
Abstract
Mesenchymal stem cells (MSCs) can differentiate into osteoblasts and hold promise for applications of bone regeneration such as bone tissue engineering. However, current approaches for in vitro osteogenesis cannot effectively induce osteogenesis and need to be modified to produce quality bone for clinical applications. Previous studies have shown that the conditioned medium (CM) from osteoblast culture enhances osteogenesis of MSCs, and soluble osteogenic factors in the CM may be involved in the regulation. However, these factors are not fully identified. In this study, we profiled soluble factors secreted from MG-63 cells using a comparative protein array and found that osteoprotegerin (OPG), known as a potent anti-osteoclastogenic protein, was at the highest relative level among 507 soluble molecules detected by the array. Furthermore, treating hMSCs with OPG before osteogenic induction significantly increased the expression of osteocalcin mRNA transcript and the production of calcium deposits compared to the untreated control cells, suggesting that OPG is capable of priming undifferentiated hMSCs for the enhancement of subsequent osteogenesis. Furthermore, we showed that the nuclear factor-kappaB (NF-κB) was activated by OPG in undifferentiated hMSCs and that blocking NF-κB activation before osteogenic induction decreased osteogenesis of OPG-pretreated cells upon receiving osteogenic stimuli. Taken together, our results suggest that OPG is a pro-osteogenic factor that can be used as an osteogenic supplement in a growth medium to prime the osteogenic capacity of undifferentiated hMSCs to enhance osteogenesis for bone tissue engineering.
Introduction
The effect of soluble factors secreted by osteoblasts on osteogenesis of MSCs has been examined through the studies investigating the conditioned medium (CM) from osteoblast 4 or MSC culture, 5 or MSC and osteoblast coculture.6–8 A previous study has reported that the CM from heat-shocked osteoblast culture improves in vitro osteogenesis of MSCs. 4 Another study using a coculture setup, in which, MSCs and osteoblasts were grown separately in transwells, has shown that cocultured MSCs increase cell proliferation and alkaline phosphatase (ALP) activity. 6 In the same study, the group further demonstrates that the MSC/osteoblast/osteocyte coculture synergistically enhances MSC osteogenesis compared to the MSC/MSC, MSC/osteoblast, or MSC/osteocyte coculture. 6 Similar to the finding reported in the study using transwell coculture, it has been shown that another coculture setup in which, MSCs and osteoblasts are grown together with direct physical contact also enhances MSC osteogenesis. 7 Another study further investigating whether the physical contact between MSCs and osteoblasts is critical for the regulation of MSC osteogenesis has shown that coculture with separately grown cells more effectively enhances osteogenesis than coculture with mixedly grown cells. 8 These previous studies evidently suggest that osteoblasts produce soluble factors that can promote MSC osteogenesis. However, the soluble osteogenic factors involved in this regulation have not been fully identified.
In this study, we aimed to identify soluble osteogenic factors present in the osteoblast-like MG-63 cell culture medium, which can be used to enhance MSC osteogenesis for the applications of bone tissue engineering. We collected the CM from the MG-63 cell culture medium and evaluated the effect of the CM on osteogenesis of bone marrow-derived hMSCs. We further identified soluble pro-osteogenic factors in the CM that induce the activity and determined their intracellular target molecules involved in the regulation.
Materials and Methods
Chemicals, proteins, culture medium, and antibodies
The chemicals, 1α, 25-dihydroxyvitamin D3 and transforming growth factor-beta1 (TGF-β1), were purchased from Enzo Life Sciences (Farmingdale, NY) and R&D systems (Minneapolis, MN), respectively. All other chemicals used for differentiation induction and nuclear factor-kappaB (NF-κB) inhibition were obtained from Sigma-Aldrich (St. Louis, MO). Recombinant human osteoprotegerin (OPG) and progranulin (PGRN) were acquired from PeproTech (Rocky Hill, NJ) and R&D systems, respectively, and rat tail collagen type 1 was acquired from BD Biosciences (Bedford, MA). The Dulbecco's Modified Eagle's Medium (DMEM), fetal bovine serum (FBS), and the antibiotic–antimycotic solution were obtained from Invitrogen (Carlsbad, CA), Atlantic Biologicals (Miami, FL), and Cellgro (Manassas, VA), respectively. An anti-CD105 antibody was obtained from AbD Serotec (Raleigh, NC) and all other antibodies used for flow cytometry analysis were acquired from BD Biosciences. The anti-TATA-box-binding protein (TBP) antibody was obtained from Abcam (Cambridge, MA) and all other antibodies for Western blotting were obtained from Cell Signaling Technology (Boston, MA).
Cell culture
With the approval from the Institutional Review Board of the University of Washington, femoral heads of the patients undergoing total hip arthroplasty were obtained through the collaboration with Dr. Paul Manner at the Department of Orthopedics, the University of Washington. Human MSCs were isolated from the femoral heads following the procedures previously described, 9 and the MG-63 cell line was obtained from Lonza (Walkersville, MD). Human MSCs and MG-63 cells were cultured in the DMEM-Low Glucose (DMEM-LG) and DMEM-High Glucose (DMEM-HG), respectively, supplemented with 10% FBS and the antibiotic–antimycotic solution and maintained at 37°C in a humidified 5% CO2 incubator. The culture medium was changed every 3 days and upon 70%–80% confluence, cells were trypsinized and passaged.
Preparation of CM
MG-63 cells were plated at a density of 5,700 cells/cm2 in the hMSC basal medium (DMEM-LG with 10% FBS and antibiotic–antimycotic solution), and the culture medium was collected every 2 days until the cells were grown to full confluence after 6 days. Then, the collected medium from all the time points was combined and filter sterilized, referred to as the 100% master CM. The control medium prepared in parallel from an acellular culture vessel following the same procedures used for the 100% master CM preparation was referred to as the 0% master CM. To prepare a working CM for hMSC culture, the 20% and 50% master CM were first made by mixing different amounts of the 100% and 0% master CM, and then the 0%, 10%, 25%, and 50% working CM were prepared by diluting the 0%, 20%, 50%, and 100% master CM with an equal amount of fresh hMSC basal medium, respectively. A schematic diagram depicting the procedure of CM preparation is illustrated in Figure 1.
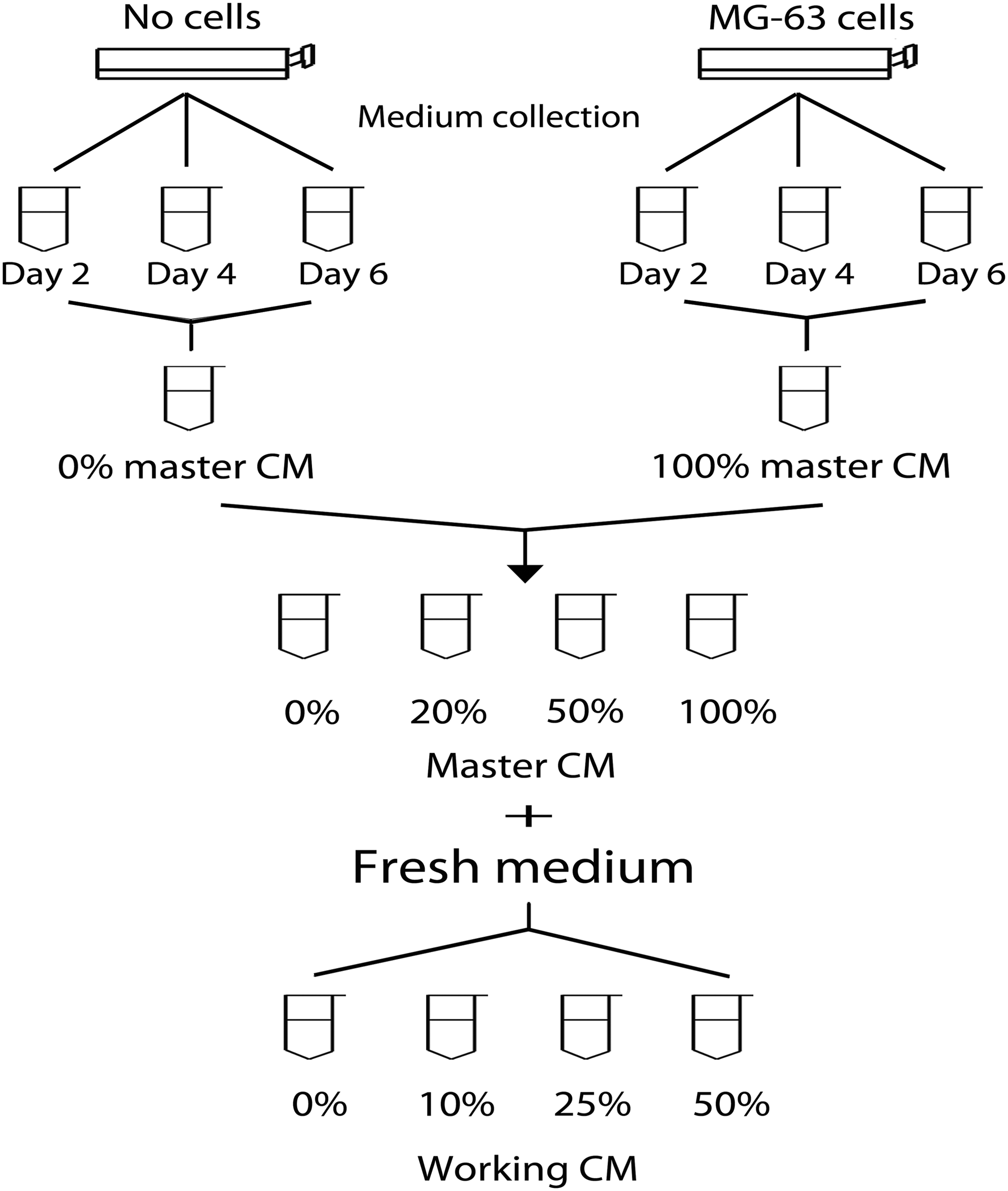
Illustration of procedures for preparation of the master and working CM. CM, conditioned medium.
Immunophenotyping by flow cytometry
Human MSCs were maintained in the 0% and 50% working CM for two passages and trypsinized, and resuspended in the fluorescence-activated cell sorting (FACS) buffer (0.5% bovine serum albumin and 0.1% sodium azide in 1× phosphate-buffered saline [PBS] solution). Five hundred thousand cells were incubated with a hMSC-specific surface marker antibody (the positive markers CD29, CD44, CD73, CD90, CD105, and CD106, and the negative markers CD34, CD45, and CD144) for 30 min at 4°C, washed twice in the FACS buffer, and fixed in 4% formaldehyde in PBS for 25 min on ice. The isotope controls were prepared in parallel following the same procedures aforementioned. Fluorescence intensity data were acquired from 10,000 cells using FACScan (BD Biosciences) and analyzed using the CellQuest software (BD Biosciences).
CM treatment and multilineage differentiation of hMSCs in vitro
The experimental design is illustrated in Figure 3A. Specifically, before multilineage differentiation induction, hMSCs were cultured in the basal medium supplemented with or without the 50% working CM for two passages. For osteogenesis, CM-pretreated cells were plated at a density of 5,000/cm2 on culture plates coated with rat tail collagen type I, and induced in the osteogenic medium (DMEM-LG basal medium with 10 mM β-glycerophosphate, 0.1 μM dexamethasone, 50 μg/mL L-ascorbic acid 2-phosphate, and 10 nM vitamin D3) supplemented with the 0% or 50% working CM. For adipogenic differentiation, CM-pretreated cells were plated at a density of 10,000/cm2 in the adipogenic medium (DMEM-LG basal medium with 1 μM dexamethasone, 0.5 mM IBMX, and 1 μg/mL insulin) supplemented with the 0% or 50% working CM. For chondrogenic differentiation, CM-pretreated cells were prepared into cell pellets with 250,000 cells per pellet and cultured in the chondrogenic medium (serum-free DMEM-HG with 1% ITS, 0.9% sodium pyruvate, 50 μg/mL L-ascorbic acid 2-phosphate, 0.1 μM dexamethasone, and 10 ng/mL TGF-β1). The control cells of each differentiation condition were prepared in parallel in the same basal and differentiation medium without the CM. The media were changed every 3 days.
Total RNA isolation and real-time PCR analysis
Total RNA was isolated using the RNAeasy Miniprep kit (Qiagene, Valencia, CA) and converted to cDNA by the high-capacity cDNA reverse transcription kit (Invitrogen). Real-time PCR was performed using the iQ™ SYBR® Green Supermix (BioRad, Hercules, CA) with the primers listed in Table 1. Ubiquitin C (UBC) was used as a reference gene and the relative mRNA expression levels of target genes were calculated using the ΔΔCT method.
Forward and reverse primers are indicated as “F” and “R,” respectively.
Quantification of double-stranded DNA
Cell numbers in culture receiving different experimental treatments were determined by quantification of double-stranded DNA (dsDNA) detected by PicoGreen Assay. Briefly, an initial density of 5,000 cells/cm2 of hMSCs seeded in osteogenic culture, whereas a number of 250,000 hMSCs per pellet prepared for chondrogenic culture were induced for 21 days before the cells were harvested for PicoGreen detection. To quantify dsDNA amounts, cells were washed with 1× Tris-buffered saline (TBS) and lysed in a cell digestion buffer (150 mM Tris base, 0.1 mM ZnCl2, and 0.1 mM MgCl2) for 30 min at 37°C, and then overnight at 4°C. Contents of dsDNA were determined using the Quant-iT PicoGreen dsDNA Assay kit (Invitrogen) following the manufacturer's instructions.
Quantification of calcium mineral deposits
Human MSCs undergoing osteogenesis were washed in 1× PBS without calcium and magnesium, and calcium mineral deposits were dissolved in 0.5 N hydrochloric acid (HCl) overnight at 4°C. The amount of calcium in the acid solution was determined using the Total Calcium LiquiColor kit (Stanbio laboratory, Boerne, TX), following the manufacturer's instructions.
Protein array assay
Culture medium supernatants of MG-63 cell and hMSC cultures were prepared following the procedures of making the 100% master CM described above and collected for the protein array assay. The culture media were then analyzed using the L-Series 507: Label-based Human Antibody Array kit (RayBiotech, Norcross, GA), according to the manufacturer's instructions. The fluorescent signals were detected by the GenePix® 4000B scanner and the data were analyzed using the GenePix Pro 6.0 software (Molecular Devices, Sunnyvale, CA). The raw data were background adjusted and a signal threshold was set at the mean value of negative controls plus four times their standard deviation.
Enzyme-linked immunosorbent assay
An enzyme-linked immunosorbent assay (ELISA) to measure the quantity of OPG, angiopoietin-1 (ANGPT1), or PRGN in the supernatants of MG-63 cell and hMSC cultures was performed using a commercial ELISA kit acquired from RayBiotech or Assay Biotechnology (Sunnyvale, CA). The cell culture supernatants were prepared in the same way described in the protein array assay, and the assay was conducted following the manufacturer's instructions.
OPG or PRGN treatment and subsequent osteogenesis of hMSCs
The experimental design is illustrated in Figure 5A. Briefly, before the differentiation induction, hMSCs were cultured in the basal medium supplemented with or without 40 ng/mL OPG for two passages. Both treated and untreated cells were further induced in the osteogenic medium with or without 40 ng/mL OPG for 21 days. The concentration of 40 ng/mL OPG used in this assay was determined based on the measurement obtained from the ELISA described above. To evaluate osteogenesis, the expression of mRNA transcripts of bone-related markers and the production of calcium deposits were analyzed. For the study investigating the effect of PRGN on osteogenesis, the experimental procedures described in Figure 5A were followed to treat hMSCs with PRGN. Briefly, hMSCs were treated with or without 4.4 ng/mL PRGN, which was previously determined by the ELISA measurement, during expansion and subsequent osteogenic culture, and then analyzed for the expression of mRNA transcripts of bone-related markers and the production of calcium deposits.
Protein extraction and Western blot analysis
To prepare whole cell lysate, cells were harvested, washed with PBS, and lysed in RIPA (50 mM Tris-HCl (pH 7.5), 1% NP-40 substitute, 0.25% sodium deoxycholate, 150 mM sodium chloride, and 1 mM ethylenediaminetetraacetic acid (EDTA)) supplemented with 1×cOmplete, EDTA-free and PhosSTOP (Roche, Indianapolis, IN). Nuclear and cytoplasmic protein samples were prepared using the NE-PER nuclear and cytoplasmic extraction kit (Thermo Scientific, Rockford, IL). Ten to 25 μg of protein samples were electrophoresed on a 4%–20% gradient gel, and then transferred to PVDF membranes. The membrane was incubated with an anti-NF-κB or glyceraldehyde 3-phosphate dehydrogenase (GAPDH) antibody in a blocking solution (5% nonfat milk in 1×TBS containing 0.1% Tween 20 [TBS/T]) overnight at 4°C, and then washed three times in TBS/T and incubated with a horseradish peroxidase-linked secondary antibody in the blocking solution at room temperature for 1 h. Western blot images were obtained using the Kodak Image Station 4000R Pro system (Carestream Health, Rochester, NY).
NF-κB inhibition and subsequent osteogenesis of hMSCs
Before the induction of osteogenesis, cells were treated with 40 ng/mL OPG and 0.1 or 1 μM 2-[(aminocarbonyl)amino]-5-(4-fluorophenyl)-3-thiophenecarboxamide (TPCA-1) for two passages. After the pretreatment, cells were induced in the osteogenic medium without OPG and TPCA-1 for 21 days. The osteogenesis of cells was analyzed at days 3, 11, and 21.
Statistical analysis
All quantitative assays were performed in triplicate. Data are expressed as the mean±SD. One-way ANOVA with the Dunnett's test was performed to determine statistical significance for the experiment to compare more than two groups. Briefly, ANOVA was first performed to test for significant differences between means of the experimental groups, and then the Dunnett's test was performed to determine statistical significance between any pairs of the control and treated groups. For the experiment to compare two groups, the Student's t-test was performed to determine statistical significance between the control and treated groups. A p-value<0.05 was considered significant.
Results
Human MSCs cultured in MG-63 CM maintain phenotypic characteristics of hMSCs
We used the MG-63 human osteoblast-like cell line, which has been widely used as a model of osteoblasts for the studies of bone biology and tissue engineering,10–16 to prepare the CM for hMSC culture. To examine the effect of the CM on hMSC phenotypes, we analyzed surface markers of hMSCs after these cells were cultured in the 50% working CM for two passages. We decided to pretreat the cells for two passages since our previous study suggests that the length of a treatment with a target pharmacological molecule for two passages is sufficient to induce effects on hMSC differentiation. 9 Expression of various surface markers (positive markers CD29, CD44, CD73, CD90, CD105, and CD106 and negative markers CD34, CD45, and CD144) of the CM-treated hMSCs was evaluated by flow cytometry and compared to that of the control cells grown in the 0% working CM (Fig. 2A). Our results showed that the expression of the surface markers between the treated and control cells was highly comparable, suggesting that the immunophenotype of the 50% CM-treated hMSCs is similar to that of the control cells. In addition, the morphology of hMSCs in cultures with CM concentrations ranging from 10% to 50% was similar to that of the control cells in the 0% working CM (Fig. 2B). Together, these data show that the CM does not cause phenotypic changes in hMSCs, suggesting that MG-63 cells are a suitable cell source for preparing the osteoblastic CM for this study. Interestingly, while the phenotypic characteristics of hMSCs were maintained in the CM cultures, cells seemed to grow rapidly in the culture with a higher percentage of the CM, suggesting that the MG-63 CM contains pro-mitotic factors that promote cell division of hMSCs (Fig. 2B).
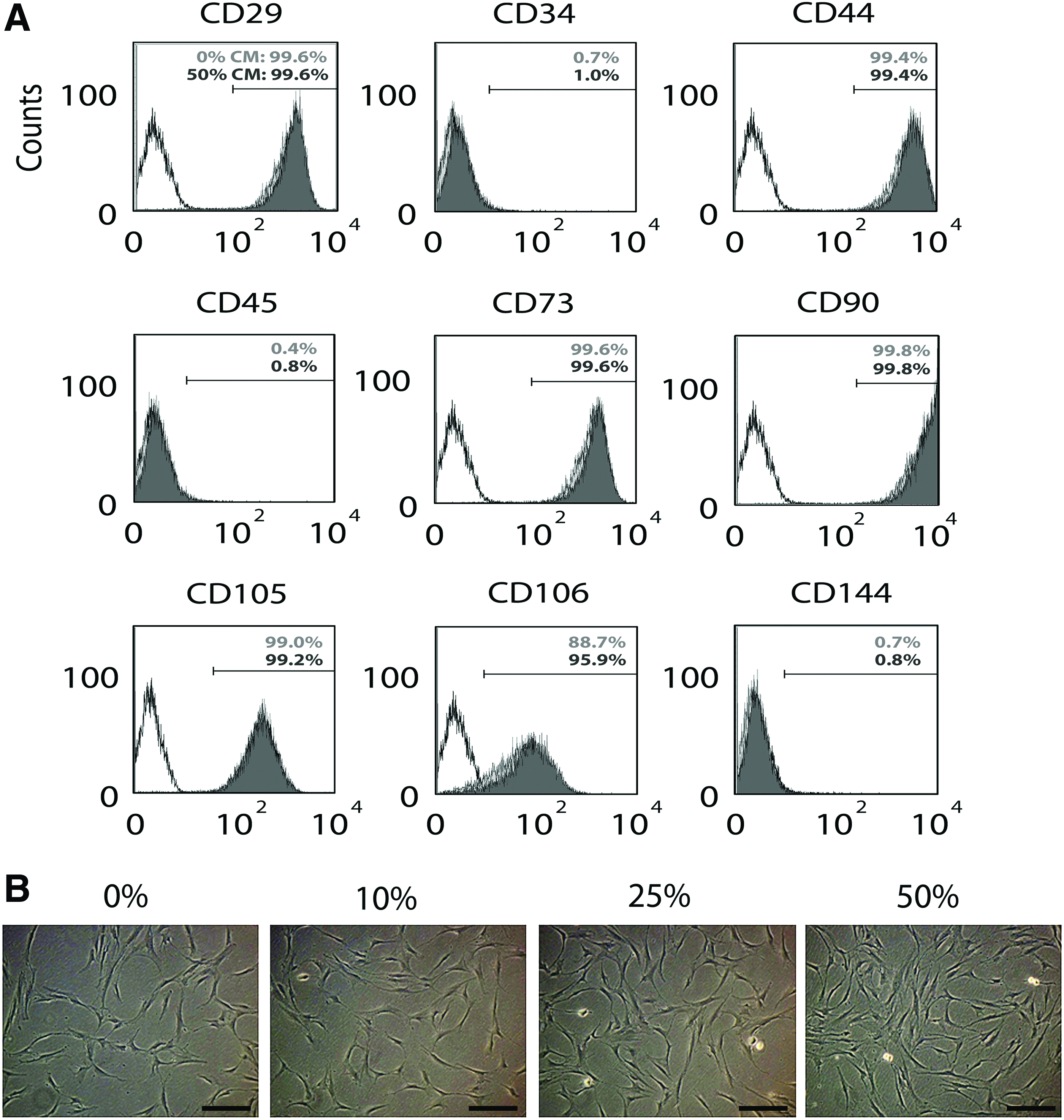
Phenotypic characteristics of hMSCs cultured with the MG-63 osteoblast CM. Isolated hMSCs were maintained in the 0%, 10%, 25%, and 50% working CM for two passages and assayed for hMSC surface markers and morphology.
MG-63 CM contains pro-osteogenic factors to enhance the osteogenic capacity of hMSCs
To test whether the MG-63 CM can modulate differentiation of hMSCs and whether the extent of modulation is affected by the presence of the CM in hMSC expansion and/or differentiation culture, we first maintained hMSCs in the basal medium supplemented with the 0% or 50% CM for two passages, and then induced osteogenesis and adipogenesis in differentiation media with or without the 50% CM (Fig. 3A). For chondrogenesis, since the standard protocol of chondrogenesis requires no serum in the chondrogenic medium, the CM treatment was performed only in the expansion culture before chondrogenesis. After 21 days of differentiation, the expression of lineage-specific mRNA transcripts and biochemical markers were analyzed. Our results showed that the mRNA transcript levels of the osteogenic marker osteocalcin (OC) significantly increased in the cells treated with the CM before differentiation (+/−), during differentiation (−/+), or both (+/+), compared to the cells expanded and differentiated in the standard basal and osteogenic medium without the CM (−/−) (Fig. 3B). Similar results were observed through the assays of the ALP activity quantification (Fig. 3C) and qualitative Alizarin Red staining (Fig. 3D), showing increases of both the ALP activity and mineralization in the CM-treated hMSCs (+/−, −/+, and +/+) compared to those of the control cells (−/−). Both real-time PCR and biochemical results suggest that soluble factors released from osteoblast-like MG-63 cells modulate the osteogenic capacity of undifferentiated hMSCs and enhance the induction of osteogenic differentiation. In contrast, CM treatment had no significant effect on adipogenesis and chondrogenesis of hMSCs (Fig. 3E–H), suggesting that the soluble proteins released from osteoblast-like MG-63 cells specifically stimulate osteogenesis and have negligible effects on adipogenesis and chondrogenesis of hMSCs.

Regulation of the MG-63 osteoblast CM on osteogenesis, adipogenesis, and chondrogenesis of hMSCs.
OPG is a potent soluble factor in MG-63 CM that stimulates hMSCs for osteogenesis
To identify active soluble factor(s) in the MG-63 cell culture supernatant that promotes hMSCs for osteogenesis, a comparative protein array assay was performed to analyze the supernatants collected from the MG-63 cell or MSC culture. We used the hMSC culture supernatant as a reference medium to compare with the MG-63 culture supernatant since hMSC culture without the CM was the control medium used in this study (Fig. 3). The array result showed that five soluble proteins (OPG, ANGPT1, angiopoietin-like 1 (ANGPTL1), crossveinless-2 (CV2), and PRGN) in the MG-63 culture supernatant were of at least twofold higher signal intensity than those in the hMSC culture supernatant (Fig. 4A). Among the five proteins, OPG showed the greatest difference of signal intensity (9.3-folds) between the hMSC and MG-63 cultures. We further quantified the concentration of OPG using an ELISA and the results showed that the concentrations in the hMSC and MG-63 cell culture supernatants were 6.4 and 35.5 ng/mL, respectively (Fig. 4B). Given that OPG is not tumorigenic and known as an important molecule for maintaining bone mass, we thus selected OPG to further investigate its role in the regulation of hMSC osteogenesis.
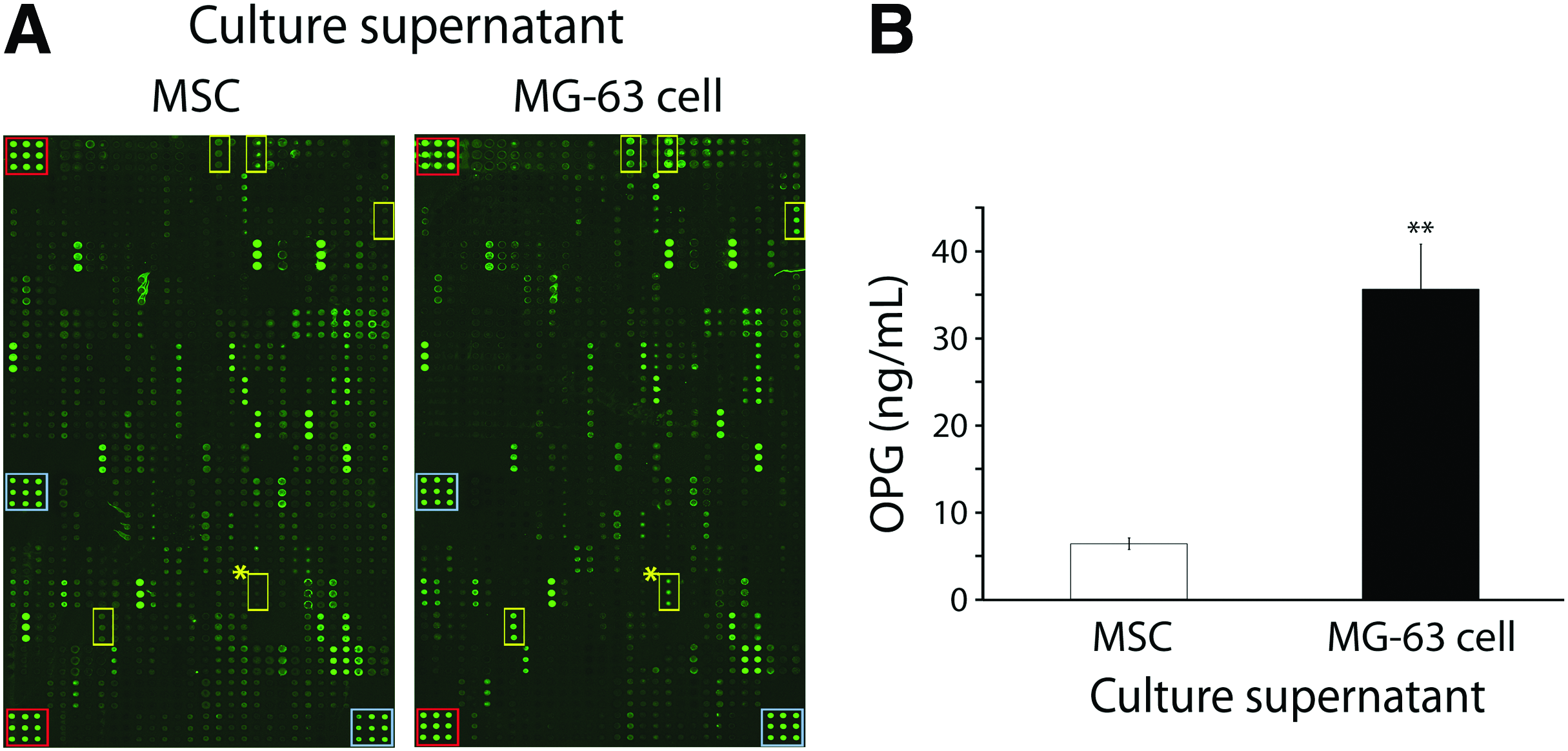
Identification of pro-osteogenic factors in MG-63 cell culture supernatant.
To evaluate the effect of OPG on regulating hMSC osteogenesis before and/or during differentiation, we maintained hMSCs in the basal medium supplemented with 0 or 40 ng/mL OPG for two passages before osteogenesis. The cells then were induced for osteogenesis in the differentiation medium with or without OPG (Fig. 5A), and then analyzed for the mRNA transcript expression of bone-related markers and the amount of calcium mineral deposits (Fig. 5B–D). The real-time PCR analysis showed that the expression level of OC mRNA transcript was significantly upregulated in the +/− treatment group, in which, cells were treated with 40 ng/mL OPG before differentiation and induced for differentiation without OPG, compared to the control group without OPG treatment in both expansion and differentiation culture (−/−) (Fig. 5B). The same OPG treatment (+/−) also increased the average expression levels of bone sialoprotein (BSP), osteomodulin (OMD), and ALP than the control group (−/−) (Fig. 5B). The calcium quantification assay showed that the production of calcium mineral deposits increased more than twofold in all the hMSC cultures treated with OPG, including the groups with the treatment before differentiation (+/−), during differentiation (−/+), or both (+/+), than the control group (−/−) (Fig. 5C). Similar results were also observed in the Alizarin Red staining assay (Fig. 5D). Taken together, these results demonstrate that OPG is a pro-osteogenic factor produced by osteoblast-like MG-63 cells. Interestingly, when comparing the OC expression levels of hMSCs stimulated by the CM (Fig. 3B) and OPG (Fig. 5B), we found that the fold change of OC expression between the treated and control groups was greater by the induction of the CM than by that of OPG alone, suggesting that the CM may contain other osteogenic factors than OPG to promote osteogenesis. In addition, the results of dsDNA quantification demonstrated that the experimental groups treated with OPG before and/or during osteogenic induction showed significantly increased cell numbers compared to the control group (Supplementary Fig. S1; Supplementary Data are available online at www.liebertpub.com/tea), indicating that OPG may be able to enhance either proliferation or viability of the cells.
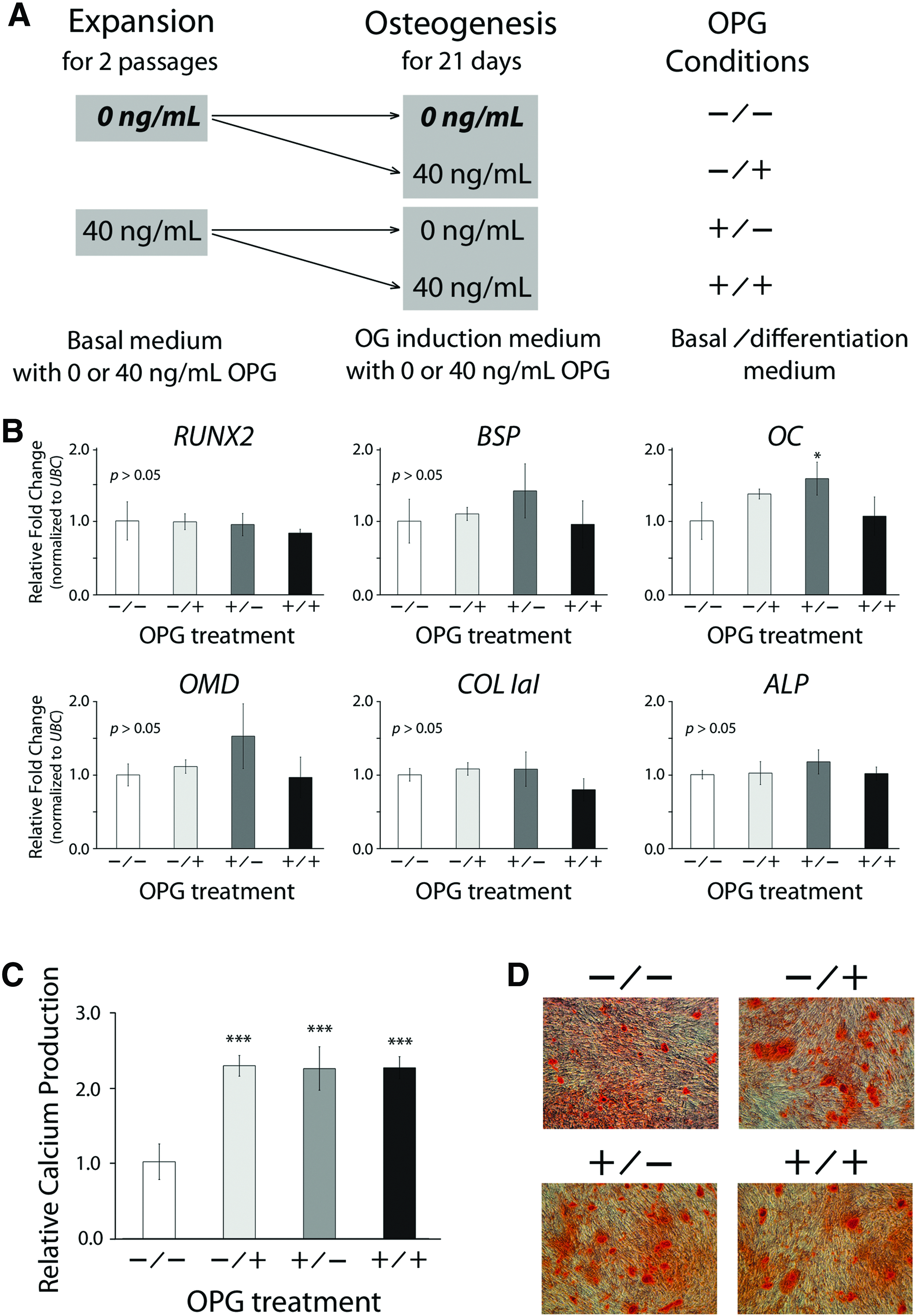
Effects of OPG stimulation in expansion and/or differentiation culture on subsequent osteogenesis of hMSCs.
PRGN is abundant in MG-63 CM, but does not affect hMSC osteogenesis
We intended to investigate the other four soluble factors (ANGPT1, ANGPTL1, CV2, and PRGN) that were previously identified by comparative protein arrays for their osteogenic effect. However, due to the availability issue of ELISA kits and recombinant proteins, we picked ANGPT1 and PRGN for further study. Our ELISA results showed that the concentration of ANGPT1 in the MG-63 culture supernatant was significantly lower than that in the hMSC culture supernatant (Supplementary Fig. S2A), different from the results measured by the protein arrays. On the other hand, compared to the protein array results, the ELISA results of PRGN concentrations showed a consistent trend, 2.9-fold higher in the MG-63 culture supernatant than in the hMSC culture supernatant (Supplementary Fig. S2B). Moreover, with the concentration confirmed by the ELISA measurement, hMSCs treated with PRGN before and/or during osteogenesis showed comparable expression levels of mRNA transcripts of bone-related markers except BSP and OC (Supplementary Fig. S3A). However, the amounts of calcium deposits produced by the cells were similar among the four experimental groups (Supplementary Fig. S3B). These results suggest that unlike OPG, PRGN is not able to enhance osteogenesis of hMSCs.
OPG-induced enhancement of hMSC osteogenesis is modulated through NF-κB activation
We further examined the role of NF-κB in the regulation of the osteogenic capacity of undifferentiated hMSCs treated with OPG. We chose NF-κB because this intracellular molecule is a downstream target of OPG, 17 and has been shown to be involved in the regulation of osteogenesis.18,19 To this end, hMSCs were cultured in the basal medium supplemented with or without 40 ng/mL OPG for two passages and collected before the induction of osteogenesis to analyze the expression of NF-κB. The results of Western blot analysis showed comparable total protein levels of NF-κB between the whole cell lysates of hMSCs treated with and without OPG (Fig. 6). In contrast, while the levels of NF-κB in the cytoplasm (the inactive form) were similar between these two groups of cells, the amount of NF-κB in the nucleus (the active form) was upregulated in the OPG-treated hMSCs compared to that in the untreated cells, suggesting that NF-κB was increasingly activated and translocated from the cytoplasm to the nucleus of the OPG-treated cells. This finding suggests that NF-κB is likely involved in the regulation of the osteogenic capacity of hMSCs stimulated by OPG.
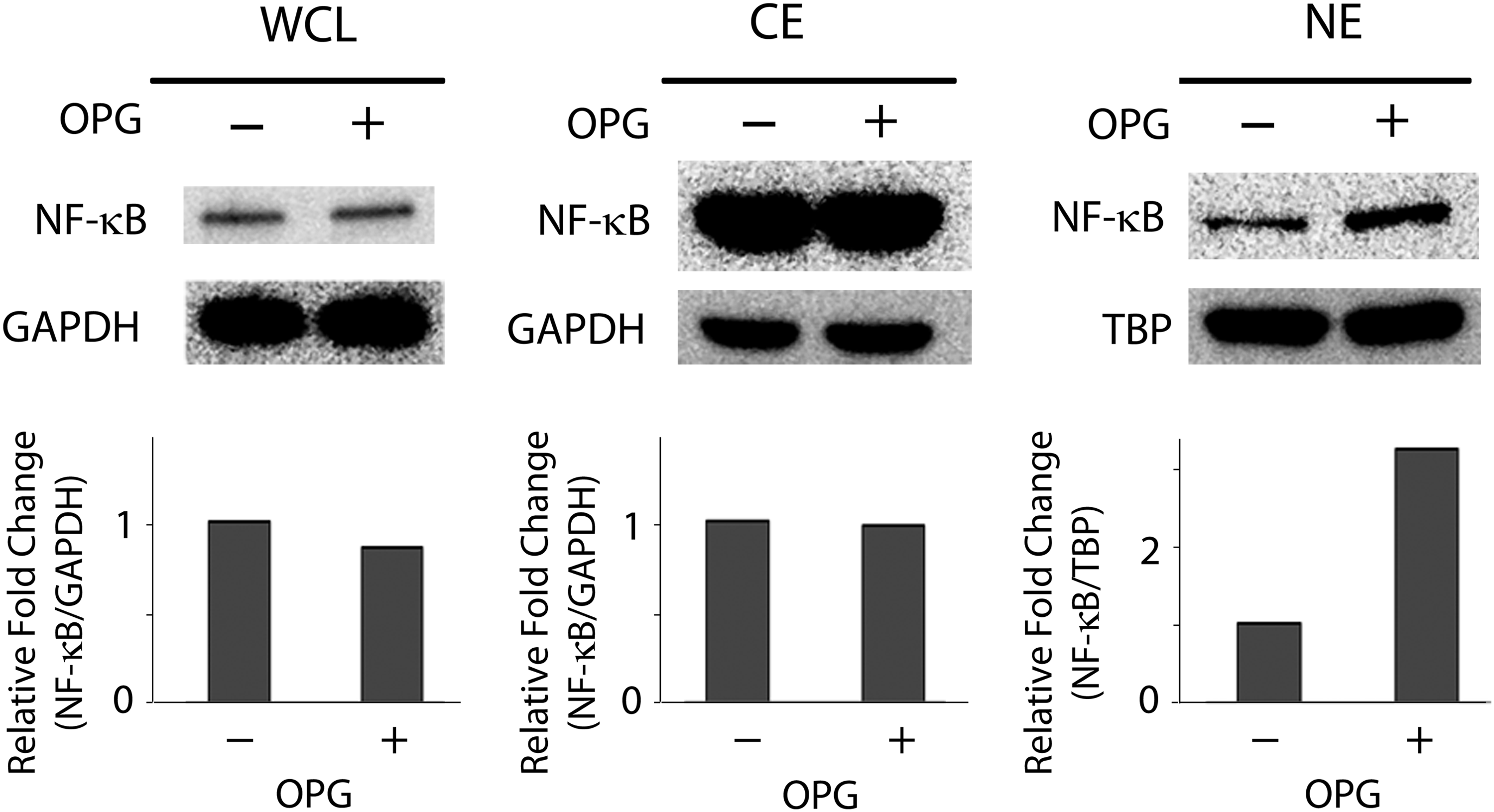
Analysis of NF-κB of OPG-treated hMSCs before osteogenic induction. Human hMSCs in expansion culture were treated with or without 40 ng/mL OPG for two passages and analyzed for the levels of NF-κB. Western blot analysis of whole cell lysate (WCL), cytoplasmic extract (CE), and nuclear extract (NE) of hMSCs showed that the levels of total and cytoplasmic NF-κB were similar between OPG-treated and -untreated hMSCs, whereas the level of nuclear NF-κB was increased in OPG-treated cells than that in the untreated cells. Densitometric measurements of the blot are shown as the fold change relative to the group without OPG treatment. NF-κB, nuclear factor-kappaB.
To investigate whether OPG modulates the osteogenic potential of undifferentiated hMSCs through the regulation of NF-κB, we treated hMSCs with both OPG and pharmacological NF-κB inhibitor TPCA-1, a potent, selective inhibitor of IκB kinase-2 (IKK-2), 20 for two passages in expansion culture before osteogenesis, and then induced in osteogenic culture without OPG and TPCA-1. At day 21, the positive control culture confirmed that the expression levels of OC mRNA transcripts (Fig. 7A) and nuclear NF-κB (Fig. 7B; lane 2) in the OPG-treated hMSCs were upregulated compared to the negative control culture with the untreated cells (Fig. 7A, B; lane 1). Moreover, the levels of nuclear NF-κB decreased with increasing TPCA-1 concentrations (Fig. 7B; lanes 2–4). To evaluate the effect of inhibiting the NF-κB activity of the OPG-treated hMSCs on the regulation of subsequent osteogenesis, cells were collected at days 3, 11, and 21 of osteogenesis and analyzed for the mRNA expression of bone-related markers. Our real-time PCR results showed that the expression of RUNX2, ALP, and OC of the cells treated with TPCA-1 was significantly downregulated at day 3. Interestingly, the cells inhibited by TPCA-1 transiently upregulated the expression of ALP and OC at day 11, but downregulated the expression of these two mRNA transcripts again at day 21 (Fig. 7C–E). Furthermore, the production of calcium deposits per dsDNA content decreased in the TPCA-1-treated cells compared to that in the untreated cells (Fig. 7F–H). These results indicate that OPG-treated hMSCs increase their osteogenic capacity through the activation of NF-κB before differentiation and upregulate osteogenesis upon receiving signals of osteogenic induction.
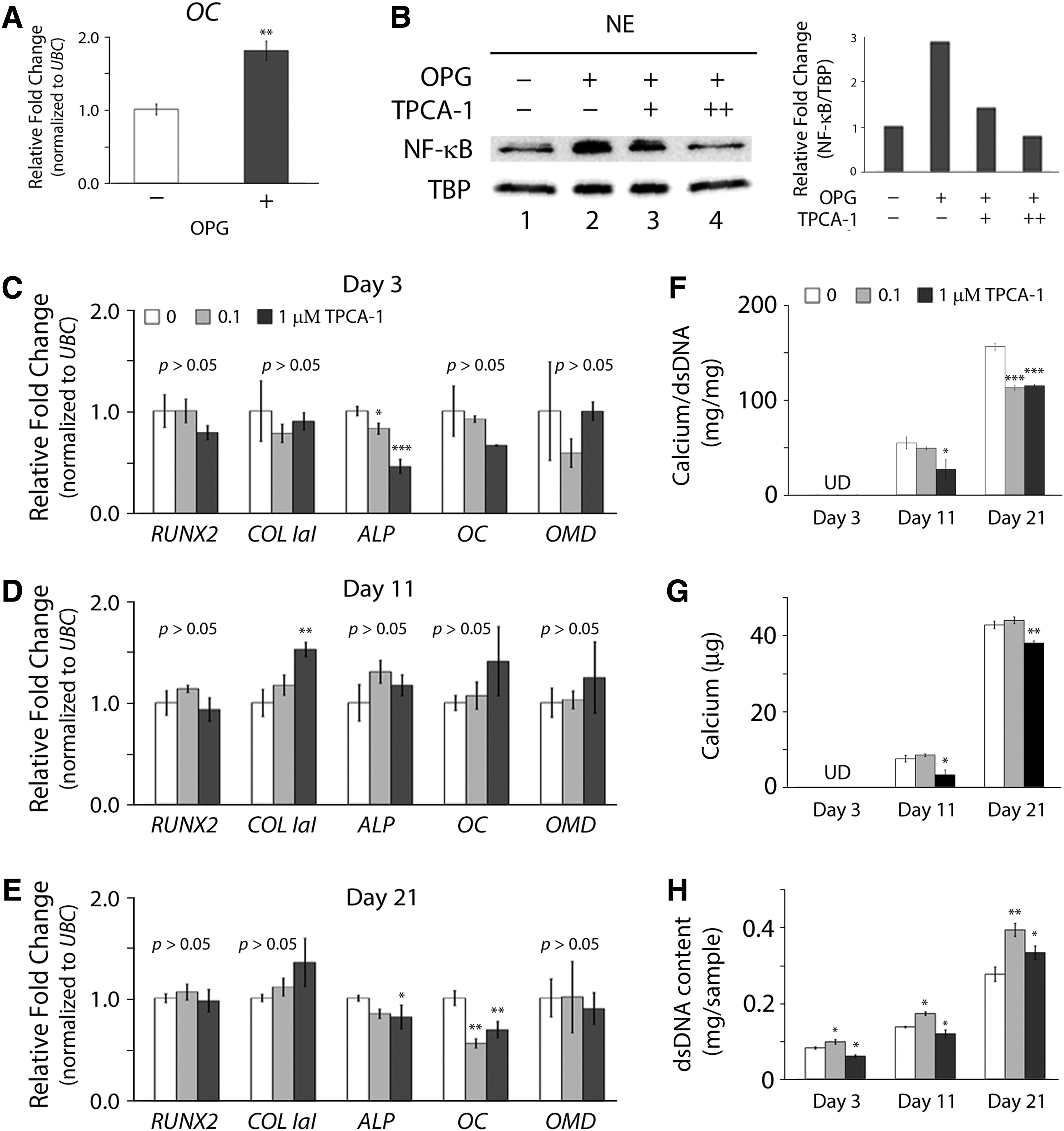
Effect of the NF-κB inhibitor TPCA-1 on osteogenesis of OPG-treated hMSCs. Human MSCs were treated with both 40 ng/mL OPG and 0.1 (+) or 1 μM (++ ) TPCA-1 for two passages before osteogenesis, and then induced for osteogenesis without OPG and TPCA-1.
Discussion
In this study, we demonstrate that hMSCs treated with the osteoblast CM increase their osteogenic capacity, and further identify OPG as one of the key soluble molecules involved in the regulation to enhance osteogenic differentiation. OPG is a decoy receptor that binds to the tumor necrosis factor-alpha (TNF-α) family members, such as the receptor activator of NF-κB ligand (RANKL), and can be produced by various types of cells, including osteoblasts, endothelial cells, and MSCs.21–23 The role of OPG in RANK/RANKL-dependent osteoclastogenesis has been well characterized as a potent anti-osteoclastogenic factor.23–26 Previous studies have shown that the efficiency of osteoclastogenesis is inversely related to the OPG level in both in vitro and in vivo models,24,25,27 and that significant bone resorption due to increased osteoclast activity is observed in OPG knockout animal models.24,26 In addition to the role of anti-bone resorption, OPG functions as an anti-apoptosis factor. Previous studies have shown that OPG inhibits TNF-related apoptosis-inducing ligand to suppress apoptosis in several types of cells.28,29
We used the human osteoblast-like cell line MG-63 as an osteoblast cell model to identify pro-osteogenic factors for enhanced bone generation. MG-63 cells are derived from osteosarcoma characterized by aggressive local bone formation. 30 Our rationale for the use of MG-63 cells was based on that MG-63 cells may release certain soluble factors, different from those released from normal osteoblasts, that are nononcogenic and are potent to enhance MSC osteogenesis. Furthermore, with several advantages over primary osteoblasts, 31 MG-63 cells have been widely used as an osteoblast model to investigate cell differentiation,13,15 protein expression, 32 and cytokine production. 33 On the other hand, primary osteoblasts may be used to produce the CM for the study of identifying pro-osteogenic molecules. It is possible that the types and production levels of soluble molecules released from primary osteoblasts are different from those of MG-63 cells. For example, a previous study by Pautke et al. has shown that primary osteoblasts produce more OPG than MG-63 cells. 34 Despite the differences of OPG production levels between these two types of cells, both their and our findings suggest that OPG is one of the soluble molecules greatly released in the culture medium.
OPG seems to have several attractive biological roles to improve bone tissue engineering. For example, its antiapoptotic role may reduce undesired cell death during bone generation within scaffolds and its anti-osteoclastogenic role may be beneficial in protecting newly synthesized mineral deposits from resorption. Recently, a study by Yao et al. has shown that OPG and bone morphogenetic protein-2 synergistically recruit MSCs to enhance bone formation in a dog model. 35 In our study, we show that undifferentiated MSCs treated with OPG in expansion culture before osteogenic induction, and then induced for differentiation can increase osteogenesis. Interestingly, our results also demonstrate that the continuous treatment of OPG throughout the period of both cell expansion and differentiation culture induces osteogenesis to a less extent compared to the treatment of OPG only used either in cell expansion or differentiation culture. The finding may be explained by that certain biological molecules can generate anabolic or catabolic effects on the regulation of biological activities, depending on the status of the cells. For example, studies have demonstrated that FGF-2 treatment before osteogenesis increases the osteogenic potential of MSCs during expansion culture, but inhibits osteogenesis of the cells during osteogenic culture.36,37 Our results show that OPG is likely to be one of the molecules that are able to prime undifferentiated hMSCs for enhanced osteogenesis.
The trend shown in the results of calcium quantification was different from that shown in the results of mRNA expression of OC in Figure 5. Both the calcium quantification and the mRNA expression analysis using real-time PCR are reliable methods to evaluate osteogenic differentiation of hMSCs. These assays, however, provide different aspects of the information regarding the process of osteogenesis. For example, the calcium quantification assay measures the amount of mineral deposits accumulated during the period of osteogenic culture, while the real-time PCR analysis shows a snap shot of expression levels of target genes at the moment of sample collection. Our results show that the mRNA expression levels of OC of the three OPG-treated groups become less different from those of the control group, while the amounts of accumulated calcium deposition of the OPG-treated groups are significantly higher than those of the control group after 21 days of culture. Overall, these results show a general trend that hMSC culture treated with OPG before and/or during osteogenic induction is able to enhance osteogenesis with the greatest enhancement occurring in the group treated with OPG only before osteogenesis.
Our results show that NF-κB is one of the intracellular molecules involved in the OPG-induced regulation of the osteogenic capacity of hMSCs. NF-κB is a transcription factor that plays a central role in inflammatory responses 38 and has been shown to regulate both MSC osteogenesis and osteoblast activities.18,19,39 For instance, Hess et al. have shown that constitutive activation of IKK greatly induces NF-κB signaling and enhances mineralization and the expression of RUNX2 and ALP in bone marrow-derived hMSCs. 18 Another study by Cho et al. have reported that NF-κB activation enhances osteogenesis of human adipose and bone marrow-derived hMSCs, and blocking the NF-κB activity using an inhibitor reverts enhanced osteogenesis in NF-κB-activated cells at the early stage of osteogenic differentiation. 19 In our study, we demonstrate a similar result showing that inhibiting the activation of NF-κB in OPG-treated hMSCs using the inhibitor TPCA-1 decreases osteogenic differentiation during the early and late stages of induction. It is worthwhile to mention that NF-κB in osteoblasts seems to play a different role in the regulation of bone formation. Chang et al. demonstrate that NF-κB activation inhibits maturation of osteoblasts and the production of mineral deposits, 39 suggesting that the role of NF-κB in the regulation of bone formation may be different between MSCs and osteoblasts, and its effect on the modulation of bone-related activities is associated with the differentiation status of MSCs in the osteogenic lineage.
Previous studies have reported that OPG suppresses NF-κB activation in osteoclast precursors through the binding of its ligand RANKL.17,40 However, our finding showed that OPG increases NF-κB activation in hMSCs. Currently, it is not clear how OPG activates NF-κB in hMSCs. One possible explanation is that the activation of NF-κB is initiated by the interaction of OPG and ligands other than RANKL. OPG contains several functional domains that can interact with various ligands, including RANKL. 41 In fact, Hakeda et al. have reported that OPG binds to an unknown ligand besides RANKL to regulate cell activities. 42 Further studies are needed to determine the detailed mechanism of OPG-stimulated osteogenesis of hMSCs.
Taken together, our results show that OPG enhances the osteogenic capacity of undifferentiated hMSCs through the activation of NF-κB before differentiation. Our findings also suggest that OPG is a pro-osteogenic molecule that can be used in MSC expansion culture to improve in vitro bone formation for bone tissue engineering.
Footnotes
Acknowledgments
We would like to thank Dr. Paul Manner at the University of Washington for providing human femoral heads for MSC isolation. We also thank Dr. Justin Palumbo for the help of proofreading the manuscript.
Disclosure Statement
No competing financial interests exist.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.