
Abstract
Myocardial tissue engineering is one of the most promising treatment strategies to restore heart function after a massive heart attack. The biomaterials, cells, and scaffold design play important roles in engineering of heart tissue. In this study, we have developed a fibrin-based multiscale electrospun composite scaffold for myocardial regeneration. Fibrin is the natural wound-healing matrix having angiogenic potential and comprehensively used for tissue engineering applications. It provides a natural environment for cell attachment, migration, and proliferation. Morphological, chemical, and mechanical characterization of the scaffolds was done by scanning electron microscopy, fibrin-specific phosphotungstic acid hematoxylin staining, and mechanical testing. The fiber diameters of fibrin nanofibers range from 50 to 300 nm and that of poly (lactide-co-glycolide) microfibers range from 2 to 4 μm, which mimics the structural hierarchy of native myocardial tissue. Our results indicate that this scaffold enhances the differentiation of mesenchymal stem cells into cardiomyocytes. The cardiac phenotype of the cells was confirmed by the presence of cardiac-specific proteins like α-sarcomeric actinin, troponin, tropomyosin, desmin, and atrial natriuretic peptide Estimation of D-Dimer in the culture supernatant for 2 weeks and analysis of scaffold for 3 weeks of in vitro culture of cardiomyocytes indicated the degradation of fibrin and presence of newly synthesized collagen respectively. Our results demonstrate the promising potential of this scaffold for myocardial tissue engineering applications.
Introduction
Hunting for an ideal scaffold, researchers have recognized the importance of extracellular matrix (ECM) in guiding the development and maintaining the characteristics of cardiac tissue that has gained tremendous attention lately.11–13 Collagen is the most abundant constituent of the native myocardial ECM that has a bundle diameter in nanoscale dimensions. The structural and functional properties of the myocytes are greatly influenced by the ECM proteins, and the size scale also has critical role in tissue modulation and development. 14 Advances in nanotechnology offer novel scaffold fabrication techniques to create replacement tissues in vitro mimicking the native myocardium.15–17 Electrospinning is one such versatile technique by which fibers of nanoscale to microscale dimensions can be fabricated from various synthetic and natural polymers.18–20 Several natural polymers such as collagen, elastin, fibrinogen, and gelatin have successfully electrospun in to nanofibers mimicking the topography of native ECM, but their use for tissue engineering applications is limited by the inadequate mechanical properties.21–23 We have reported the effects of micro- and nanofibers of polycaprolactone on the attachment of osteosarcoma cells and the osteogenic differentiation from bone marrow-derived human mesenchymal stem cells (hMSCs).24–25 These multiscale scaffolds combine the benefits of microfibers such as open pore structure and mechanical integrity and nanofibers such as cell attachment and proliferation, which is not otherwise achievable with single scale scaffolds. 26
Since adult cardiomyocytes lack intrinsic regenerative capacity, a suitable stem cell source is another crucial requirement. Among the different stem cell sources, embryonic stem cells demonstrated a higher degree of success in providing a large number of functional cardiomyocytes, but their clinical application is limited by teratoma formation, immunologic rejection, ethical concerns, and transdifferentiation in to other lineages. 27 If suitable microenvironment is provided, MSCs can also be differentiated into cardiomyocytes. Alhough the number of MSCs in bone marrow and cord blood is quite low, the differentiation and expansion potential of MSCs are very high. 28 Therefore, a scaffold favoring the differentiation of MSCs in to adequate number of cardiomyocytes, and to promote vascularization to meet the high metabolic demand of the myocardial tissue is the need of the hour.
Thus, the main focus of this study was to fabricate and characterize a multiscale composite scaffold that combines the mechanical properties of microfibers and the bioactive properties of nanofibers. We have previously reported a novel single-step electrospinning method for the fabrication of nanofibrous fibrin scaffolds for tissue engineering applications. 29 Several biodegradable polymers have been extensively studied for myocardial tissue engineering applications, such as polyglycolic acid, poly(3-hydroxybutyrate-co-3-hydroxyvalerate), and Poly(1,8-octanediol-co-citric acid).30–33 We chose the biodegradable polymer poly (lactide-co-glycolide) (PLGA) for the microscale component because it is FDA-approved and has suitable degradation profile and mechanical properties for myocardial tissue engineering applications. 34 In addition to these properties, the degradation products of PLGA such as lactic acid and glycolic acid have a natural course of degradation in the body. Also, the degradation rate and mechanical properties of PLGA can be easily tuned by varying the co-polymer ratio, in order to match with tissue specific applications. We have selected PLGA 75:25 because it undergoes complete degradation in 5–8 months, and within this period, complete regeneration of the tissue might take place. 35 The resultant multiscale scaffolds of PLGA (micro) and fibrin (nano) were evaluated for cell viability, proliferation, and cellular infiltration using umbilical cord blood-derived mesenchymal stem cells (UCBMSC). The ability of our scaffold to differentiate UCBMSCs into cardiomyocytes and support the growth of heart cells were also assessed in terms of cardiac differentiation marker expression and matrix deposition.
Materials and Methods
Materials
Human fibrinogen was prepared from pooled fresh frozen plasma of healthy volunteers by cryoprecipitation.36–37 Bovine thrombin was purchased from Merck. Poly vinyl alcohol (PVA) (MW 50,000), calcium chloride, chloroform, and acetone were obtained from Qualigens. PLGA (LA/GA: 75:25) (MW: 65,000 and i.v. 0.50–0.65) was from Polysciences. Texas red-Phalloidin, Live/Dead viability/cytotoxicity kit, Sytox Orange, DNA quantification kit, Iscove's modified Dulbecco's medium (IMDM), MSC-specific fetal bovine serum (MSC-FBS), and antibiotics were from Gibco, Invitrogen. Masson's trichrome staining kit and mouse anti-collagen I was from Sigma. Cardiomyocyte characterization kit was from Millipore. D-Dimer kit was from Diagnostica Stago.
Fabrication of electrospun scaffolds
Electrospinning of nanofibrous fibrin scaffolds were carried out as described previously. 24 Briefly, fibrinogen (50 mg/mL) and thrombin (100 U/mL) were separately mixed with equal volume of 12% PVA solution. These two solutions were taken separately in two 2-mL syringes and connected to a delivery device having a common needle at the end of the syringes. This delivery device is placed in a KD scientific syringe pump to be dispensed at a rate of 0.4 mL/h. A high-voltage power supply (Zeonics Systech Electrospin-2A) was used to apply a voltage of 15 kV to the 18-gauge blunt-end needle fixed to the Y-shaped connector attached with the syringes containing the solutions. The tip target distance was 10 cm. PLGA 75:25 was dissolved in chloroform:acetone mixture (4:1) at a final concentration of 40%. The polymer solution was taken in a 10-mL syringe and connected to an 18-gauge blunt-end needle and loaded in the electrospinning setup. The flow rate was adjusted to 1 mL/h and the voltage applied was 12 kV to get beadless microfibers. The distance between the needle tip and collection mandrel was 12 cm. Solutions were electrospun simultaneously on to the grounded rotating mandrel from both sides to get composite scaffolds. The speed of rotation was adjusted at ∼100 rpm so that no aligned fibers are formed. The schematic representation of the electrospinning setup used is shown in Figure 1.

Diagrammatic representation of the simultaneous electrospinning of fibrin and poly (lactide-co-glycolide) (PLGA) with rotating mandrel.
Structural and morphological characterization
Scanning electron microscopy
Morphological features of the electrospun scaffolds were characterized by scanning electron microscopy (SEM; JSM-6490 LA; JEOL). The scaffolds were fixed on to an aluminium stub and sputter-coated with platinum using JEOL (model JFC-1600) auto fine coater and observed at an accelerating voltage of 15 kV.
Water contact angle measurement
Water contact angles indicating the surface wettability of the materials were measured using drop-shape analyzer (KRUSS DSA-100). Electrospun fiber mats were cut into small pieces. A drop of deionized water was placed on the mat surface using a microsyringe attached with the drop-shape analyzer. The contact angles of the water drops on the electrospun fiber mats were measured using drop-shape analyzer followed by image processing of the sessile drop with the software. Triplicates were analyzed for each sample and the results were averaged.
Phosphotungstic acid hematoxylin staining
Mallory's phosphotungstic acid hematoxylin (PTAH) staining technique was used to confirm the presence of fibrin in the electrospun scaffolds. Briefly, the electrospun mats were fixed on cover slips and soaked in phosphate buffered saline (PBS) overnight to remove PVA. The samples were then stained with PTAH and observed under light microscope.
Mechanical characterization of the scaffolds
For mechanical characterization, scaffolds were cut into 5-cm-long dumb-bell-shaped pieces. The thickness of the scaffolds was measured with a digital screw gauge having a precision of 0.01 mm. A tensile test was conducted as per ASTM D412 using a tabletop uniaxial testing machine (Instron 3365) equipped with a 5-kN load cell under a crosshead speed of 50 mm/min at ambient conditions. The samples were uniaxially stretched to evaluate tensile properties. Four samples were tested for each type of scaffold.
Cell behavior on electrospun scaffolds
Initial cell interaction studies were carried out using MSCs isolated from umbilical cord blood. Cells after third passage were used for all experiments.
Isolation, culture, and characterization of MSCs from umbilical cord blood
UCBMSC isolation, culture, and characterization were done as described previously. 29 Briefly, mononuclear cells from umbilical cord blood were isolated by Ficoll-Paque (Amersham-Pharmacia) density gradient centrifugation (1.077 g/cm3), and plated in noncoated tissue culture flasks (Becton Dickinson) in MSC-specific culture medium. Cells were allowed to adhere overnight and nonadherent cells were washed out with medium changes. Media changes were carried out twice weekly thereafter. MSC-specific medium consists of IMDM and 20% MSC-specific FBS supplemented with 100 U/mL penicillin and 100 U/mL streptomycin (Invitrogen).
Cell attachment and spreading by SEM and actin staining
2-cm2 electrospun scaffold pieces were taken for cell interaction studies. UCBMSCs at fourth passage were seeded on to the scaffolds at a seeding density of 25,000 cells/scaffold. Cell–fiber constructs were then cultivated in MSC-specific culture medium in a humidified incubator at 37°C with 5% CO2 for 24 h. For SEM analysis, the medium was removed after 24 h and the scaffolds were washed with PBS before fixing in 2% buffered gluteraldehyde for 1 h at room temperature. After fixing, samples were washed with PBS and dehydrated through sequential rinses of 30%, 50%, 70%, 90%, and 100% ethanol for 15 min each. Samples were then fixed on to an aluminium stub and sputter-coated with platinum using JEOL (model JFC-1600) auto fine coater and observed at an accelerating voltage of 8 kV using a scanning electron microscope (SEM; JSM-6490 LA; JEOL). Pure PLGA fibers were also tested for comparison of cell attachment.
For the analysis of cytoskeletal actin arrangement, cells were stained using Texas red-Phalloidin after 48 h of cell seeding. Scaffolds were fixed in 4% paraformaldehyde (PFA) in PBS for 15 min and permeabilized with 0.5% Triton X-100 in PBS for 5 min with thorough washing with PBS between each step. Staining was then done with a 50 μg/mL Texas red-conjugated phalloidin solution in PBS for 40 min at room temperature, and then washed thoroughly with PBS to remove unbound phalloidin conjugate. Nuclei were visualized after counter staining with DAPI. Samples were then washed, dried, and mounted for imaging using a confocal microscope (Leica TCS SP5 II).
Cell viability and proliferation
A cell viability/cytotoxicity assay based on the simultaneous determination of live and dead cells with two probes such as calcien AM and ethidium homodimer (Eth D-1) that measure the intracellular esterase activity and plasma membrane integrity, respectively, was employed for the determination of cell viability on the scaffolds. Cells were seeded on the scaffolds at a seeding density of 25,000 cells/scaffold and cultured for two periods (24 and 72 h). After each time point, the scaffolds were washed with PBS and stained with the live/dead reagent containing 1 μM calcein AM and 1 μM EthD-1 as per the manufacturer's protocol. After 30 min the samples were washed with PBS and viewed under a confocal microscope (Leica TCS SP5 II). The Ex/Em for calcein was 494/517 nm and that for EthD-1 was 528/617 nm.
Cell proliferation on the scaffolds was evaluated by quantification of DNA content using Quant-iT picogreen dsDNA kit. About 25,000 cells were seeded on to each scaffold and cultured for 24 h, 72 h, 120 h, 10 days, and 14 days. After each time point, cells were trypsinzed from the scaffolds and DNA was extracted from the cells by three repeated cycles of freezing, thawing, and sonication. Equal volume of picogreen reagent and samples were added in to black 96-well plates, mixed well, and incubated for 20 min in dark. Fluorescence was measured using a fluorescence plate reader (Beckmann Coulter) with 485/520 Ex/Em filters. DNA in the samples was quantified using a standard curve plotted with known quantity of DNA ranging from 100 to 1000 ng. DNA content was converted to cell numbers using standard curve plotted against DNA content from known number of cells. Six replicates were tested for each sample and the results were averaged.
Cardiomyocyte differentiation on electrospun scaffolds
Induction of cardiomyocyte differentiation
UCBMSCs between third and fifth passage were used for the studies. Cells were seeded on to 2-cm2 electrospun scaffold pieces at a seeding density of 25,000 cells per scaffold and supplemented with cardiac differentiation induction medium containing 5 μM 5-azacytidine for 24 h. After 24 h, the medium was replaced with specific growth medium consisted of 10% FBS containing IMDM with 100 μg/mL of insulin and 100 μg/mL transferrin. Media change was given every alternate day thereafter. Another group of cells were maintained in a similar way on electrospun PLGA for comparison.
Cardiomyocyte characterization using flow cytometry and immunofluorescence
The cells were phenotyped after 7 and 14 days of differentiation induction using cardiomyocyte characterization kit by flow cytometry and fluorescent microscopy. Briefly, for flow cytometry, the cells were trypsinized from scaffolds after each culture period, washed with PBS, and fixed with 4% PFA for 15 min at room temperature. Cells were then permeabilized using 0.5% Triton X for 10 min and blocked with 1%BSA for 30 min with PBS washing between each step. Cells were then incubated with primary antibodies against α-sarcomeric actinin, troponin, tropomyosin, and atrial natriuretic peptide (ANP) for 1 h at room temperature. Unbound antibodies were washed out with PBS and cells were incubated with flourochrome-conjugated secondary antibodies for 1 h and again washed with PBS. Cells were then resuspended in PBS and analyzed using a flowcytometer (FACS Aria II, BD Biosciences) equipped with 530/30, 585/42, and 616/23 band pass filters for collecting fluorescence signals from FITC, PE, and PerCP, respectively. For microscopy, antibody treatment was done on the cell-seeded scaffolds in a similar way as described above. After staining, the scaffolds were washed, dried, and mounted for viewing under a confocal microscope (Leica TCS SP5 II).
Fibrin degradation and ECM deposition
Cells were seeded on to 2-cm2 pieces of PLGA-Fibrin scaffold at a seeding density of 25,000 cells and cultured for 24 h, 72 h, 120 h, 7 days, 14 days, and 21 days to estimate the degradation of fibrin and cell-synthesized ECM. Cardiomyocyte differentiation was induced as described above and the cells were maintained in cardiac-specific growth medium. Culture medium was collected at the above-mentioned intervals and a semiquantitative determination of D-Dimer was done using D-Dimer assay kit (Diagnostica stago) as per manufacturer's protocol.
For evaluation of ECM deposition, cells were removed by trypsinization followed by gentle washing and matrices were recovered for histochemical staining using Masson's trichrome kit (Sigma) and immunostaining for collagen Type I. For masson's trichrome staining, the recovered matrices were treated with Bouin's solution (Sigma) for overnight at room temperature. The scaffolds were then thoroughly washed in running tap water to remove the yellow color. After rinsing in deionized water, a step-wise staining for 5 min each with Biebrich Scarlet-acid fucshin, phosphotungstic/phosphomolybdic acid, and aniline blue solution was done. After staining the scaffolds were kept in 1% acetic acid for 2 min followed by dehydration with sequential rinses with alcohol. Scaffolds were then dried and mounted for viewing under a microscope (Olympus BX 51) in bright field.
For immunostaining, the recovered matrices were fixed in 4% PFA for 15 min, washed with PBS, and incubated with the primary antibody against collagen type I for 1 h at room temperature. After thorough washing with PBS, 1 h incubation with Texas red-conjugated secondary antibody was done before viewing with confocal microscope.
Statistics
Statistical analysis of the data was performed using student's t-test. Data are expressed as mean±standard deviation (SD) and p<0.05 is considered as statistically significant (*).
Results and Discussion
Scaffold fabrication, morphological, chemical, and mechanical characterization
Simultaneous electrospinning of PLGA and fibrin from both sides of a rotating mandrel was conducted to obtain a multiscale membrane of PLGA and fibrin. The spinning parameters were optimized to get beadless PLGA microfibers (Supplementary Fig. S1; Supplementary Data are available online at www.liebertpub.com/tea) and fibrin nanofibers. Figure 2 shows the SEM micrographs of electrospun PLGA fibrin composite along with their fiber diameter distribution. The fiber diameter of fibrin nanofibers ranges from 50 to 500 nm as previously reported. 29 The diameter of PLGA microfibers ranges from 2 to 4 μm. This hierarchical structure resembles the native myocardial ECM and has been shown to favor cell–matrix interactions.38–39

Scanning electron microscopy (SEM) micrographs of PLGA–fibrin electrospun membrane
Synthetic scaffolds lack sufficient cell recognition signals and exhibit high hydrophobicity, which will lead to poor cell attachment and survival. The water contact angles were analyzed for the assessment of hydrophilicity/hydrophobicity of electrospun scaffolds. The water contact angles of electrospun mats are mostly depend on the diameter, pore size, and surface chemistry of fibers. 34 The mean water contact angle observed for the scaffolds is shown in Figure 3 with images of water droplets. The data indicate that the addition of fibrin to PLGA significantly increased the hydrophilicity of pure PLGA scaffold. The decreased contact angle value of the composite scaffold suggested the increased hydrophilicity and enhanced capability for cell attachment.

Contact angle data of
The formation of fibrin nanofibers and presence of fibrin in the composite scaffold were verified by fibrin-specific PTAH staining. PTAH is a polychrome stain that gives blue color to fibrin. It can be seen from Figure 4a that, when PVA is removed from the PVA fibrin scaffold, even the fibers are clearly seen, they are not uniformly distributed may be due to the poor structural stability of the scaffold. However, as evident from Figure 4b, in the PLGA-Fibrin composite scaffold after the removal of PVA, uniform distribution of fibrin nanofibers can be visualized.
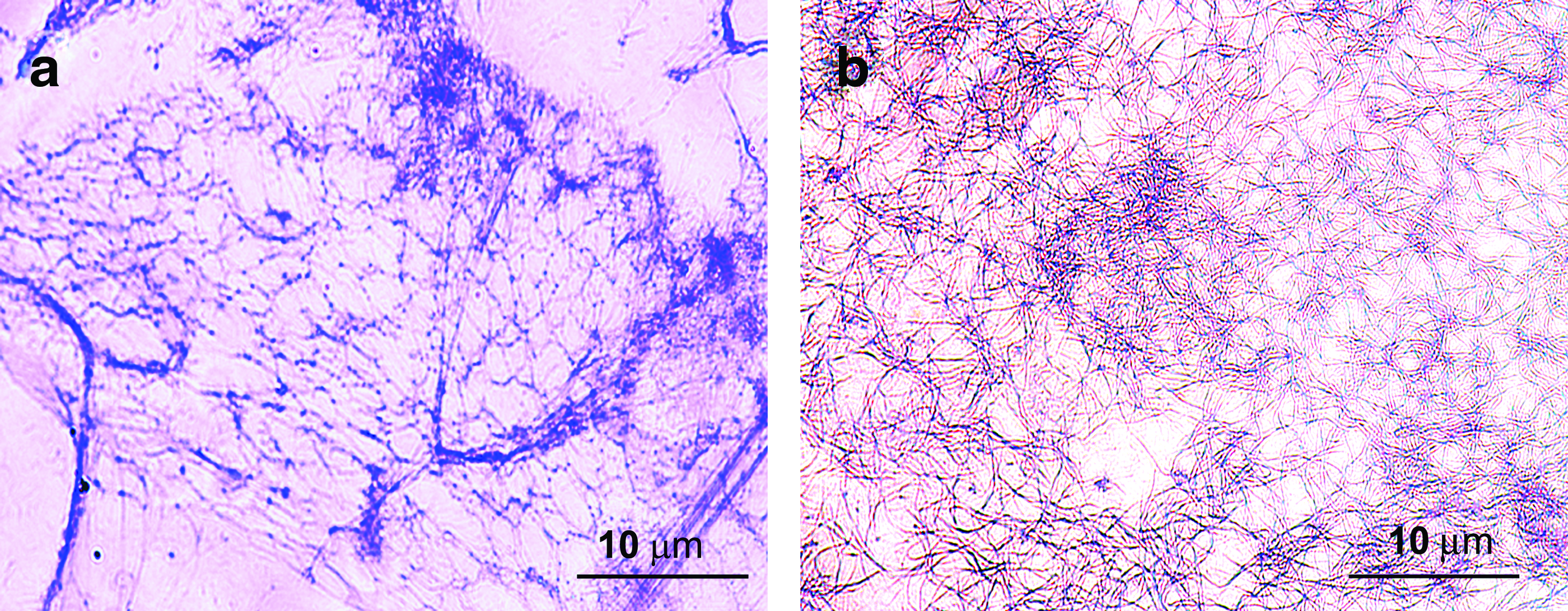
Light microscopic image of phosphotungstic acid hematoxylin-stained electrospun
Mechanical properties of electrospun scaffolds are summarized in Table 1. Maximum tensile stress has been found to be 0.7412±0.07 MPa for multimodal PLGA fibers and 0.651±0.24 MPa for PLGA–fibrin fibers. The breaking strains for PLGA and PLGA–fibrin were 86±17 and 105±10, respectively. These results are matching with the tensile properties of human myocardium. 40 Directional growth or anisotropy is a definite feature of cardiomyocytes and so methods to create such anisotropic property has been of extensive research focus in recent years.34,41 Since the fibers are deposited on the collection mandrel, which is on continuous rotation, some sort of fiber alignment might happen during electrospinning. This was confirmed by evaluating the tensile strength of PLGA–fibrin scaffold opposite to the direction of fiber alignment. The value was found to be 0.43±0.06 MPa, which is 33% less than the tensile strength along the direction of fiber formation. This indicates the scaffold is slightly anisotropic in nature.
Cell behaviour on electrospun scaffolds
Cell attachment, spreading and infiltration
Cell attachment and spreading on the electrospun scaffolds was observed using SEM and immunofluorescence. It is evident from the SEM micrographs that there is a greater propensity of cell attachment on PLGA–fibrin fibers compared to pure PLGA fibers. After 24h of cell culture, MSC spreading was more appreciable on PLGA-fibrin fibers (Fig. 5b) compared to pure PLGA fibers (Fig. 5a). This clearly shows that the scaffold composition has important role in initial cell attachment and survival.

SEM images of human mesenchymal stem cells (hMSCs) grown on
In nanometer-scale electrospun structures, even though there is excellent cell attachment and spreading, majority of cells adhere only to the surface of the scaffold resulting in poor cellular infiltration, inadequate waste and nutrient transport, and poor vascularization. 42 To overcome the limitations of poor cellular infiltration, simultaneous or sequential electrospinning of micro- and nano-fibers has been employed to enhance the pore size and thereby increase the cell infiltration. 43 Figure 6a shows the confocal image of the cells grown on the scaffold stained with Texas red phalloidin. From the reconstructed image of several Z sections of confocal microscopy data, migration of cells down to several layers of fibers was observed (Fig. 6b). The results indicate that the multiscale nature of the scaffold allowed the migration of the cells into the interior of the scaffold and the uniform distribution of fibrin fibers in the composite scaffold enhanced the proliferation of infiltrated cells throughout the scaffold volume with the maintenance of good cytoskeletal organization. The major limitation of the tissue-engineered cardiac construct is the nonviability of cells beyond a thickness of 100 μm due to insufficient oxygen and nutrient supply. Human myocardium is ∼1 cm thick and fabrication of ∼1-cm-thick viable construct in vitro is the biggest challenge. 44 The cell infiltration data seem promising that upon stacking several layers of cell-seeded membranes thick viable tissue construct could be fabricated in vitro.
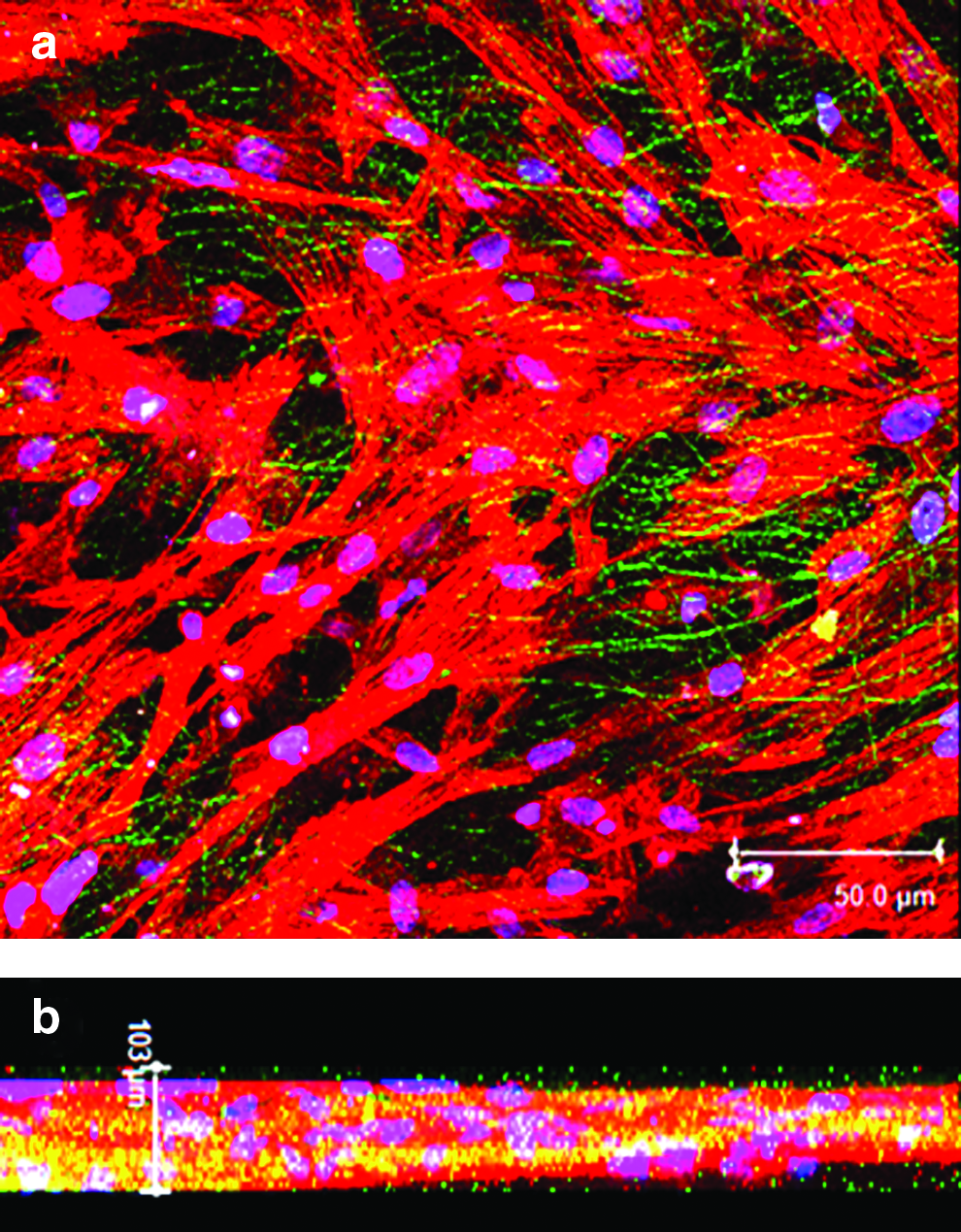
Confocal microscopy images of hMSCs grown on elcetrospun PLGA fibrin over a period of 48 h.
Cell viability and proliferation
Survival of MSCs on the electrospun scaffolds was determined by cell viability assay. Figure 7 shows the confocal microscopy images of MSCs grown on PLGA and PLGA fibrin for 24 and 72 h. Cells maintained a normal morphology on both types of scaffolds after 24 h. However, as evident from the figure, cells survived and proliferated well on the composite scaffold compared to the pure PLGA scaffold and exhibited a uniform distribution. Very less number of dead cells was observed in the composite scaffold at both time points. This result is substantiated by DNA quantification assay. The graphical representation of the quantitative determination of DNA and cell number is shown in Figure 8.
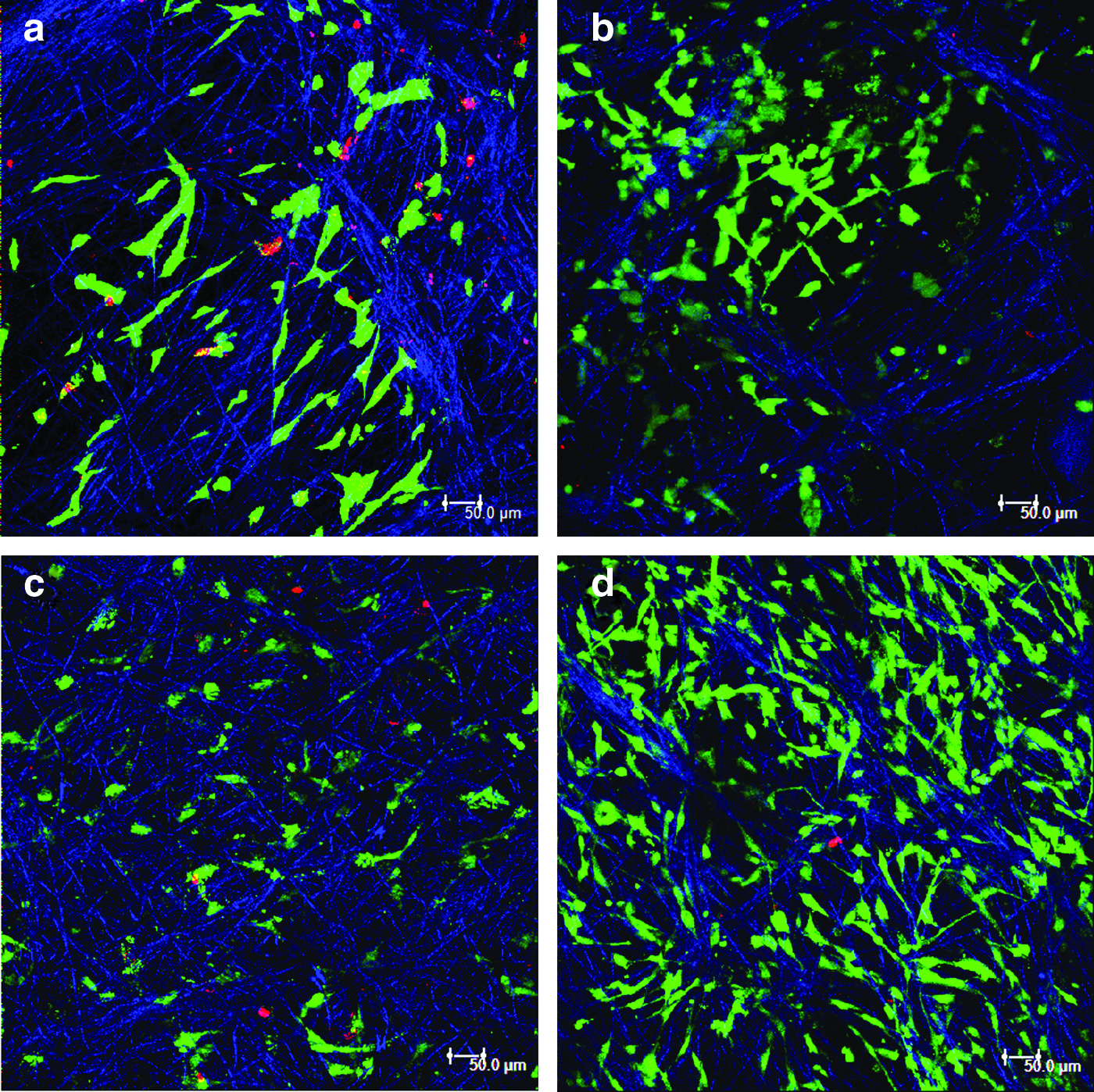
Cell survival and morphology of hMSCs grown on electrospun fibers as determined by live/dead assay. Confocal microscopy images of cells grown on PLGA
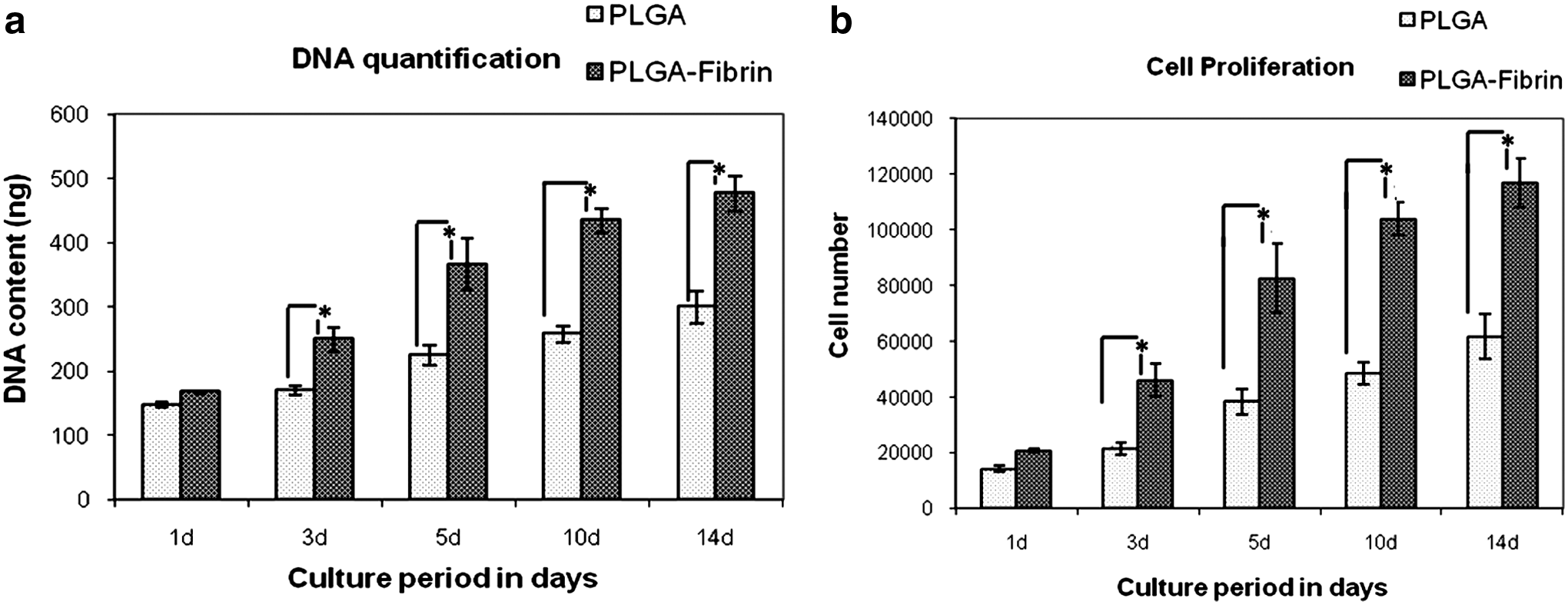
DNA quantification by picogreen assay.
PLGA–fibrin showed significantly higher proliferation of MSCs at all time points compared to pure PLGA. A significantly high proliferation was observed in the initial days up to 10 days, and the proliferation rate declined thereafter and maintained a steady state, indicating that cells became confluent.
Cardiomyocyte differentiation
To investigate whether the multiscale PLGA–fibrin scaffold stimulate the differentiation of UCBMSCs, expression of cardiac-specific proteins was determined by immunofluorescence. After 7 and 14 days of culture in specific growth medium for cardiac differentiation, the cells from both types of scaffolds were analyzed for the expression of cardiac-specific proteins, α-sarcomeric actinin, cardiac troponin, tropomyosin, desmin, and ANP by flowcytometry. Figure 9 is the graphical representation of differentiation marker expression in cells grown on both types of scaffolds after 7 and 14 days of differentiation induction by 5-azacytidine. Almost half of the cells grown on PLGA fibrin showed the expression of marker proteins except ANP after 7 days. After 14 days almost 80% of the cells expressed the differentiation markers. The protein expression was significantly low in cells grown on pure PLGA scaffold at both time points compared to that on the composite scaffold.
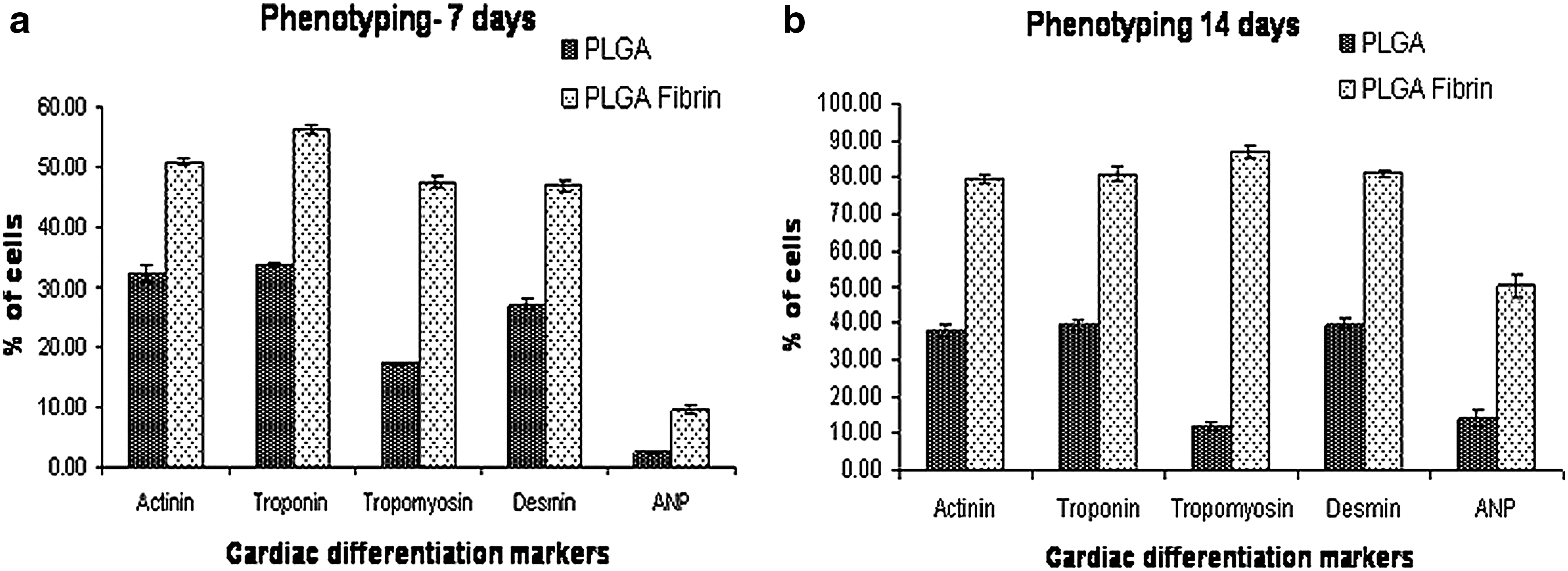
Flow cytometry data of cardiac differentiation markers. Graphical representation of percentage of cells expressing the differentiation markers after
Figure 10 is the confocal microscopic image of the MSCs grown on PLGA fibrin in differentiation medium for 14 days, which substantiate the above findings. The results indicate that the cellular microenvironment has significant role in stimulating the differentiation of MSCs into myogenic cells. 45
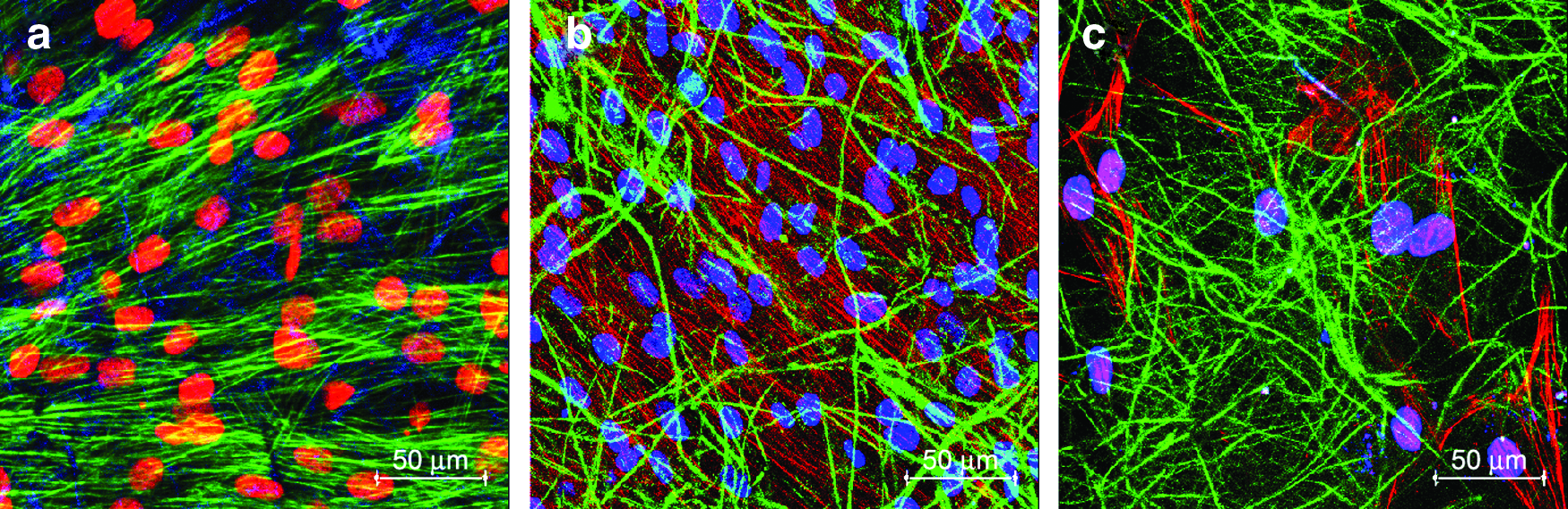
Confocal microscopy images of MSCs grown on PLGA–fibrin composite fibers after 14 days of induction of cardiac differentiation expressing
Fibrin degradation and ECM deposition
The success of an ideal myocardial construct depends on the ability of the biomaterial to degrade over time in a rate similar to synthesis of ECM by the growing cells. Fibrin is the natural wound healing matrix that serves as a temporary scaffold during wound healing and degraded in the body by matrix metalloproteases to fibrin degradation products. D-Dimer is a degradation product formed during fibrin degradation. A semiquantitative determination of D-Dimer in the culture supernatant at different time points was carried out to determine fibrin degradation. 46 Figure 11 shows the graphical representation of the results obtained. The maximum release of D-Dimer into the culture medium was observed in day 5, and the degradation product was detectable up to 14 days in very less quantity. Being a semiquantitative data, statistical analysis could not be done for this observation.
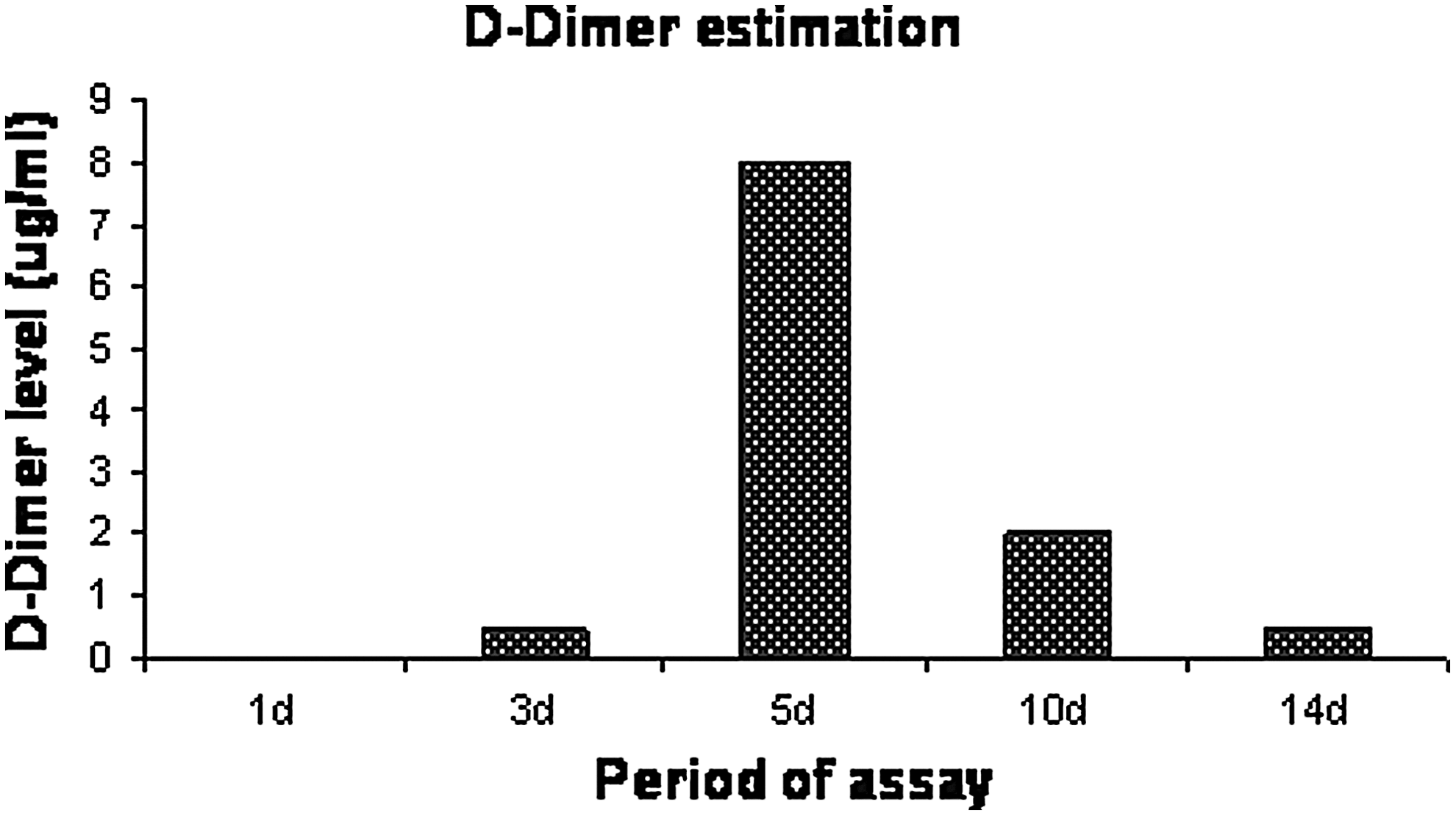
D-Dimer assay in the culture supernatant. MSCs were grown on the scaffolds in cardiac differentiation medium for 14 days and the culture supernatant collected at different time points were evaluated for the presence of D-Dimer.
Whether natural or synthetic material, it is generally accepted that it should act as a temporary scaffold for tissue regeneration and as the new tissue is formed it should degrade. Here we have used PLGA and fibrin, both are degradable, leaving no toxic degradation products. The cell-seeded scaffolds retrieved after 7, 14, and 21 days of MSC culture in cardiac differentiation medium on staining with Masson's trichrome showed degradation of fibrin and presence of newly synthesized collagen.
Figure 12 shows the collagen deposition at 7, 14, and 21 days. Up on staining with Masson's trichrome fibrin shows a red color and collagen appears blue. Figure 12a shows that fibrin is not degraded completely in 7 days, and Figure 11b shows the presence of collagen after 14 days along with some amount of fibrin. However, in 21 days as evident from Figure 12b, fibrin is almost degraded completely leaving behind the newly synthesized collagen. The presence of collagen is further confirmed by immunostaining with anti-collagen type I. Figure 12d is the matrix retrieved after 21 days labeled with anti-collagen type I. It is evident from the immunostaining data that the collagen formed is mainly of type I, which is the major component of the myocardial ECM.
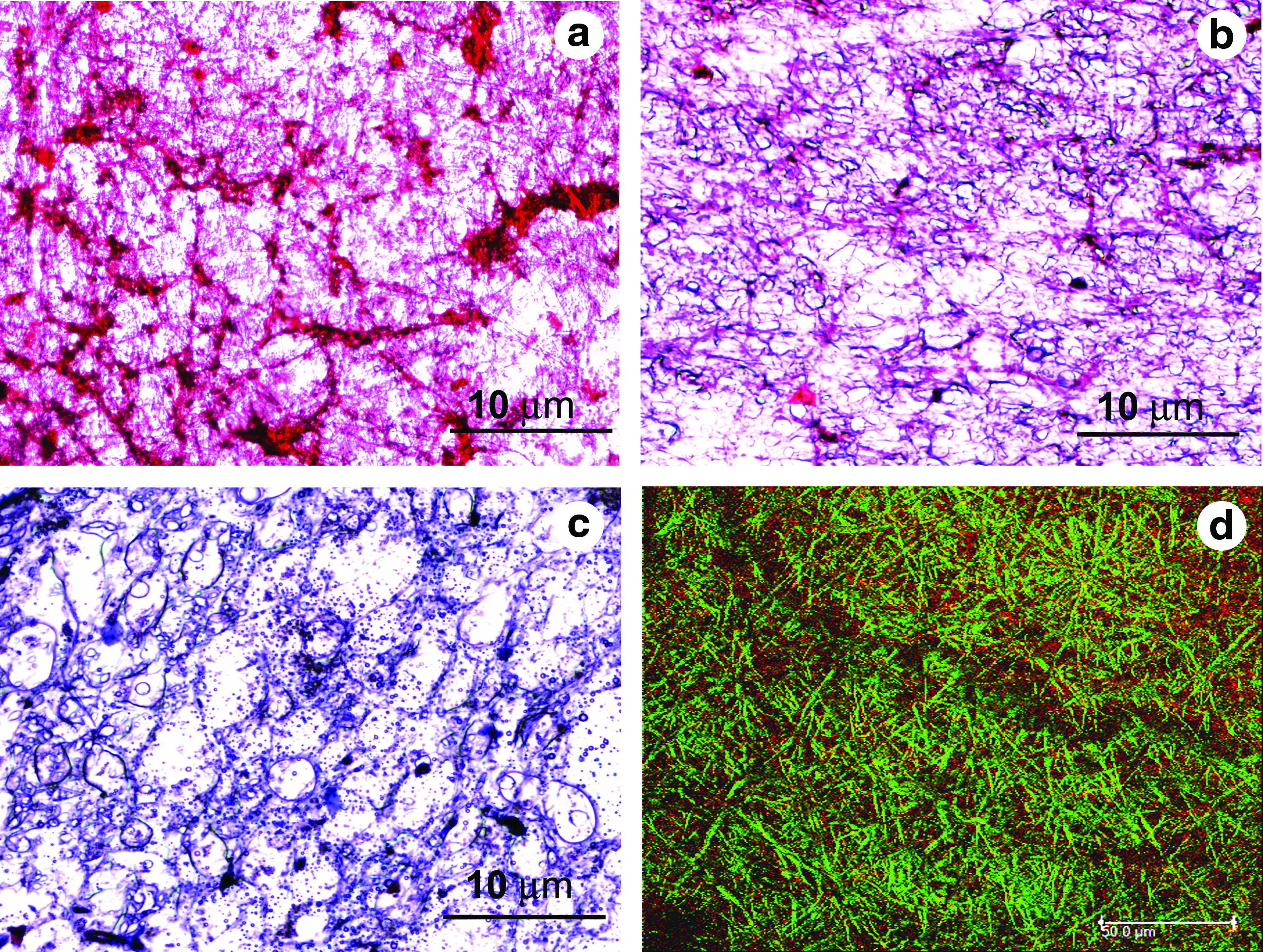
Light microscopy images of the PLGA–fibrin matrices retrieved after
Conclusion
In the present study we have demonstrated the methods of fabrication and characterization of a fibrin-based multiscale scaffold for cardiac tissue engineering applications. In vitro preliminary cell studies with UCBMSCs showed that the scaffold supports attachment, viability, proliferation, and cellular infiltration. The synergistic effect of the PLGA microfibers and fibrin nanofibers provided a favorable microenvironment for the differentiation of UCBMSCs in to a cardiac phenotype. Future work will explore the possibility of using vascular cells along with cardiomyocytes in promoting neovascularization to generate force bearing cardiac tissue. In the past, different types of bioreactors were used to provide mechanical and electrical conditioning to develop thick viable cardiac tissue suitable for clinical application.47–55 Possibly, stacking several layers of the cell seeded membranes in concert with a perfusion bioreactor in vitro will help in the formation of thick viable cardiac construct for in vivo applications.
Footnotes
Acknowledgments
The authors are grateful to the Department of Science and Technology for funding this work through the National Nanoscience and Nanotechnology Initiative monitored by Professor C. N. R. Rao under Thematic Unit of Excellence (TUE) grant. The authors gratefully acknowledge the help from Mr. Sajin P Ravi for SEM analysis and Mr. Sharath for confocal microscopy analysis.
Disclosure Statement
No competing financial interests exist.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.