of a panel of commonly used osteogenic genes, we observed that there was no significant difference in the expression of either BMP-2 or osteocalcin at the different time points. However, ALP gene expression was highest at 1 week with a significant decrease at the 2-week time point. Collagen 1 expression was the highest following 1 day in culture, but thereafter decreased significantly. Thus, we concluded that prolonged in vitro culturing did not enhance the overall osteogenic gene expression (Fig. 3D). To determine if the in vivo bone formation paralleled our in vitro findings, 1-day-, 1-week-, and 2-week-old aggregates generated using MSCs from the same donor that was used for the in vitro study, were implanted subcutaneously for 6 weeks in a nude mouse model. Previously in our laboratory, on culturing unaggregated MSCs with ceramics and the PRP gel for 2 weeks in vitro, we obtained bone in vivo, although the amounts were significantly lower compared to constructs employing aggregated cells (manuscript submitted). To determine whether unaggregated MSCs could also generate bone via the minimally invasive approach, we combined 1.5 million unaggregated cells with microceramics and the PRP gel and introduced these TECs in parallel with the TECs employing aggregated MSCs cultured for 24 h, 1 week, and 2 weeks into the subcutaneous pocket of nude mice. For a fairer comparison, the MSCs for both aggregated and unaggregated TECs were from the same donor. After 6 weeks of in vivo implantations, the amount of bone was determined histomorphometrically in all the TECs. In the unaggregated group, no bone formation was observed. The average amount of bone generated for the total scaffold area varied from 6.6%, 7.1%, and 4.4% for 1-day-old aggregates, 1-week-old aggregates, and 2-week-old aggregates, respectively (Fig. 3E). Statistical evaluation indicated that the differences between these three groups were not significant. These results suggest that while aggregation is essential for bone formation, a longer in vitro culture time is not beneficial for in vivo bone formation.
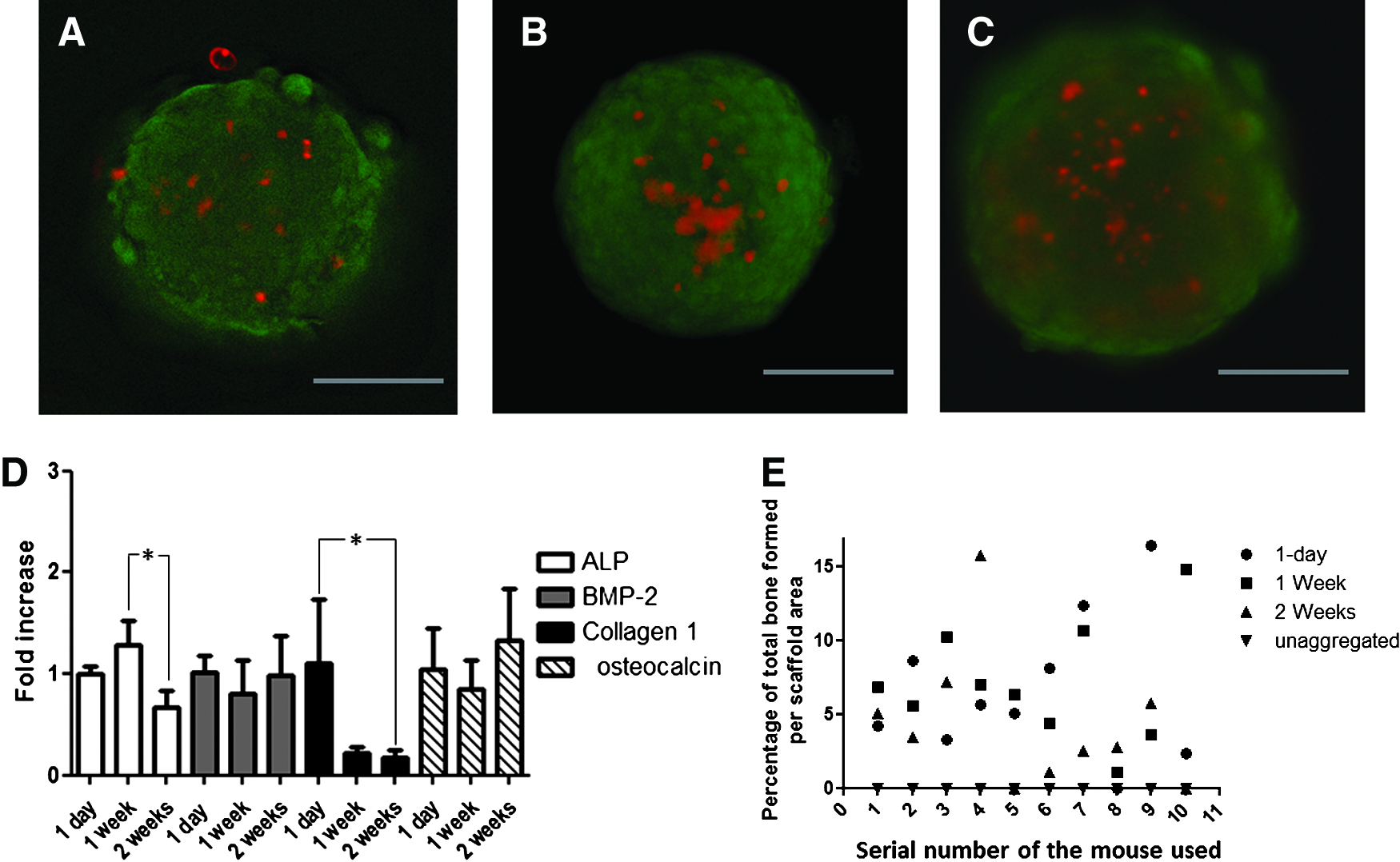
Effect of prolonged in vitro culturing of cell aggregates on the cell viability and osteogenic gene expression. 1.5 million cells were seeded on each agarose chips and cultured for 1 day, 1 week, or 2 weeks in vitro. The medium was refreshed every alternate day. At the end of
Evaluation of in vivo bone formation using MSCs from multiple donors
TECs were generated using MSC aggregates from three different donors. The aggregates from all the three donors were cultured in vitro for 24 h before combining with microceramics and the PRP gel. These resulting TECs were introduced into the subcutaneous pocket on the dorsum of 10 nude mice via a small opening just wide enough to hold the pipette tip. Histological analysis of constructs after 6 weeks demonstrated abundant bone formation within the TECs generated using MSCs from all the three donors (Fig. 4A). More specifically on average, 10.03%, 3.1%, and 5.6% of the total scaffold area was occupied with newly formed bone in the constructs using commercially obtained MSCs, donor 236, and donor 240 (D236 and D240), respectively (Fig. 4B). This data suggest that our TECs introduced into the defect via a minimally invasive route can reliably generate bone in vivo.
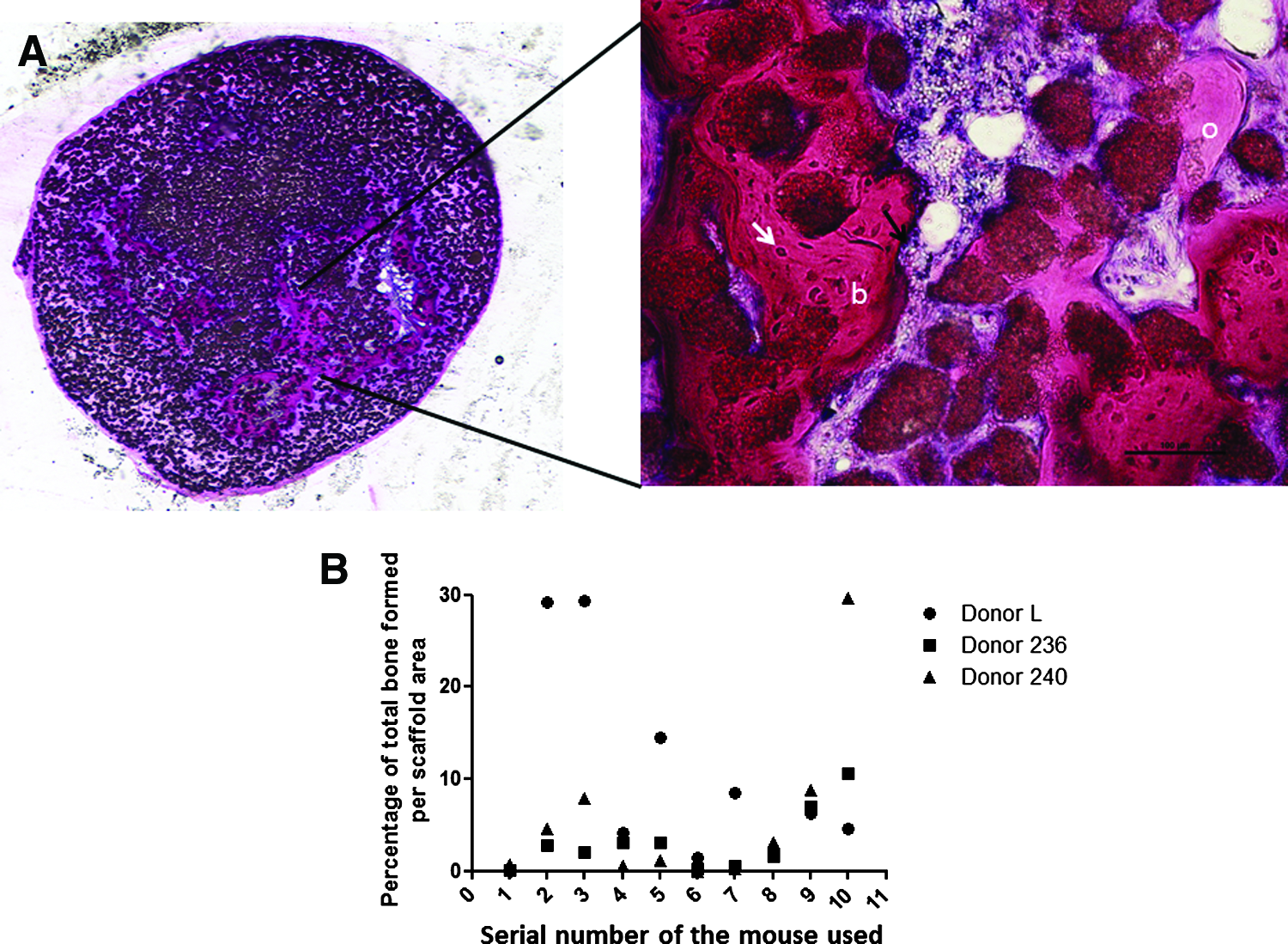
Evaluation of the in vivo bone-forming capacity of injectable TECs using multiple donors. TECs were generated using PRP gel and microceramic particles combined with 1-day-old MSC aggregates from three different human donors.
Comparison of the injectable system with the open approach in terms of in vivo bone formation
Conventionally TECs are introduced into a defect site via an open incision. Our results indicate that in our system, a minimally invasive approach also results in new bone formation in vivo. To investigate if there are significant differences in the amounts of bone obtained when the constructs are gelled ex vivo at 37°C for 15 min, and then inserted via an open incision of around 3 mm, or introduced directly into the subcutaneous space of the animal using an opening, big enough to just fit the pipette tip and allowed to gel in vivo, we implanted both groups in parallel within the same immune-deficient mouse. The study was performed with cells from two human MSC donors, D236 and D240. In the open approach, the constructs comprising the MSC aggregates, microceramic particles, and the PRP gel were generated in vitro on six-well plates before introducing them into the subcutaneous pocket via a 2-mm incision. In the minimally invasive approach, the components of the construct were directly introduced into the subcutaneous pocket via a small hole, just big enough to fit the pipette tip. On histological evaluation of the two types of samples after 6 weeks, the constructs employing cells from D236 had on an average 2.9% and 3.1% of the scaffold area filled with newly formed bone in the case of the open and injectable approach, respectively. In the case of constructs employing cells from D240, on average 5.4% of the scaffold area was covered with bone in the open approach versus 4.8% for the injectable approach. In both the donors, the differences were not statistically significant, (Fig. 5) indicating that in our system, the minimally invasive approach is a viable alternative to the open approach for the purpose of bone tissue engineering.

Comparison of the conventional open approach for introducing constructs into a defect with the minimally invasive approach. Cell aggregates mixed with microceramic particles and fibrin–thrombin gel were either generated ex vivo and introduced into the nude mice through an open incision or directly delivered into the subcutaneous pocket through a pipette tip. Both sets of constructs were implanted in parallel in the same animals. The study was repeated with two separate donors (D 236 and D 240). In both the donors tested, no significant difference was observed in the amount of bone obtained using the open-incision approach versus the minimally invasive approach of construct delivery via the pipette tip.
Discussion
Injectable bone TECs that are self-setting in situ and compatible with the use of MSCs can bring significant benefits in several clinical situations, such as mandibular atrophy, scaphoid nonunions, spinal disease, and craniofacial defects, where endoscopic efforts at placing conventional bone grafts are already under investigation.33,34 In this study, we developed a TEC suitable for clinical use and amenable to minimally invasive approaches by combining the human PRP-derived gel, hMSC aggregates, and microceramic particles. We verified the bone-forming capacity of the TEC using MSCs from multiple donors. The MSCs used in this study are of human origin and the microceramics are composed of calcium phosphate compounds that are commonly used for orthopedic and dental applications.35–37 The gelling of the PRP gel, in addition to being temperature dependent only occurs after the two components, that is, fibrin and thrombin are mixed together giving the surgeon time and flexibility during the surgical procedure.
Tisseel® (Baxter Biosciences) is a commercially available fibrin–thrombin-based sealant that is already approved for clinical use as an adhesive sealant in controlling bleeding during surgical procedures. 38 However, the commercially available product, unlike that used in this study, is not suitable for tissue engineering applications as the high cross-linking density of the fibrin network prevents cell migration within the construct.39,40Moreover, the PRP gel used in our current study can be an autologous preparation, which can be obtained easily in large quantities, at no extra cost. 26 Because of its autologous origins, there are no risks or concerns about disease transmission and immunogenic reactions as is the case for allogeneic or xenogeneic preparations. Furthermore, unlike the commercially available fibrin glue, the PRP gel is derived by activating the release of the native concentration of fibrin within the platelets with calcium chloride and thrombin. During the activation of the platelets, a myriad of growth factors are released, which can potentially enhance bone regeneration and vascularization of the construct.41,42
A study by Yamada et al. involved the use of a similar autologously derived fibrin–thrombin gel in conjunction with canine MSCs for repair of critical-sized defects in dogs. 26 In this study, the gel in addition to being the delivery vehicle also doubled up as the scaffolding material. According to studies reported in literature, a preformed solid matrix helps with improved retention of cells at the defect site, provides the cells with a substrate for bone deposition, acts as filler at the defect site, and in many cases, the osteoconductive and osteoinductive properties of the scaffold promote faster bone healing.18–21 In fact, we have preliminary results, which indicate that the calcium phosphate ceramics are necessary to generate bone in our system (data not shown).
A study by Mankani et al. described the use of single-cell suspension of hMSCs in combination with 0.5–1 mm HA/TCP particles and a mouse fibrinogen and thrombin mixture to generate bone in vivo. 10 Our study differed from this study with respect to the use of fibrin–thrombin gel of human origin and smaller ceramics, which aided the delivery of the construct into the defect site. Reports in literature suggest that the use of microparticles in place of bigger granules increases the surface area available for cell attachments and calcium release, both of which can influence in vivo bone formation. 43 The microparticles can also easily adapt to the contour of the defect without compromising the use of the minimally invasive delivery route for the graft placement. Finally, as the gel is very soft, the ceramics help to increase the mechanical strength of the tissue-engineered graft. It can be argued that use of even smaller sized particles can further aid the placement of constructs via the minimally invasive approach. According to literature evidence, at least in case of HA and TCP particles, 44 μm represents the lowest threshold below which bone marrow-derived hMSC–ceramic combinations do not yield any bone in subcutaneous locations in nude mice at the two time points tested, that is, 4 weeks and 10 weeks. However, while this provides an indication, it would be interesting to confirm the validity of these findings using particles of different sizes within our system.43,44
A salient feature of our study was the use of cell aggregates instead of single cells. We observed in the past that in vitro aggregation of the cells before in vivo administration, significantly improved their performance. Within this study, we observed no bone formation when unaggregated cells from the same donor were combined with the ceramics and gel and implanted in vivo. These results are in accordance with studies from other groups that have reported the beneficial effects of cell aggregation on the expression of osteogenic genes.29–31 It is believed that the behavior of the cells within the aggregate can be influenced by the improved integrin-specific signaling as a result of improved cellular cross talk within the aggregate.45,46 In addition to cell–cell signaling, culturing cells as aggregates also improves the interaction of the cells with the extracellular matrix which in addition to functioning as an adhesive substrate also acts as a reservoir of growth factors, which play a role in maintaining the differentiation potential of the cells. 47 Surprisingly, our results indicate that although the initial aggregation was beneficial for in vivo bone formation, neither the expression of the osteogenic genes nor the amount of bone obtained rose proportionately to an increase in the in vitro culture duration of the cell aggregates. One of the possible explanations was the increase cell death within the aggregates over time. This can be improved by either generating smaller aggregates or through the use of dynamic culturing techniques, which will ensure a better nutrient supply to the cells in the core of the aggregates.
In conclusion, we describe here, a clinically applicable system using human MSCs for the generation of TECs for delivery via a minimally invasive route. Future studies aimed at scaling up the size of the implants, implantations at orthotopic sites in immunocompetent animals, and investigations into the mechanical properties of the newly deposited bone in the minimally invasive approach compared to the conventional surgically placed implants, can all prove beneficial in the clinical translation of this technique. Possibilities to replace the need for aggregation of expanded MSCs with aggregation of the mononuclear fraction obtained directly from a fresh bone marrow or fat tissue may be of interest to further streamline the technique for clinical applications.
Footnotes
Acknowledgments
The authors gratefully acknowledge the support of the TeRMSmart Mix Program of The Netherlands Ministry of Economic Affairs and The Netherlands Ministry of Education, Culture and Science.
Disclosure Statement
No competing financial interests exist.