
Abstract
Human embryonic stem cells (hESCs) and induced pluripotent stem cells (iPSCs) provide an unlimited source for the generation of human hepatocytes, owing to their indefinite self-renewal and pluripotent properties. Both hESC-/iPSC-derived hepatocytes hold great promise in treating liver diseases as potential candidates for cell replacement therapies or as an in vitro platform to conduct new drug trials. It has been previously demonstrated that the initiation of hESC differentiation in monolayer cultures increases the generation of definitive endoderm (DE) and subsequently of hepatocyte differentiation. However, monolayer culture may hinder the maturation of hESC-derived hepatocytes, since such two-dimensional (2D) conditions do not accurately reflect the complex nature of three-dimensional (3D) hepatocyte specification in vivo. Here, we report the sequential application of 2D and 3D culture systems to differentiate hESCs to hepatocytes. Human ESCs were initially differentiated in a monolayer culture to DE cells, which were then inoculated into Algimatrix scaffolds. Treatments of hESC-DE cells with a ROCK inhibitor before and after inoculation dramatically enhanced their survival and the formation of spheroids, which are distinct from HepG2 carcinoma cells. In comparison with monolayer culture alone, sequential 2D and 3D cultures significantly improved hepatocyte differentiation and function. Our results demonstrate that hESC-DE cells can be incorporated into Algimatrix 3D culture systems to enhance hepatocyte differentiation and function.
Introduction
Over the last decade, significant progress has been made in developing methods to differentiate pluripotent stem cells efficiently to hepatocyte-like cells (HLCs), through the sequential application of growth factors that are known to play an important role at various stages of liver development.6–9 The resulting HLCs exhibit a typical hepatocyte morphology, express almost all the hepatocyte markers, and perform a wide range of hepatocyte functions. However, the functionality of in vitro differentiated hepatocytes is lower than in primary hepatocytes. This is hardly surprising, given that functional performance of primary hepatocytes deteriorates after a short period in culture, 10 indicating that the culture conditions in these experiments are suboptimal for promoting and maintaining hepatocyte functionality. Therefore, there is a need to improve hepatocyte differentiation and culture conditions to enhance their functionality.
The majority of studies to date involving hepatocyte differentiation have been carried out in two-dimensional (2D) monolayer culture conditions throughout,6,7,11 or have been initiated with embryoid body (EB) formation, followed by 2D cultures. 12 Monolayer culture is thought to have certain advantages over EB formation for the efficient differentiation of pluripotent stem cells to specific lineages, as it enables all the cells to receive a similar level of stimuli from the culture medium and neighboring cells, compared with EB initiation, where cells receive a diverse range of signal inputs depending on their location within the EB. This difference may be especially crucial for the efficient generation of definitive endoderm (DE), since high levels of the growth factor Activin A through supplementation in the culture medium are necessary for the specification of hESCs to DE cells. 13 However, since liver development is also regulated by cell–cell contact, the absence of suitable cell contacts in the 2D culture conditions may contribute to the reduced functionality. Indeed, it has been demonstrated that a three-dimensional (3D) culture can reduce the hepatocyte functional decline that is seen using the 2D culture system. 14 Therefore, we hypothesized that transference from an initial 2D culture condition to a 3D system would significantly improve hepatocyte maturation and concurrently their functionality. Here, we report the differentiation of hESCs to hepatocytes utilizing a combination of 2D and 3D culture systems. We demonstrate that DE cells derived from monolayer differentiation of hESCs have the ability to form aggregates in Algimatrix scaffold 3D culture when treated with a Rho-associated kinase (ROCK) inhibitor. Residing in this 3D culture microenvironment therefore enhances subsequent hepatocyte maturation, leading to a clear improvement in their functionality.
Materials and Methods
Culture and differentiation of hESCs
The hESC line H1 was routinely cultured and propagated in Matrigel-coated plates with a mouse embryonic fibroblast-conditioned medium supplemented with basic fibroblast growth factor as previously described. 15 hESCs were differentiated into hepatocytes in monolayer culture using the protocol previously established in our laboratory (Fig. 1A). 7 Briefly, differentiation was initiated by culturing for 24 h with 100 ng/mL Activin A and 1 mM sodium butyrate (NaB), followed by 2 days with 100 ng/mL Activin A and 0.5 mM NaB. The cells were then cultured for 5 days in a stage II medium containing 20% KO serum replacement and 0.1% dimethyl sulfoxide (DMSO), followed by 4–5 days in stage III medium (L15 medium containing 8.3% fetal bovine serum, 8.3% tryptose phosphate broth, 10 μM hydrocortisone 21-hemisuccinate, 1 μM insulin and 2 mM glutamine, 10 ng/mL hepatocyte growth factor, and 20 ng/mL Oncostatin M). For 3D differentiation, hESCs were differentiated in the same medium as in the monolayer cultures until the second day of stage II when they were inoculated into the Algimatrix plate (Invitrogen 12684-023) (Fig. 1A). Before inoculation, cells were treated with 10 μM ROCK inhibitor Y-27632 (ROCKi) for 2 h before they were detached from the culture dish with accutase. Viable cells were transferred into a 24-well Algimatrix plate by centrifugation at 200 g for 3 min at either 0.5×106 or 1×106 cells per well in a medium also supplemented with ROCKi, which was withdrawn after 48 h.

Differentiation of human embryonic stem cells (hESCs) to hepatocyte-like cells by sequential application of two-dimensional (2D) and three-dimensional (3D) culture systems.
Analysis of the spheroids
Cell spheroids were released from the scaffold by 5-min incubation with 1 mL dissolving buffer (Invitrogen A11340-01) and were then harvested by centrifugation at 200 g for 4 min. Spheroids were stained by 90 s of incubation with propidium iodide (PI) resuspended at 40 μg/mL in phosphate-buffered saline (PBS), and images were captured by a Nikon E2000 phase-contrast fluorescent microscope. Dimensional analysis of the spheroids was performed using images taken under a phase-contrast microscope and using the medium axis of the spheroid. To determine the viable cell number, spheroids were dissociated into single cells by 8–10-min incubation with trypsin–ethylenediaminetetraacetic acid at 37°C, during which time the spheroids were gently resuspended twice using a pipette. Single cells were then stained with trypan blue and counted with a hemocytometer.
RNA extraction and reverse transcription–polymerase chain reaction
Total RNA was extracted using TRI reagent solution (Sigma T9424) following the manufacturer's instructions. Remaining traces of DNAs were removed by DNase I treatment. First-strand cDNA was synthesized from 2 μg total RNA using SuperScript II reverse transcriptase (Invitrogen) with oligo-dT primer (Invitrogen) in a 20-μL volume. All primer sequences and polymerase chain reaction conditions used are listed in Table 1.
CYP3A4 activity assay
Cells cultured in both monolayer and Algimatrix scaffold conditions were treated with either 10 μM rifampicin or 0.1% DMSO (vehicle only) for 48 h. The spheroids were then released from the scaffold as described above into a new culture dish. Subsequently, the cells in both spheroids and monolayer culture were subjected to the P450-Glo CYP3A4 assay using a Luciferin-IPA kit (Promega V9001) according to the manufacturer's instructions. Briefly, cells were incubated for 1 h at 37°C with a phenol-free DMEM supplemented with 10% fetal bovine serum containing 3 μM Luciferin-IPA after washed with PBS. A medium containing IPA substrate alone was used as background control. About 50 μL of the resulting medium was transferred to a 96-well plate containing an equal volume of Luciferin Detection Reagent and incubated at room temperature for 30 min to initiate the luminescent reaction. The remaining cells were enzymatically dissociated and quantified via cell count. Activity was measured on a Perkin Elmer Victor 2 luminometer. Values were corrected to account for background. For each treatment, three independent experiments were performed to allow statistical analysis.
BrdU incorporation assay
hESC-DE or HepG2 cells were inoculated into an Algimatrix scaffold with 0.5 million cells per well. After 24 h, BrdU was added into the medium to a final concentration of 10 μM and incubated for 48 h. The spheroids were then released from the scaffold, dissociated as described above, and placed onto coverslips. After 24 h culture without BrdU, cells were then fixed by 10-min incubation with 4% paraformaldehyde, washed with PBS, and treated with 2M HCl for 1 h at 37°C. Cells were then washed and blocked for 1 h in PBS containing 10% goat serum, incubated with mouse anti-BrdU antibody (DSHB, 1:1000 dilution) for 1 h, followed by 30-min incubation with goat anti-mouse IgG Alexa Fluor 568 secondary antibody (Invitrogen; 1:400 dilution) at room temperature. Images were taken using a Leica SP5 fluorescent microscope.
Urea secretion assay
Urea secretion was assessed by a colorimetric assay (DIUR-500 BioAssay Systems) according to the manufacturer's instructions. Briefly, upon a quick thaw of the frozen supernatants, 50 μL water (blank), 50 μL standard (5 mg/dL), or 50 μL samples were transferred into triplicate wells of a clear-bottom 96-well plate. Then, 200 μL working reagent was added, and samples were mixed by gentle tapping. These were then incubated for 50 min at room temperature. A spectrophotometer plate reader was used to read absorbance at 430 nm, and urea secretion was calculated following the manufacturer's instructions.
Results
ROCK inhibitor promotes the survival and spheroid formation of hESC-DE cells in Algimatrix scaffold
Since monolayer culture condition can efficiently differentiate hESCs into DE cells, an essential early stage in the generation of hepatocytes,7,13 and 3D culture provides a better cell–cell contact, which enhances hepatocyte differentiation and function, 14 it is plausible to hypothesize that a combination of the two conditions would greatly enhance the generation of functional hepatocytes from hESCs. To test this hypothesis, we initiated DE differentiation in hESCs using monolayer culture previously established in our laboratory 7 (Fig. 1A) and then incorporated the resulting DE cells into 3D culture. We have previously shown that using this monolayer culture, a majority of the cells were differentiated into DE cells by the end of the stage I differentiation. 7 However, due to considerable cell death that occurs during the 24-h NaB (1 mM) treatment, it was difficult to obtain a sufficient number of cells for the 3D culture at end of the stage I. Therefore, we continued the differentiation on monolayer into the stage II for 1 day before subjecting them into the 3D culture in a bid to increase the number of viable cells (Fig. 1A). These cells are very similar to those found at the end of stage I, as they express high levels of the DE markers, Sox17 and FoxA2, but significantly reduced expression of the pluripotent markers, Oct4 and Nanog, with no expression of the neural marker Pax6 (Fig. 1B). These cells will henceforth be referred to as hESC-DE cells.
To incorporate cells into 3D culture, hESC-DE cells were first disassociated into single cells from the culture plate and then inoculated into a 24-well Algimatrix 3D culture plate at 0.5–1×106 cells per well. HepG2 cells were also inoculated into the plate in the same way as hESC-DE cells. Interestingly, spheroids were observed in HepG2 cells after 5 days in the 3D culture, but none were visible in hESC-DE cultures (Fig. 2A). PI staining revealed considerable cell death in the hESC-DE cultures (Fig 2B-upper panel). As the HepG2 cells grew well in the 3D culture plate, we surmised that the scaffold itself does not induce any detrimental effect on the cells that leads to their death. As such, we reasoned that dissociation of the hESC-DE cells might be the cause of the high levels of cell death observed postinoculation. It has been reported previously that dissociation of hESCs into single cells increases ROCK-dependent hyperactivation of actin–myosin contraction, which induces apoptosis,16,17 and that treatment of hESCs with the ROCK inhibitor Y-27632 (ROCKi) significantly reduces apoptosis after dissociation. 18 To test whether use of ROCK inhibitor could reduce hESC-DE cell death after dissociation, the cells were pretreated with 10 μM ROCKi for 2 h before disassociation, and the treatment was continued for further 48 h after inoculation. With this treatment, spheroids were clearly observed 24 h after inoculation and were well formed after 9 days with no signs of detectable apoptosis (Fig. 2B). By contrast, ROCKi treatment did not show a significant effect on their ability to form spheroids in HepG2 cells. These results therefore suggest that hESC-DE cells are different from HepG2 cells and require the inhibition of the ROCK signaling pathway to survive and to form 3D structures in Algimatrix scaffold.

Effect of ROCK inhibitor (ROCKi) on cell survival and spheroid formation of hESC-DE cells in Algimatrix scaffold.
hESC-DE cells form spheroid in the 3D culture via cell aggregation
Since different growth behaviors were observed with hESC-DE cells and HepG2 cells upon their inoculation into the Algimatrix scaffold, we next examined how spheroids are formed in these two cell types, by monitoring the cells immediately after the inoculation. To our surprise, compact 3D aggregates were already visible in hESC-DE cells 3 h after inoculation, with larger aggregates formed when more cells were inoculated, whereas HepG2 cells still appeared as single cells or loose-contacted cell clusters at the time (Fig. 3A, upper panel). By 24 h in the 3D scaffold, hESC-DE aggregates appeared as spheroids, and similar spheroids were also apparent in HepG2 cells (Fig. 3A, middle panel). The size of spheroids in the hESC-DE cells did not change significantly, whereas the spheroids in the HepG2 cells gradually increased in size during the first 48 h in 3D culture (Fig. 3A, lower panel). These data suggest that hESC-DE cells may form spheroids predominantly via cell aggregation, which can occur within a relatively short time period and may be promoted by a ROCK inhibitor, whereas HepG2 cells may form spheroids through cell aggregation as well as cell proliferation.
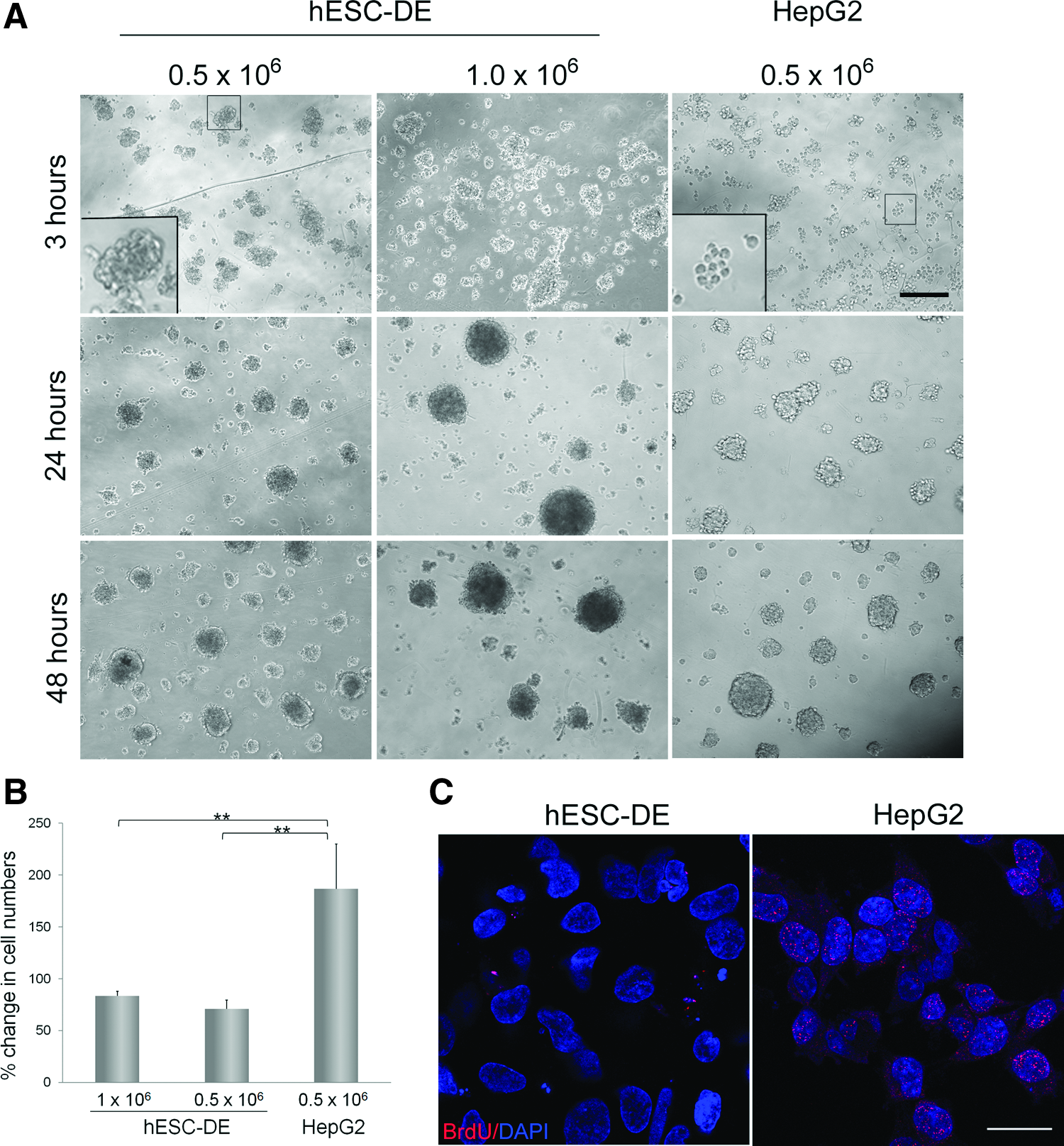
Spheroid formation and cell proliferation in Algimatrix 3D cultures.
To validate this observation, we compared the total viable cell numbers seeded at inoculation with those after 9 days of 3D culture in both cell types. As expected, the number of viable HepG2 cells in the 3D culture notably increased during this time period, although not to the same extent as in the 2D culture, whereas the number of hESC-DE cells showed a decrease in numbers, most likely due to cell death (Fig. 3B). To further confirm the proliferative properties of the two cell types, we incubated the cells with BrdU for 48 h after 3D inoculation, and then stained the cells with an anti-BrdU antibody. HepG2 cells showed positive signals with the BrdU antibody, whereas positive staining was not detected in the nuclei of hESC-DE cells. Therefore, these results confirm that hESC-DE cells do not proliferate after inoculation into the Algimatrix scaffold.
Cell density affects the size of spheroids in the Algimatrix scaffold
Spheroid size has been reported to affect hepatocyte viability and function in the 3D culture. 14 Since the cell spheroids in culture do not have a vascular network, cells constituting the center of the spheroids become hypoxic and have a reduced uptake of oxygen from the cell medium if they are larger than 100 μm in size. On the other hand, if the spheroid size is too small, they produce significantly less albumin as a result of poor maturation. 19 To study factors that affect the spheroid size, we categorized the spheroids produced by our 3D culture into three groups according to their size: small with spheroids of <50 μm in diameter, medium between 50 and 100 μm in diameter, and larger over 100 μm in diameter, and examined the effect of initial inoculation seeding density on the spheroid size. We measured and categorized ∼100 spheroids from each inoculation condition 9 days after the cells were inoculated. The results show that in hESC-DE cells, a majority (84%) of spheroids produced from the lower seeding density of 5×105 cells/well fell into the medium-size bracket, with <10% of spheroids falling into the larger- or smaller-size group. However, at higher seeding density of 1×106 cells/well, almost 20% and 30% of the spheroids were in the large- and smaller-size group, respectively, with <60% in the medium-size category (Fig. 4). In HepG2 cells, the lower seeding density of 5×105 cells/well produced very few spheroids (<5%) of the large size. These results suggest that the initial seeding density of hESC-DE cells has an effect on the sizes of the spheroids formed in the 3D scaffold.
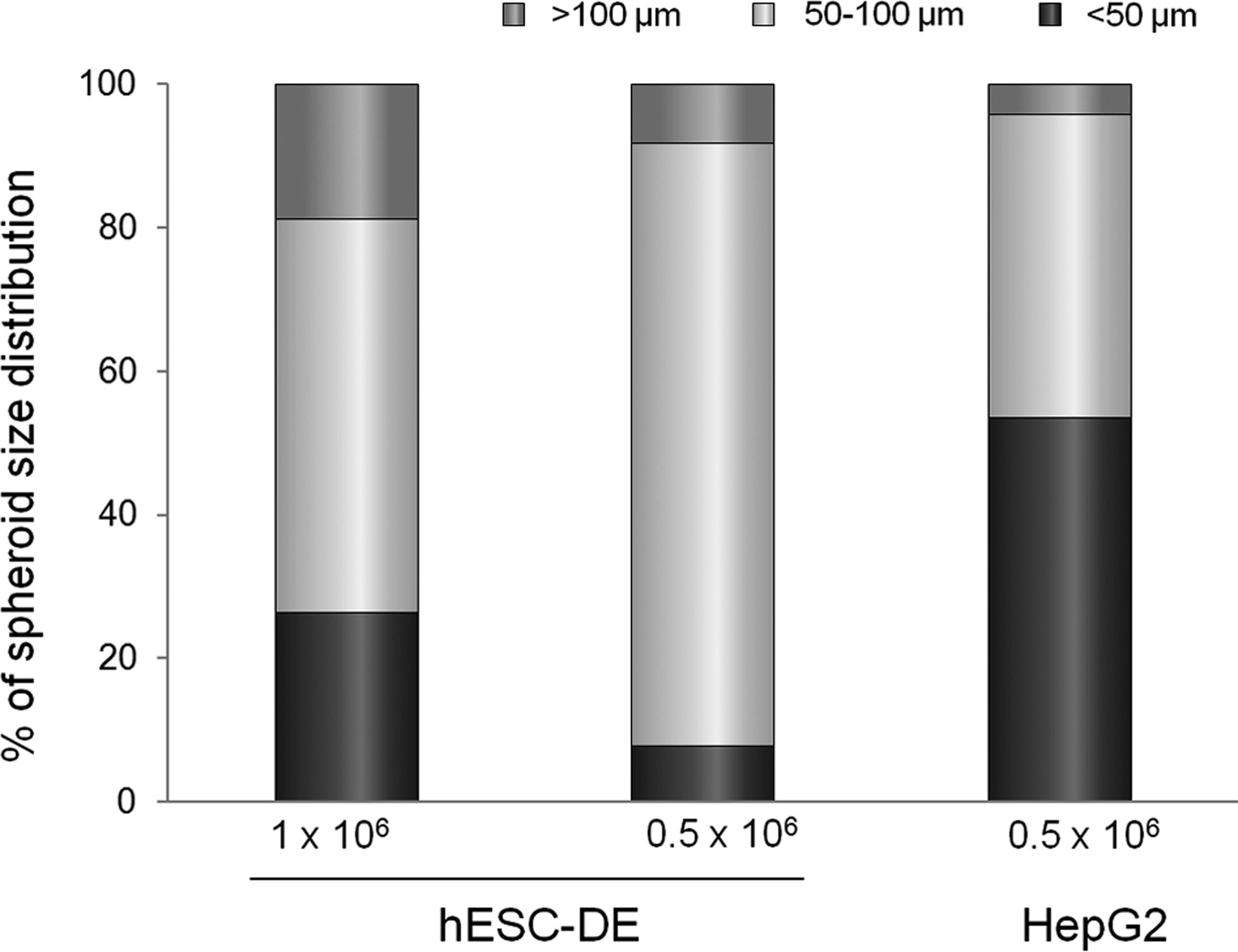
Effect of seeding density on spheroids' sizes. Spheroids from each culture were released after 9 days in the Algimatrix scaffold, and their size was measured from images taken with a phase-contrast microscope as described in the Materials and Methods section. At least 100 randomly selected spheroids were measured from each culture condition and grouped according to their size.
Algimatrix 3D culture promotes hepatocyte differentiation and function
Alginate scaffold has been found to promote cells from the liver of newborn rats to differentiate into functional liver tissues. 20 To evaluate whether the Algimatrix 3D culture has any advantage over monolayer culture in differentiating DE cells into hepatocytes, we compared the hepatic gene expression profiles in the 3D culture to those in monolayer as well as to undifferentiated hESCs and the hESC-DE cells used at the 3D inoculation stage. Unsurprisingly, undifferentiated hESCs exhibited high levels of the pluripotent gene Oct4, but showed no expression of hepatic markers. Expression of the DE marker, Sox17, was apparent in hESC-DE cells, but neither Oct4 nor any hepatic genes were detectable. In contrast, hESC-DE cells that had been further differentiated in both 2D and 3D culture conditions expressed the hepatocyte genes, albumin, apolipoprotein F (ApoF), tryptophan dioxygenase (TO), and cytochrome P450 family members, CYP3A4 and CYP7A1 (Fig. 5A). Significantly, the cells differentiated in the Algimatrix 3D scaffold exhibited considerably higher levels of hepatocyte-specific markers than the cells differentiated in monolayer, particularly those that are expressed predominantly in the more mature hepatocytes, such as ApoF, TO, and CYP3A4. 21
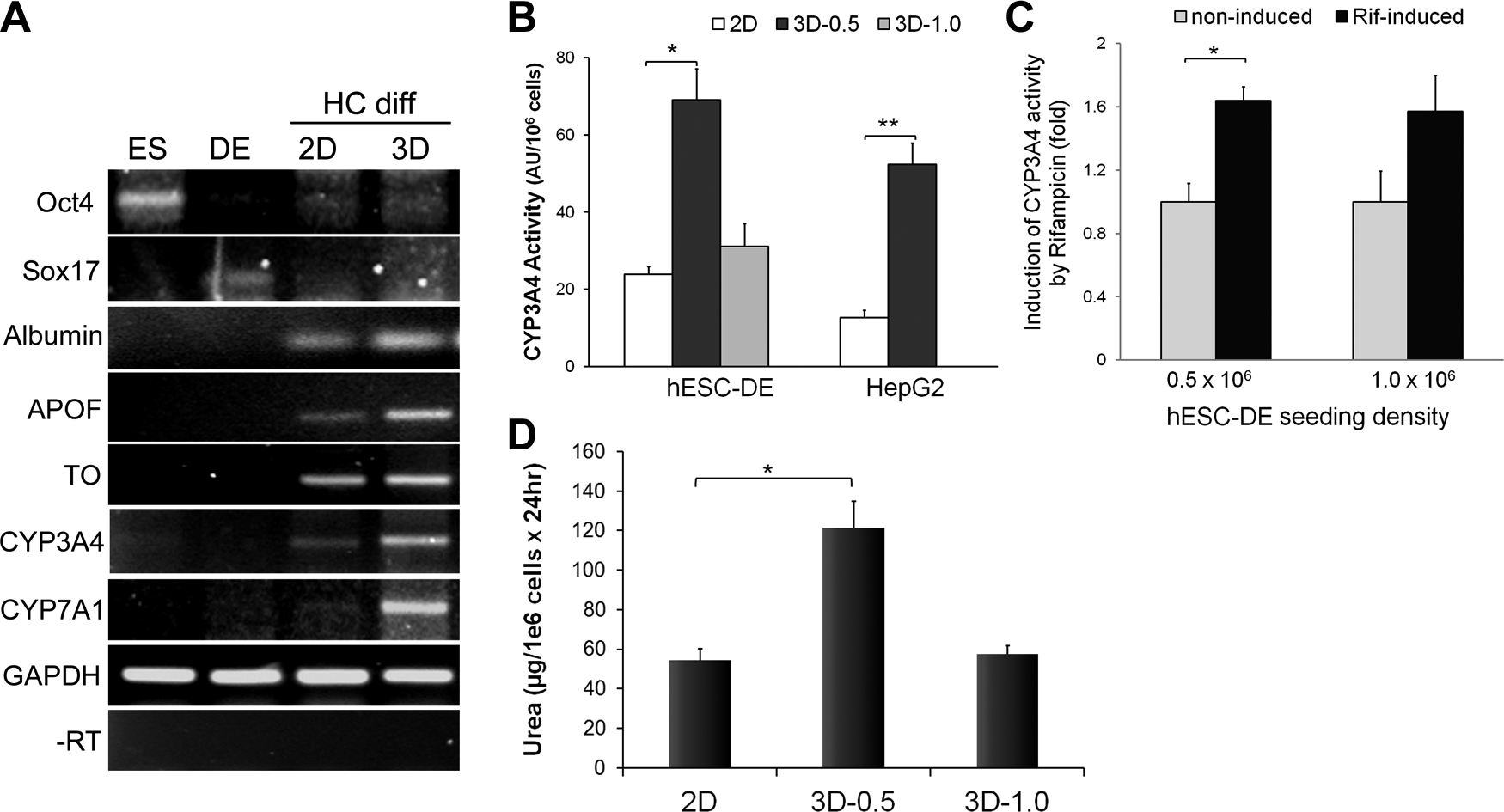
Comparison of the effect of 2D (monolayer) and 3D culture conditions on hepatocyte marker expression and functionality.
Since CYP3A4 is the most crucial P450 enzymes in the human liver, metabolizing about half of all drugs on the market today, we examined CYP3A4 activity in hESC-DE and HepG2 cells that were either cultured in 3D conditions or differentiated in monolayer to further determine the improvement of 3D culture on the differentiation and function of hepatocytes. In comparison with the monolayer culture system, at the lower seeding density of 0.5×106 cells/well, Algimatrix 3D culture significantly increased basal levels of CYP3A4 activity in both hESC-DE cells and HepG2 cells (Fig. 5B). The higher inoculation density also increased activity, but to a lesser extent, probably due to a suboptimal spheroid size. Furthermore, the cells cultured in the 3D condition also exhibited increased CYP3A4 activity in response to rifampicin induction (Fig. 5C). To further validate the improvement of 3D culture on hepatocyte differentiation and function, we measured urea secretion in hESC-derived hepatocytes from both 2D and 3D cultures (Fig. 5D), which showed that Algimatrix 3D culture significantly enhanced urea secretion when hESC-DE cells were seeded at 0.5×106 cells/well. Taken together, we have shown that Algimatrix 3D culture system improves the differentiation of hESC-DE cells to hepatocytes, and as a result, enhances the expression of hepatocyte markers and certain functions.
Discussion
We have demonstrated here that DE cells derived from hESCs are able to form spheroids in an Algimatrix scaffold. In comparison with the monolayer culture, Algimatrix 3D culture enhances hepatocyte differentiation and function. These results correspond with the previous findings from hESCs. 9 However, in the previous studies, the 3D culture was directly applied to the hESCs, in which hESCs were detached into small clumps and cultured in a low-adherent plate to form EBs. Since pluripotent hESCs in the EBs receive variable signals depending on their position, this method may have an increased chance of inducing the differentiation of cells to other lineages, which consequently reduces the final yield of hepatocytes. Our previous studies, as well as those of others, have shown that monolayer culture supplemented with high-dosage Activin A can differentiate hESCs efficiently to the DE cells that are a prerequisite for the generation of hepatocytes.7,13 Therefore, to increase hepatocyte production, we incorporated the 3D culture at a later stage in the differentiation protocol, once the hESCs were committed to DE. We differentiated hESCs for 4 days in monolayer culture with our established method and demonstrated that the cells were primarily differentiated into DE (Fig. 1B) before incorporating them into 3D culture.
Here, and in accordance with others, we have demonstrated that an alginate-based 3D culture system can enhance the functionality of HepG2 cells and has no toxic effects on the cells.22,23 Interestingly, we have also shown that hESC-DE cells behave differently from HepG2 cells when cultured in an Algimatrix 3D scaffold. Firstly, hESC-DE cells required the treatment of ROCKi to survive in the 3D culture after dissociation, whereas HepG2 cells do not require ROCKi to survive or to form spheroids. In addition, treatment of HepG2 cells with ROCKi showed no effect on their survival and proliferation. This phenotype of hESC-DE cells is similar to that reported in undifferentiated hESCs. 24 In hESCs, apoptosis upon dissociation is thought to be caused by ROCK-dependent actomyosin hyperactivation, which is triggered by the loss of E-cadherin-dependent intercellular contact. 17 However, hESC-DE cells stained with anti-E-cadherin antibody exhibited no clear positive signals (data not shown), indicating that the apoptosis induced by dissociation in hESC-DE cells is not a downstream effect of E-cadherin. Nonetheless, one cannot exclude the possibility that this phenotype could still be related to the loss of attachment-dependent actin–myosin contraction. 16 Further experiments are required to elucidate the underlying mechanisms. Secondly, hESC-DE cells aggregate more rapidly than the HepG2 cells. Thirdly, unlike HepG2 cells, hESC-DE cells were unable to proliferate in the Algimatrix 3D scaffold, which was demonstrated by the fact that there was no significant increase in the cell numbers after 9 days in the 3D culture, and that BrdU labeling showed no incorporation (Fig. 3). This was also different from their behavior in monolayer culture, in which hESC-DE cells were able to divide and incorporate BrdU. Although the exact mechanisms remain unclear, it has been demonstrated previously that the growth properties and differentiated functionality of rat hepatocytes are reciprocally modulated in culture by cell–cell contact, and that this reciprocal modulation is lost in hepatoma cells.25,26 Therefore, it is likely that increased cell–cell contact may account for the phenotype, which may also contribute to the improved differentiation and functionality.
We have analyzed spheroid formation and hepatocyte function of hESC-DE cells at two different seeding densities. The two different seeding densities did not seem to have a clear effect on the timing of spheroid formation, but did appear to impact the size of spheroids formed in the Algimatrix scaffold. However, this effect was not a simple positive correlation. Surprisingly, the higher seeding density not only resulted in a higher proportion of the larger spheroids but also increased the number of smaller spheroids. In addition, our results showed that lower seeding densities that generated significantly more medium-sized spheroids had higher levels of CYP3A4 activity and a significant induction upon drug stimulation as well as increased urea secretion, indicating that the size of hESC-DE spheroids is important for the hepatocyte differentiation and function. It is not surprising that it is the medium-sized spheroids that exhibit better functionality, as the larger size reduces the nutrition/oxygen accessibility of the cells found at the center and affects the survival of these cells; while the smaller spheroids do not form the proper cell–cell contacts, which may subsequently hinder hepatocyte differentiation and function. 19 In summary, our study has demonstrated that hESC-DE cells can be incorporated into an Algimatrix scaffold 3D culture upon treatment with the ROCK inhibitor. The 3D culture enhances hESC-DE cell differentiation to HLCs and improves the resulting hepatocyte function, such as CYP3A4 activity.
Footnotes
Acknowledgments
We thank Dr. Sam Coward and Dr. Isobel Massie for their technical help and discussion at early stage of the project and thank Dr. Fiona Lamont for their helpful discussion on the manuscript. T.S.R. was supported by a studentship from the University of Malaya/Ministry of Higher Education, Malaysia. This work is supported by the Genesis Research Trust.
Disclosure Statement
No competing financial interests exist.