
Abstract
Neovascularization represents an important issue in tissue-engineering applications, since survival of implanted cells strongly relies on sufficient oxygen and nutrient supply. We have recently observed that human bone marrow-derived mesenchymal stem cells (MSCs) support neovessel formation originating from coimplanted endothelial cells (ECs) in vivo, suggesting that MSCs may function as perivascular cells by investing and stabilizing nascent EC-derived neovessels. In this study, we investigated EC-induced mural cell differentiation of MSCs in vitro. For this purpose, endothelial progenitor cells (EPCs) from two different origins, namely adult peripheral blood (pbEPCs) and neonatal cord blood (cbEPCs), or human umbilical vein endothelial cells (HUVECs), were cocultured with human MSCs to analyze the effect on MSC differentiation toward a smooth muscle cell (SMC)/pericyte phenotype. EPCs as well as HUVECs increased alpha-smooth muscle actin expression in MSCs upon cocultivation in a time-dependent manner. This effect was strongly dependent on direct cell-to-cell contact and extracellular signal-regulated kinase (ERK) signaling, but was not mediated by heterotypic gap junction communication. Beyond enhanced SMC marker gene expression in MSCs, EPCs also enhanced the functional characteristics of cocultured MSCs by increasing their ability to attach to EC tubes in vitro. In conclusion, our study has shown that EPCs from adult peripheral blood as well as from cord blood commit cocultivated MSCs toward an SMC/pericyte phenotype in a cell-contact- and ERK-dependent manner.
Introduction
Especially, EPCs have gained much interest, because these cells are capable of self-renewal, possess EC characteristics, and can be easily isolated noninvasively from peripheral blood.11,12 EPCs have already been used as a cell source for enhancing angiogenesis in regenerative medicine. In this context, it was reported that EPCs supported the regeneration of infarcted myocardium.13,14 The supportive function of EPCs in terms of neovascularization of tissues seems to be mainly due to a proangiogenic effect caused by secretion of angiogenic growth factors, whereas the vasculogenic potential of these cells seems to be rather low.15,16 However, it was shown that coimplantation of perivascular cells such as smooth muscle cells (SMCs) 17 or 10T1/2-cells 10 strongly enhances the vasculogenic potential of EPCs in vivo.
Recently, it was shown that a similar effect could be created by coimplantation of mesenchymal stem cells (MSCs),16,18 suggesting that coimplanted MSCs support EPC, as well as human umbilical vein endothelial cell (HUVEC)-mediated neovessel formation, most probably by acting as perivascular mural cells. This poses the question as to whether the interaction between EPCs and MSCs induced the differentiation of MSCs toward an SMC/pericyte phenotype, as it was previously shown for the interaction between macrovascular ECs and mesenchymal cells.19–21
To address this question, we monitored the cellular fate of MSCs upon coculture with EPCs from two different origins, namely from adult peripheral blood (pbEPCs) and postnatal umbilical cord blood (cbEPCs) in vitro. Our experiments demonstrate that EPCs differentiate cocultured MSCs toward an SMC/pericyte phenotype in a process that is strictly cell contact and ERK dependent and that does not involve gap junction communication between EPCs and MSCs.
Materials and Methods
Cell culture
Human pbEPCs were isolated and expanded as previously described.22,23 In brief, peripheral blood mononuclear cells were isolated from 50 mL of peripheral blood from human adult volunteers by density-gradient centrifugation with the Biocoll separating solution (Biochrom). Cells were plated on 25-cm2 culture flasks coated with rat type I collagen (50 μg/mL) in EGM-2 (Lonza) supplemented with single quots (human epidermal growth factor, vascular endothelial growth factor, human fibroblast growth factor-B, R3-IGF-1, ascorbic acid, heparin, hydrocortisone, and gentamicin/amphotericin-B) and 10% fetal calf serum (FCS) and cultivated at 37°C, 5% CO2 in a humidified atmosphere. After 4 days in culture, nonadherent cells were removed by washing with phosphate-buffered saline (PBS), and a new medium was applied, and the culture was maintained for another 4–5 weeks. Media were changed every 3 days. After reaching about 80% confluence, cells were trypsinized and seeded into 75-cm2 culture flasks coated with human fibronectin (50 μg/mL) (Sigma) for expansion. pbEPCs from passages 2–7 were used for experiments. To confirm the endothelial phenotype, EPCs were analyzed for cell surface expression of endothelial markers. EPCs were positive for CD31, von-Willebrand-factor (vWF), VEGFR-2, VE-Cadherin, CD105, and CD145, whereas the hematopoietic cell surface markers CD45 and CD14 were not expressed. Moreover, EPCs showed cobblestone morphology and were able to incorporate Dil-labeled acetylated low-density lipoprotein.
Human umbilical cbEPCs were purchased from Lonza and grown in EGM-2 (Lonza) supplemented with single quots and 10% FCS at 37°C, 5% CO2 in a humidified atmosphere. cbEPCs from passages 2–6 were used for experiments.
HUVECs were purchased from Promocell and were cultured in an EC growth medium (ECGM; Promocell) supplemented with 10% FCS and a supplement mix (epidermal growth factor, hydrocortisone, and basic fibroblast growth factor) at 37°C, 5% CO2 in 75-cm2 tissue culture flasks. HUVECs from passages 2–6 were used for experiments.
Human MSCs were isolated and expanded as described before. 24 In brief, mononuclear cells (MNCs) were purified by density-gradient centrifugation with Biocoll separating solution (Biochrom AG) from human bone marrow aspirate (Lonza). MNCs were seeded in culture flasks at a density of 5×105 cells/cm2 in an expansion medium (alpha-Minimal Essential Medium, 10% FCS, 50 μg/mL gentamicin, 5 ng/mL bFGF) at 37°C, 5% CO2. The medium was changed twice weekly, removing all nonadherent cells. Once adherent cells had grown to confluence, they were detached and reseeded at a density of ∼2000 cells/cm2 and cultivated for two further passages.
MSCs were routinely analyzed for characteristic cell surface marker expression. MSCs expressed CD105, CD90, and CD73, whereas CD45, CD34, and CD14 were not expressed. Moreover, MSCs were able to differentiate into osteoblasts, adipocytes, and chondrocytes in vitro.
Human dermal fibroblasts (hFBs) were isolated from resections during plastic surgical procedures by collagenase digestion following standard protocols. Fibroblasts were cultured in Dulbecco's Modified Eagle Medium containing 10% FCS and 1% penicillin/streptomycin at 37°C, 5% CO2.
Coculture of MSCs with different human primary cell types
MSCs were either grown in monoculture or cocultured in a 1:1 ratio with HUVECs, pbEPCs, cbEPCs, or hFBs in an ECGM (Promocell) supplemented with 10% FCS and supplement mix (EGF, hydrocortisone, and bFGF) at 37°C, 5% CO2 in 75-cm2 tissue culture flasks or six-well plates for different time periods (1–11 days). Gap junction communication was inhibited by adding 18α-glycyrrhetinic acid (18α-GA) (Sigma) to the cell culture medium at a final concentration of 50 μM. The extracellular signal-regulated kinase (ERK) pathway was inhibited by adding the MEK inhibitor PD98059 at a final concentration of 30 μM (Cell Signaling Technology).
For transwell culture, 8×104 MSCs were plated per well in six-well plates, and 8×104 HUVECs, pbEPCs, or cbEPCs were plated onto a 1-μm-pore membrane insert (BD Biosciences) that was placed above the wells.
Immunomagnetic separation
To separate MSCs and ECs after cocultivation, an immunomagnetic separation system (Invitrogen Dynal AS; Invitrogen) was used. In brief, cells were detached from the culture dishes by trypsin/ethylenediaminetetraacetic acid treatment. Enzymatic digestion was stopped by addition of 2 mL PBS and 5% FCS. Thereafter, cells were centrifuged (1000 rpm, 5 min) at room temperature and washed once with PBS and 0.1% bovine serum albumin (BSA). Cells were resuspended in 1 mL of PBS and 0.1% BSA, mixed with 25 μL of magnetic beads (Dynabeads; Invitrogen) coated with an anti-CD31 antibody, and incubated on a rotator for 30 min at 4°C. The EC binding to the CD31-coated Dynabeads was separated using a magnetic particle concentrator (Invitrogen Dynal AS; Invitrogen). The unbound cells (MSCs) were transferred to a new Eppendorf tube, and the separation process was repeated. Thereafter, MSCs were centrifuged (1000 rpm, 5 min) at room temperature and used for further experiments.
Western blot analysis
Cells were lyzed on ice by the addition of a radio-immunoprecipitation assay buffer (PBS, 1% NP-40, 0.5% sodium deoxycholate, 0.1% sodium dodecyl sulfate (SDS), 0.2 mM phenylmethane sulfonylfluoride, 1 μg/mL aprotinin, 1 μg/mL leupeptin, 1 mM Na-Orthovanadate). The particulate material was removed by centrifugation. The protein concentration of the supernatants was determined using the BCA protein assay kit (Thermo Scientific). Ten micrograms of total protein was separated by SDS–polyacrylamide gel electrophoresis, transferred to Hybond ECL nitrocellulose membranes (GE Healthcare), and probed with antibodies against alpha-smooth muscle actin (αSMA, monoclonal mouse, 1:250; Dako), calponin (monoclonal mouse, 1:1000; Sigma), or connexin43 (Cx43, monoclonal mouse, 1:250; Millipore). To confirm equal loading of the gel, the membrane was stripped and reprobed with monoclonal antibodies against β-actin (Sigma) or GAPDH (Abcam). The protein bands were visualized using enhanced chemiluminescence (ECL; GE Healthcare) and captured on Hyperfilm ECL (GE Healthcare).
Quantitative real time reverse transcription polymerase chain reaction
TaqMan reverse transcription polymerase chain reaction was carried out as previously described. 25 Total RNA was prepared using an RNeasy Mini Kit (Qiagen), according to the manufacturer's instructions. Total RNA (3 μg) was treated with 3 U of deoxyribonuclease I (DNase I) (Invitrogen) to digest genomic DNA contamination. Random-primed cDNA synthesis was performed using 3 μg of DNase I-treated total RNA and 50 U of StrataScript reverse transcriptase according to the manufacturer's instructions (Stratagene). TaqMan PCR assays were performed in 384-well optical plates on a LightCycler (Roche) using the Absolute QPCR ROX Mix (Abgene) according to the manufacturer's instructions. Oligonucleotide primers and probes for human GAPDH (GADPH forward: 5′-TGGGCTACACTGAGCACCAG-3′; GAPDH reverse: 5′-CAGCGTCAAAGGTGGAGGAG-3′, GAPDH probe: 5′-FAM-TCTCCTCTGACTTCAACAGCGACACCC-TAMRA-3′) were designed using Primer Express (Applied Biosystems) according to the company guidelines. Oligonucleotide primers and the TaqMan probe for αSMA were purchased from Applied Biosystems (Cat. No.: Hs00426835_g1). The thermal cycling conditions were 95°C for 15 min, followed by 50 cycles at 95°C for 15 s and at 60°C for 1 min. Data were analyzed using the relative standard curve method, with each sample being normalized to GAPDH to correct for differences in RNA quality and quantity. Results from three experiments are expressed as mean arbitrary units±standard deviation (SD).
Immunostaining
Cells were fixed with ice-cold methanol and incubated for 30 min with a blocking solution (5% goat serum; Sigma), followed by incubation with the respective primary antibodies, monoclonal mouse anti-αSMA antibody (diluted 1:200; Dako), monoclonal mouse anti-calponin antibody (diluted 1:2000; Sigma), and monoclonal mouse anti-smooth muscle myosin antibody (diluted 1:100; Sigma) in 5% goat serum overnight at 4°C. After washing with PBS, endogenous peroxidase activity was blocked by incubation in 0.3% H2O2 for 30 min at room temperature (RT). After three additional washing steps with PBS, the corresponding secondary antibody (ready-to-use horseradish peroxidase-conjugated goat anti-mouse immunoglobulin; Dako) was applied and incubated for another 30 min at RT. Thereafter, cells were washed three times with PBS and then exposed to the DAB chromogen substrate (ready to use; Dako) for approximately 7 min. Thereafter, cells were washed twice in H2O bidest and weakly counterstained with hematoxylin. Cells were photographed using an inverted microscope.
Modified Matrigel assay
A modified Matrigel assay was performed as described before. 20 In brief, growth factor-reduced Matrigel (Becton Dickinson) was thawed on ice overnight and spread evenly over each well (30 μL) of a 24-well plate. The plates were incubated for 30 min at 37°C to allow the Matrigel to solidify. Approximately 1.5×104 MSCs grown in monoculture or in coculture with HUVECs, pbEPCs, or cbEPCs for 7 days were isolated by negative immunomagnetic cell separation, labeled with PKH26 according to the protocol provided by the manufacturer (Sigma), and seeded with unlabeled HUVECs (3×104) in triplicate into each well in 500 μL of ECGM (Promocell) supplemented with 10% FCS and supplement mix. Plates were then incubated at 37°C, 5% CO2 in a humidified atmosphere for 20 h. Thereafter, cells were fixed with ice-cold methanol. Images were captured at a 100-fold magnification under bright field and red fluorescence. Images were merged, and numbers of red PKH26-labeled MSCs that associated with HUVEC sprouts were calculated for each region of interest (ROI). Nine ROIs per experimental group were analyzed using the freeware image analysis program ImageJ (http://rsb.info.nih.gov). Results for the different experimental groups were expressed as means±SD from nine randomly selected ROIs per group.
Scrape-loading assay
The presence of functional gap junctions in MSC/HUVEC cocultures grown in the presence or absence of 18α-GA (Sigma) for 2 and 5 days was investigated by a scrape-loading assay as described previously 26 using Lucifer Yellow, a small molecule that is able to diffuse intercellularly via gap junctions. In brief, MSCs and HUVECs were seeded in a 1:1 ratio in 12-well cluster plates in triplicates. Thereafter, cells were grown in ECGM, 10% FCS and supplement mix for 2 or 5 days either in the presence or absence of 18α-GA (50 μM). The medium was changed on day 3. At the end of the incubation period, cells were washed twice with PBS and overlayed with 1 mL PBS containing 1 mg/mL Lucifer Yellow (Sigma) plus or minus 18α-GA (50 μM). Using a scalpel blade, one scrape per well allowed the dye to enter the damaged cells. Five minutes later, the cells were washed three times with PBS and fixed in 3.7% formalin. Cells were photographed at a 100-fold magnification at bright field and fluorescence using an inverted fluorescence microscope.
Statistical analysis
Statistically significant differences between groups were determined by using an unpaired Student's t-test. Statistical significance was defined when p<0.05.
Results
To investigate whether cocultivated HUVECs increase the expression of SMC markers in MSCs, HUVECs were cocultivated with human MSCs for 7 days at a 1:1 ratio. Thereafter, MSCs were isolated by negative immunomagnetic cell separation and analyzed for αSMA and calponin expression by Western blot analysis. As shown in Figure 1A, HUVEC cocultivation leads to an increase in αSMA as well as calponin expression in relation to MSCs grown in monoculture. As a positive control, MSCs grown in monoculture were treated with TGF-β1, which also leads to a strong upregulation in the expression of the SMC markers αSMA and calponin, as previously reported.27,28 Upregulation in the expression of the SMC markers αSMA, calponin, and smooth muscle myosin by cocultivated HUVECs or by TGF-β1 treatment was also confirmed by immunohistochemistry as shown in Figure 1B.
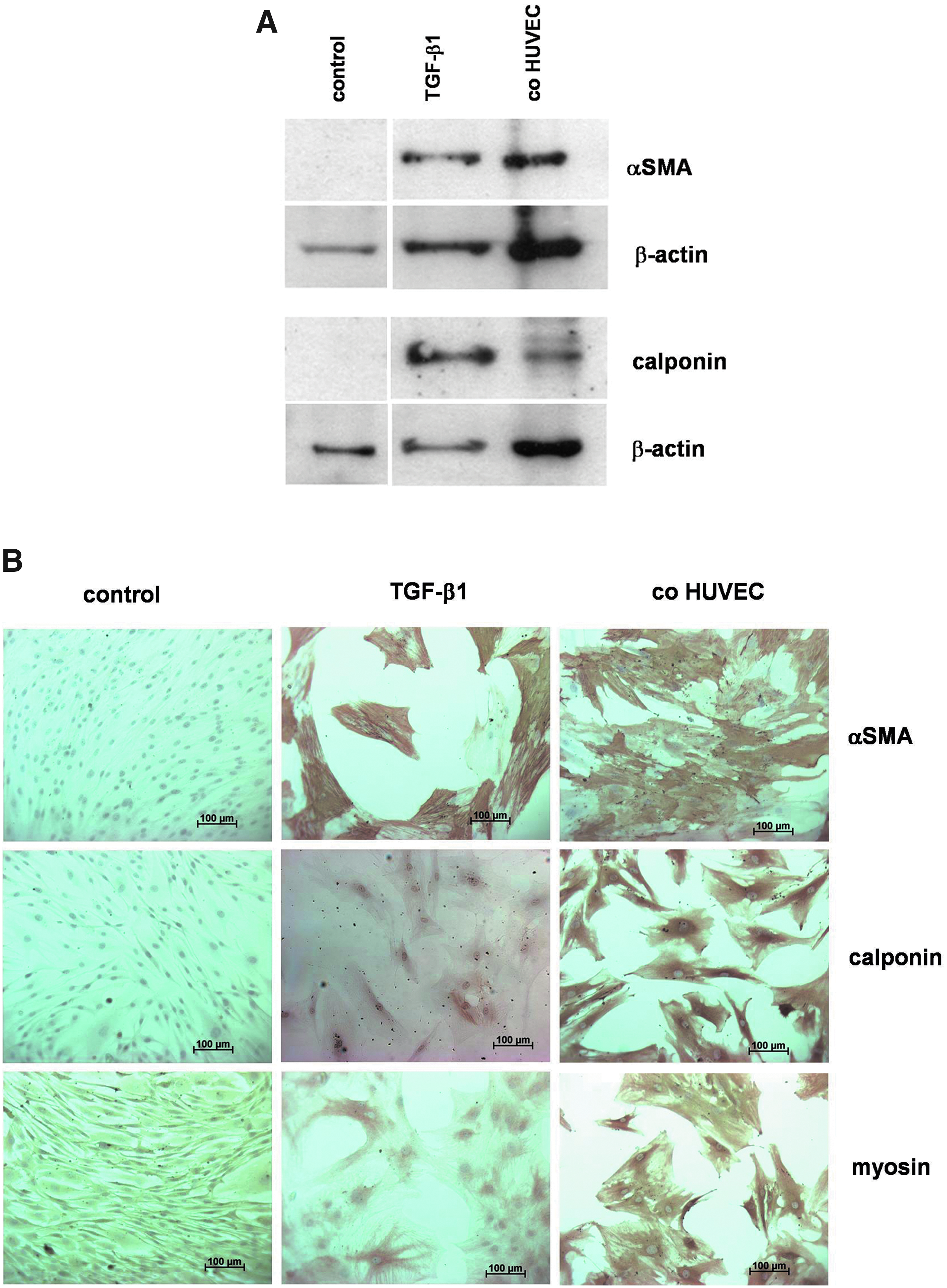
Induction of SMC markers in MSCs upon cocultivation with HUVECs.
We next investigated whether αSMA expression in MSCs could also be induced by cocultivation with EPCs. For this purpose, EPCs from two different origins were used, namely EPCs from peripheral blood (pbEPCs) or from cord blood (cbEPCs). As demonstrated by Western blot analysis, both EPC populations induce αSMA expression in cocultured MSCs to a similar extent as cocultured HUVECs (Fig. 2A). The inducing effect on αSMA expression was also detected in immunohistochemical stainings. Interestingly, human dermal fibroblasts (hFBs) were unable to induce αSMA expression in cocultivated MSCs, suggesting that the ability to induce SMC differentiation of MSCs seems to be cell type-specific and is probably restricted to cells of endothelial origin (Fig. 2B).

Effects on αSMA expression in MSCs upon cocultivation with different types of ECs or hFBs.
Time-course experiments revealed a time-dependent increase in αSMA expression in MSCs cocultivated with either HUVECs or pbEPCs (Fig. 3). The inductive effect of EC cocultivation on αSMA expression could first be seen after a 3–5-day cocultivation period and lasted for at least 11 days. Notably, HUVECs and pbEPCs displayed a very similar time course in the induction of αSMA expression in MSCs upon cocultivation.

Time course of HUVEC- and pbEPC-induced αSMA expression in cocultured MSCs. MSCs were grown in monoculture (control) or in coculture with HUVECs (co-HUVECs) or pbEPCs (co-pbEPCs) for the indicated time periods. Thereafter, MSCs were isolated by negative immunomagnetic cell separation and analyzed for αSMA expression by Western blotting. Cellular proteins (10 μg/lane) were separated by 7.5% SDS-PAGE, blotted on nitrocellulose membranes, and probed with an antibody against αSMA. Equal loading of the gel was confirmed by reprobing of the membrane with an antibody against β-actin. Color images available online at www.liebertpub.com/tea
It was previously reported that the effect of cocultured ECs on the expression of SMC markers in mesenchymal cells is mediated by direct cell contact. 19 To investigate whether this also holds true for our cocultivation system, we employed a transwell chamber system to analyze αSMA expression in MSCs. This cocultivation method permits diffusion of soluble molecules while preventing direct cell contact between the cocultured cell populations. In this experimental setting, HUVECs, pbEPCs, or cbEPCs were unable to induce αSMA expression in cocultivated MSCs. In contrast, direct cocultivation permitting heterotypic cell contacts between the different EC types and MSCs strongly induced αSMA expression in MSCs (Fig. 4).
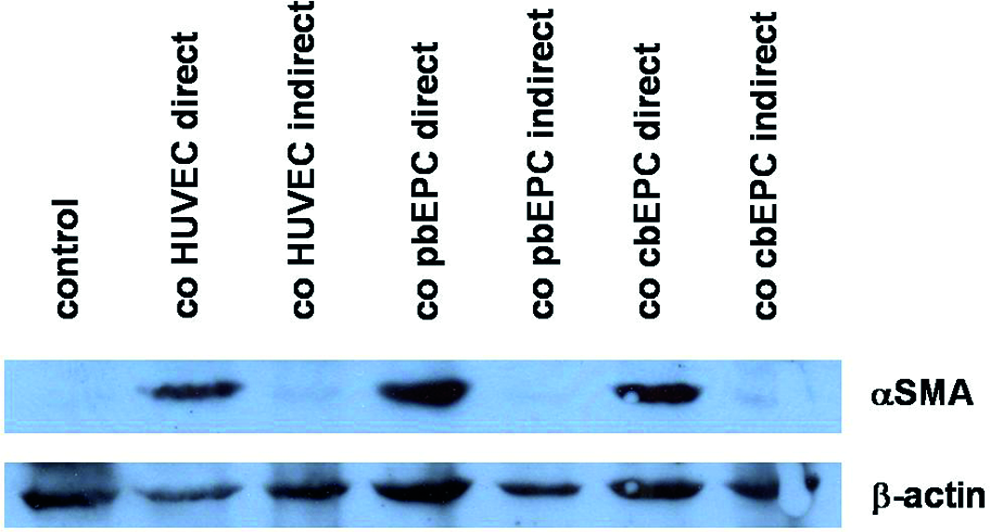
EC-induced αSMA expression is dependent on direct cell contact. MSCs were grown in monoculture (control) or in direct or indirect coculture with HUVECs, pbEPC, and cbEPCs. Thereafter, MSCs were isolated by negative immunomagnetic cell separation and analyzed for αSMA and β-actin expression by Western blotting. Color images available online at www.liebertpub.com/tea
It was previously shown in experiments with embryonic fibroblasts from undifferentiated mesenchyme of Cx43 knockout mice that gap junction communication is involved in EC-induced mural cell differentiation. 29 We therefore investigated whether inhibition of gap junction communication may interfere with EC-induced αSMA expression in MSCs. For this purpose, we used 18α-GA, a highly specific inhibitor of gap junctions.30,31 As shown in Figure 5A, coculture of HUVECs and cbEPCs induced a strong upregulation of αSMA mRNA expression in MSCs in relation to MSC monoculture (control). This upregulation was also seen in MSC/pbEPC cocultures, although to a lesser extent. However, 18α-GA treatment had absolutely no effect on αSMA mRNA expression, neither in MSC monoculture nor upon cocultivation with HUVECs, pbEPCs, or cbEPCs. Similarly, αSMA protein expression in MSCs was also completely unaffected by 18α-GA treatment as evidenced by Western blot analysis (Fig. 5B), indicating that the inductive effect of endothelial cell cocultivation on αSMA expression in cocultured MSCs is not modulated by inhibition of gap junction communication. To prove that the gap junction inhibitor 18α-GA is really functional, we performed scrape-loading dye transfer assays on MSC/HUVEC cocultures (Fig. 5C). Cells were grown for 2 or 5 days in the presence or absence of 18α-GA (50 μM). After wounding the cell monolayer by scraping with a scalpel blade, Lucifer Yellow is transferred via gap junctions from the wounded cells to neighboring nonwounded cells. This diffusion was almost completely inhibited in 18α-GA treated cells.

Effect of functional inhibition of gap junction communication between EC and MSCs by 18α-GA. MSCs were either grown in monoculture (control) or in coculture with HUVECs (co-HUVECs), pbEPCs (co-pbEPCs), or cbEPCs (co-cbEPCs) for 5 days either in the absence or the presence of 18α-GA (50 μM). Thereafter, MSCs were isolated by negative immunomagnetic cell separation.
To further confirm functionality of gap junction inhibition by 18α-GA, we investigated the effect of this inhibitor on Cx43 phosphorylation, since it was published previously that 18α- as well as 18β-GA induce dephosphorylation of connexins, which is associated with disassembly of gap junction plaques and subsequent disruption of gap junction communication.32,33 As shown in Figure 5D, 18α-GA treatment decreased the amount of the hyperphosphorylated form of Cx43 (P2) in control MSCs as well as in MSCs cocultivated with HUVECs or pbEPCs. In this Western blot, we have used the same protein lysates that have already been used in the αSMA Western blot.
These results confirm not only that cells respond to 18α-GA but also that this gap junction inhibitor is effective in inhibiting gap junction signaling in MSC/EC cocultures. We conclude from these results that heterotypic gap junction communication is not involved in HUVEC- or EPC-induced differentiation of MSCs toward an SMC phenotype.
To determine the role of the ERK pathway in EC-induced differentiation of MSCs, we examined the effect of the MEK inhibitor PD98059 on the expression of αSMA in MSCs in response to cocultivation with the different EC types by Western blot analysis (Fig. 6). Inhibition of the ERK pathway attenuated HUVEC-, pbEPC-, as well as cbEPC-induced αSMA expression in cocultivated MSCs (Fig. 6A). Quantification of αSMA signal intensities and normalization to GAPDH revealed between 87% (co-cbEPCs) and 73% (co-pbEPCs) inhibition of EC-induced αSMA expression in cocultured MSCs (Fig. 6B).
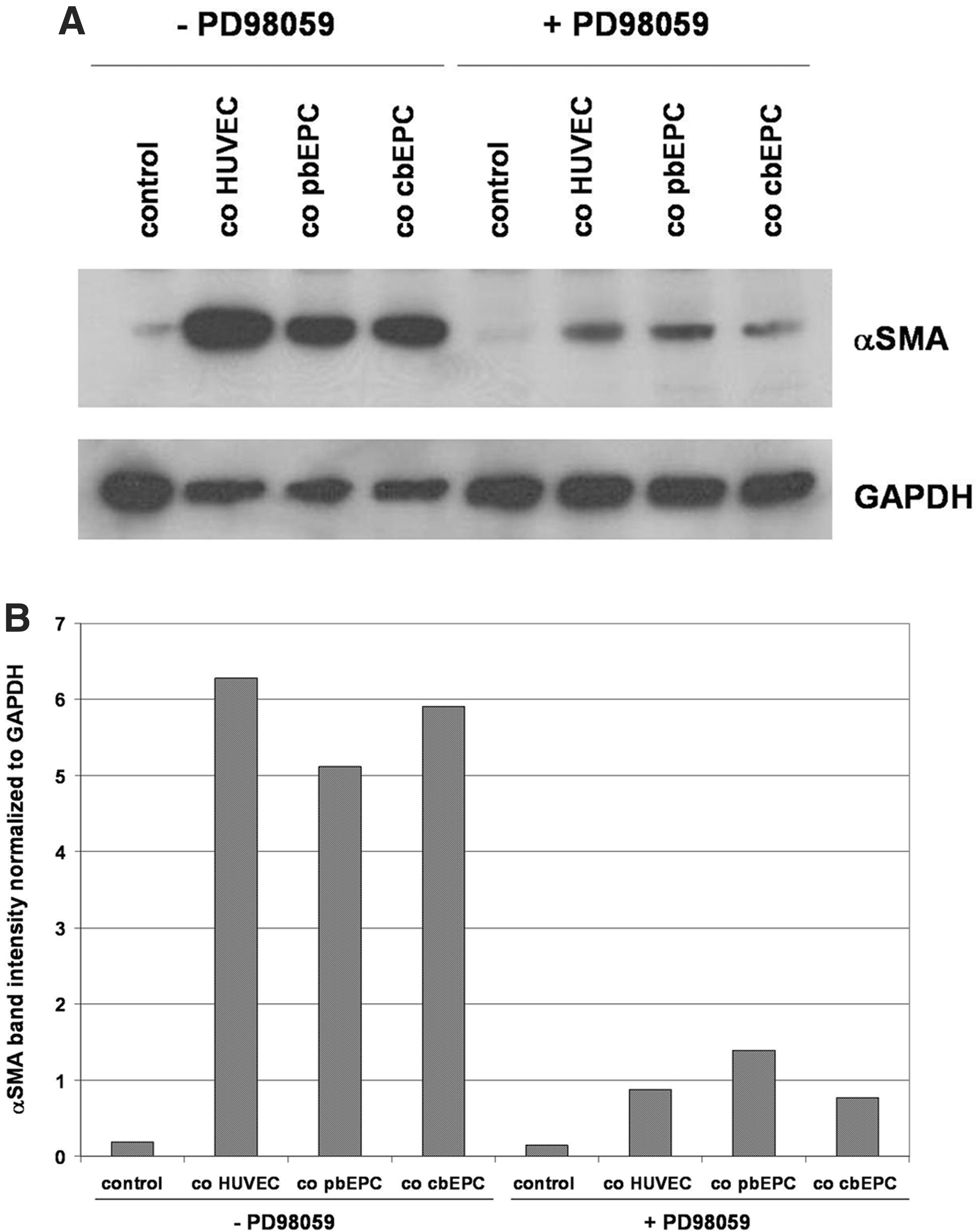
Effect of ERK inhibition on EC-induced αSMA expression in cocultivated MSCs.
To investigate the functional characteristics of MSCs that were shifted toward an SMC phenotype by EC precocultivation, a modified Matrigel assay was employed. MSCs grown in monoculture or in coculture with HUVECs, pbEPCs, or cbEPCs for 7 days were isolated by negative immunomagnetic cell separation, labeled with PKH26, mixed with HUVECs, and seeded on Matrigel-coated cell culture dishes. In this assay, co-seeded unlabeled HUVECs formed vascular sprouts on Matrigel. As shown in Figure 7A, red PKH26-labeled MSCs from cocultures with HUVECs, pbEPCs, or cbEPCs showed enhanced attachment to HUVEC sprouts in comparison to MSCs grown in monoculture. Quantification of the number of red PKH26-labeled MSCs that associated with HUVEC sprouts revealed statistically significant increased coverage rates in groups where MSCs were precultured with the different EC populations in comparison to the control group (Fig. 7B).

Functional characteristics of MSCs differentiated to SMCs by coculture with ECs. MSCs from monoculture (control) or from cocultures with HUVECs (co-HUVECs), pbEPCs (co-pbEPCs), or cbEPCs (co-cbEPCs) were labeled with PKH26 (red), mixed with HUVECs, and seeded on Matrigel.
These results strongly suggest that EC-mediated cell-contact-dependent differentiation of MSCs toward an SMC phenotype significantly enhanced their functional properties in terms of binding to in vitro formed HUVEC tubes.
Discussion
MSCs represent a very promising cell type in tissue-engineering applications, because these cells can be easily isolated from bone marrow or fat tissue with little or no donor-site morbidity. Moreover, these progenitor cells display a high proliferation rate ex vivo while preserving their multilineage differentiation potential and can be differentiated into several mesenchymal cell types such as chondrocytes, osteoblasts, and adipocytes.34,35 It was also reported that MSCs can be committed to an SMC/pericyte cell fate in vitro. This can be accomplished by treatment with TGF-β1 28 or by cocultivation with ECs. In this context, it was shown that cocultivation of MSCs with MS1-cells (a mouse pancreatic EC line) leads to an upregulation of the cardiac-/smooth muscle-specific gene myocardin. 36 Similarly, it was also demonstrated that cocultivation of human MSCs with human macrovascular endothelial cells (HUVECs) increased expression of αSMA in MSCs. 20
In our study, we investigated the effect of endothelial progenitor cells from two different origins (adult peripheral blood and postnatal cord blood) on MSC differentiation toward an SMC/pericyte phenotype in direct comparison to vascular-derived ECs, represented by HUVECs. In our experiments, HUVEC cocultivation induced SMC marker gene expression in MSCs to a similar extent as TGF-β1 treatment, which was used as a kind of positive control for the differentiation of MSCs toward SMCs. Interestingly, both pbEPCs as well as cbEPCs induced SMC marker gene expression in cocultured MSCs to a similar extent as HUVECs. Moreover, the time course of αSMA expression in MSCs in response to cocultivated HUVECs or EPCs was very similar. In both cases, an inductive effect could first be seen after 3–5 days and lasted for at least 11 days. This relatively delayed temporal expression pattern is in agreement with a previous report dealing with HUVEC-induced αSMA expression in bone marrow-derived MSCs. 20
It was previously reported that SMC maker gene expression in mesenchymal cells upon cocultivation with ECs is dependent on direct cell–cell contact and is not mediated by soluble factors.19–21,36 Interestingly, the same holds true for EPC-mediated upregulation of αSMA in MSCs. Upregulation of this SMC/pericyte marker can only be detected in MSCs upon direct cocultivation with HUVECs, pbEPCs, or cbEPCs, but not when MSCs and ECs are cocultivated in a transwell system that permits diffusion of soluble molecules while preventing heterotypic cell contact.
An important mechanism of a homotypic, as well as heterotypic contact-dependent, cell interaction is represented by gap junction communication. Studies based on mural cell progenitors isolated as embryonic fibroblasts from undifferentiated mesenchyme from mice deficient for Cx43 revealed that gap junction communication plays an important role in the upregulation of SMC markers by cocultivation with bovine aortic ECs. 29 In our experiments, we used 18α-GA to study the gap junction communication between human MSCs and human ECs. 18α-GA is a highly specific inhibitor of gap junction communication.30,31 Unexpectedly, 18α-GA was completely unable to inhibit HUVEC- or EPC-induced αSMA upregulation in cocultured MSCs on mRNA as well as on the protein level. In contrast, by investigating the effect of 18α-GA on gap junction-dependent Lucifer Yellow dye transfer in a scrape-loading assay, we have seen that 18α-GA severely inhibited intercellular gap junction communication. In an additional control experiment, we also analyzed the effect of 18α-GA on Cx43 phosphorylation by Western blot analysis, because it was previously reported that 18α- as well as 18β-GA induce the dephosphorylation of connexins.32,33 In this control experiment, we used the same protein lysates that were used for the determination of αSMA expression. As expected, 18α-GA strongly decreased the hyperphosphorylated form of Cx43 in MSC monocultures as well as in MSCs cocultivated with ECs. These results are of particular interest, because they confirm that 18α-GA is biologically effective in inhibiting gap junction signaling without significantly inhibiting HUVEC- or EPC-mediated upregulation of αSMA expression in cocultured MSCs. Our inhibition experiments with 18α-GA demonstrated that gap junction communication seems not to be important for HUVEC- and EPC-mediated differentiation of human MSCs toward an SMC/pericyte phenotype. However, the reason for the discrepancy between our data and the data published by Hirschi et al. 29 is not clear so far, but it is tempting to speculate that the conflicting results may be attributed to differences in the cell types used for the cocultivation experiments. It seems to be quite obvious that the mural cell progenitors isolated as embryonic fibroblasts from undifferentiated mesenchyme from Cx43 knockout mice are not identical to the human MSCs used in our studies. Moreover, Hirschi et al. 29 have used bovine aortic ECs, whereas we have used ECs from human umbilical vein or EPCs from cord blood or adult peripheral blood. Therefore, it seems to be very likely that the conflicting results concerning the involvement of gap junction communication may be the result of using different EC types and/or different mesenchymal precursor cells that could exhibit phenotypic or physiological differences.
To unravel the intracellular pathways that are responsible for EC-mediated upregulation of αSMA in MSCs, we analyzed the contribution of the ERK pathway by using the MEK inhibitor PD98059. Our experiments showed an at least 73% inhibition of EC-induced αSMA expression in cocultivated MSCs. It was previously published that MEK inhibition blocked TGF-β3-induced differentiation of adipose tissue-derived MSCs into an SMC lineage. 37 In another report, inhibition of the ERK pathway abrogated thromboxane-induced differentiation of MSCs into SMCs, evidenced by decreased expression of the SMC marker αSMA. 38 Moreover, the ERK pathway is also important for TGF-β1-induced αSMA expression in human lung fibroblasts and for the transition of lung fibroblasts to myofibroblasts.39,40 Our results demonstrated that the ERK pathway is also important for EC-induced differentiation of MSCs toward an SMC phenotype.
The in vivo function of SMCs/pericytes, as mural cell types, is to invest endothelial tubes during blood vessel assembly and remodeling. Attachment of mural cells is of great importance for the stabilization of blood vessels 41 and for EC survival. 42 This in vivo function can be recapitulated in vitro by a modified Matrigel assay. In this assay, HUVECs form vascular sprouts on Matrigel-coated culture dishes. We have seen in our experiments that co-seeded undifferentiated MSCs from monocultures display only a very weak attachment to HUVEC sprouts. In contrast, MSCs precultivated with HUVECs, pbEPCs, or cbEPCs displayed significantly enhanced attachment characteristics. This result further suggests that not only macrovascular ECs such as HUVECs but also EPCs from adult or postnatal origin are able to commit cocultured MSCs toward an SMC phenotype.
In this study, we investigated the effect of ECs on MSC differentiation. However, it would also be interesting to explore whether HUVECs or EPCs may also change their phenotype upon cocultivation with MSCs. We have seen in previous studies that cocultivation of HUVECs and MSCs severely increased proliferation and survival of MSCs, whereas proliferation and survival of HUVECs were completely unaffected. 43 However, this does not necessarily mean that HUVECs are completely unresponsive toward MSCs or other cell types. For example, in expression profiling assays, we recognized profound transcriptomic changes in HUVECs upon cocultivation with human primary osteoblasts with a striking upregulation of transcripts related to extracellular matrix components and proangiogenic factors (article in preparation). This suggests that HUVECs enter a proangiogenic state upon cocultivation with osteoblasts and possibly also upon cocultivation with MSCs.
In summary, our experiments have shown that EPCs enhance SMC marker expression in cocultivated human MSCs in a time-, cell-contact- and ERK-dependent manner without involvement of heterotypic gap junction communication. Enhanced SMC marker expression was also associated with enhanced mural cell function in vitro as evidenced by enhanced EC tube coverage.
Footnotes
Acknowledgments
We thank Beate vom Hoevel and Beate Schmitt for excellent technical assistance. This work was supported by funding through the Else Kröner-Fresenius-Stiftung (2010_A160).
Disclosure Statement
No competing financial interests exist.