
Abstract
Application of autologous cells is considered for a broad range of regenerative therapies because it is not surrounded by the immunological and ethical issues of allo- or xenogenic cells. However, isolation, expansion, and application of autologous cells do suffer from variability in therapeutic efficacy due to donor to donor differences and due to prolonged culture. One important source of autologous cells is mesenchymal stromal cells (MSCs), which can differentiate toward endothelial-like cells, thus making them an ideal candidate as cell source for tissue vascularization. Here we screened MSCs from 20 donors for their endothelial differentiation capacity and correlated it with the gene expression profile of the whole genome in the undifferentiated state. Cells of all donors were able to form tubes on Matrigel and induced the expression of endothelial genes, although with quantitative differences. In addition, we analyzed the effect of prolonged in vitro expansion on the multipotency of human MSCs and found that endothelial differentiation is only mildly sensitive to expansion-induced loss of differentiation as compared to osteogenic and adipogenic differentiation. Our results show the robustness of the endothelial differentiation protocol and the gene expression data give insight in the differences in endothelial differentiation between donors.
Introduction
Mesenchymal stromal cells (MSC), a much used source of autologous cells, can be isolated relatively easily from multiple sources, including adipose tissue, tibia, femur, lumbar spine, and trabecular bone.1–4 They can be then expanded in vitro and are characterized by their multipotency. Among others, they can differentiate into the adipogenic, osteogenic, and chondrogenic lineages.5,6 What is less well documented is the ability of MSCs to differentiate toward other cell types such as skeletal muscle cells and neural cells.7–9 Moreover, it was shown lately that MSCs can also be a source of endothelial-like cells,10–12 which would qualify them as candidate cells for therapies aimed at improved tissue vascularization, such as treatment of ischemic tissues and various strategies concerning large graft engineering.
We and others have shown that when human MSCs are grown in an endothelial differentiation medium, they express endothelial markers such as CD31, the von Willebrand factor (vWF), and VE-cadherin, both at the mRNA and protein level.11–17 In addition, they actively take up acidic low-density lipoprotein, another hallmark of endothelial cells. When grown on Matrigel, the cells will form tube-like structures. Moreover, when these cells are implanted in immune-deficient animals, CD31-positive human cells can be seen in the vessel wall of perfused blood vessels, demonstrating that human cells can functionally contribute to blood vessels. Nevertheless, further characterization of the cells is needed, and for that reason, we refer to the cells as endothelial-like cells.
To apply MSC-derived endothelial-like cells, it is essential to establish a stable protocol for MSC isolation, culture, and differentiation, because it will affect the therapeutic effect of the cells, as demonstrated previously for the application of MSCs in bone tissue engineering.18–20 We have recently described a robust protocol to induce endothelial-like cells from bone marrow-derived MSCs in vitro and demonstrated their ability to contribute to the vasculature upon implantation in a mouse model. What remains to be addressed to bring endothelial-like MSCs to the clinic is both the large interdonor variation in multilineage potential1,21 and the phenomenon of loss of multipotency upon culture expansion of MSCs. We and others have observed striking differences between hMSCs of different donors with regard to the growth rate, expression of both lineage-specific and nonspecific markers such as alkaline phosphatase (ALP) and STRO-1 and their response to in vitro differentiation and ectopic bone formation.1,21–23 Similar situations can occur upon endothelial differentiation, which can explain the controversy concerning the ability of MSCs to acquire endothelial characteristics. A complicating fact is that therapeutic efficacy of MSCs is often not directly linked to the marker gene or protein expression in vitro.24–26
Because the yield of hMSCs upon isolation is very low, expansion in culture is an essential step in their application. Unfortunately, this is associated with culture-induced loss of multipotency, as described by several researchers.1,27 It was demonstrated that in vitro expanded MSCs acquire a phenotype characterized by loss of multipotency already at early passages27–29 followed by replicative senescence at later stages of expansion. It appears that a hierarchy exists among the different lineages with respect to the number of population doublings at which, loss of the particular differentiation route comes in effect.1,30
Although donor variability and loss of multipotency has been well-described for differentiation into the osteo- and adipogenic lineages, no such data are available for endothelial-like differentiation. To address this deficiency, we have created a bank of MSCs isolated from 62 donors (patients who were undergoing orthopedic surgery) and collected data about their in vitro expansion and differentiation capacity as well as their gene expression profiles. Using a microarray study, we have identified a diagnostic bone-forming classifier 23 capable of indicating the in vivo bone-forming capacity of hMSCs from different donors (unpublished data). In this manuscript, we have selected 20 donors of this bank and evaluated their propensity to differentiate into endothelial-like cells to sketch interdonor variability and to evaluate whether gene expression in undifferentiated hMSCs correlated to it. Moreover, we have studied the loss of the endothelial-like differentiation potential during culture expansion and compared it to loss of adipo- and osteogenic differentiation.
Materials and Methods
Isolation and culture
Human MSCs (hMSCs) were isolated from human bone marrow from donors who provided us with written informed consent. 31 Aspirates were resuspended using a 20G needle and plated at a density of 0.5 million mononucleated cells/cm2. Cells were grown in the MSC proliferation medium, which contains the minimal essential medium (alfa-MEM; GIBCO) supplemented with 10% fetal bovine serum (FBS; Lonza), 100 U/mL penicillin (GIBCO), 10 μg/mL streptomycin (GIBCO), 2 mM L-glutamin (GIBCO), 0.2 mM L-ascorbic acid 2-phosphate magnesium salt (ASAp; Sigma Aldrich), and 1 ng/mL basic fibroblast growth factor (bFGF; Instruchemie) at 37°C in a humid atmosphere with 5% CO2. Cells were expanded up to passage 2. For further experiments, hMSCs from 23 different donors and one immortalized clone (iMSCs, courtesy of Ola Myklebost) were cultured in the basic medium (alfa-MEM supplemented with 10% FBS, 100 U/mL penicillin, 10 μg/mL streptomycin, 2 mM L-glutamin, 0.2 mM ASAp). Human umbilical vein endothelial cells (HUVEC; Lonza) were cultured in the endothelial growth medium (EGM-2; Lonza).
ALP analysis
To analyze the activity of ALP, hMSCs from one donor were seeded at a density of 5,000 cells/cm2 in six-well plates. The osteogenic medium (alfa-MEM, 10% FBS, 100 U/mL penicillin, 10 μg/mL streptomycin, 2 mM L-glutamine, 0.2 mM ASAp, 0.01 M BGP, 10−8 M dexamethasone) was changed twice a week and cells were cultured for 1 week. The ALP activity was analyzed using the Sigma kit #85 as per the manufacturer's instructions. Briefly, the culture medium was aspirated, cells were washed twice with calcium- and magnesium-free phosphate-buffered saline (PBS) and fixed in acetone/citrate. After washing in deionized water, cells were stained with Fast Blue RR/naphthol. As a result, naphthol AS-MX is liberated and immediately coupled with a diazonium salt forming an insoluble, visible pigment at sites of phosphatase activity. Cells were photographed using a Nikon SMZ 10A camera.
To confirm the results, an ALP biochemical assay (CDP-Star; Roche) was performed on selected samples. Briefly, cells were washed with PBS and lysed with the lysis buffer (100 mM potassium phosphate, pH 7.8, 0.2% Triton-X-100) for 10 min at room temperature. Ten microliters of the lysate was put in an optiplate together with 40 μL of CDP-Star, incubated for 30 min in darkness at room temperature, and then measured with a Victor Light Luminescence Plate Reader (Perkin Elmer).
Adipogenesis
Adipogenic differentiation capacity of hMSCs was determined as described previously. 32 Briefly, cells from one donor were cultured for 3 weeks in the adipogenic medium (DMEM, 10% FBS, 100 U/mL penicillin, 10 μg/mL streptomycin, 0.5 mM IBMX, 1 μM dexamethasone, 10 μM insulin, and 200 μM indomethacin). After that, lipid formation was visualized by staining with Oil Red O. Cells were photographed using a Nicon Eclipse TE300 and the adipocytes were counted in the picture frame. At least 3 different locations of each well were included in the quantification.
Endothelial induction of MSCs
hMSCs from passage 3 and iMSCs from passage 25 were used for endothelial induction protocol as described previously. 10 Briefly, cells were seeded at a density of 3,000 cells/cm2 on tissue culture plastic in the EGM-2 and cultured for 10 days. After 1 day in static culture, shear force was applied using an orbital shaker. For induction on Matrigel, wells on six-well plates were covered with 1 mL of growth factor reduced Matrigel (BD Bioscience) diluted 1:1 in the EGM-2 without growth factors. Cells were seeded at the density of 30,000 cells per cm2 (60,000 cells per cm2 in case of HUVECs due to their smaller size) and cultured in a humid atmosphere with 5% CO2. After 24 h of culture on Matrigel, hMSCs start to express endothelial markers and are referred to as endothelial-like MSCs (EL-MSCs).
Matrigel assay
The assay was performed as described above. The formation of capillary-like structures (CLS) on Matrigel was observed over time using an inverted microscope (Nikon Eclipse TE300). Cells from all 20 donors were used in this study. Pictures were taken at different time points (4, 8, 16, 24, 48, and 96 h after seeding) using a Nikon DS-L2 camera. The experiment was performed in triplicate.
TubeCount
CLS formation was quantified based on images taken at a 24-h time point using TubeCount software, as described previously. 10 As a result, we gathered valuable statistics such as the total and average tube length, average tube width, number of tube branching points, and total tube area. A minimal number of 3 pictures per condition were analyzed.
RNA isolation and quantitative PCR
Total RNA was isolated using the TRIZOL reagent according to the manufacturer's protocol. In short, 1 mL of Trizol reagent was added per T25 flask (cells cultured in the basic medium) or per well (cells cultured on Matrigel in six-well plates). Samples were incubated for 5 min at room temperature to allow complete dissociation, and phase separation was performed by adding chloroform. After that, samples were centrifuged at 12,000 g for 15 min. RNA was precipitated by mixing the aqueous phase with isopropyl alcohol followed by a 10-min incubation at room temperature. Samples were centrifuged again and the remaining RNA pellet was washed with 75% ethanol. The obtained samples were dissolved in water, and after that, the quantity and quality of RNA were analyzed using spectrophotometry (ND-1000 spectrophotometer). The OD 260/280 nm ratios >1.8 were observed for all samples indicating high purity.
Five hundred nanograms of RNA was used for first strand cDNA synthesis using Superscript II (Invitrogen) according to the manufacturer's protocol. One microliter of 3×diluted cDNA was used for PCRs performed in a Light Cycler real time PCR machine (BioRad). Data were analyzed using Bio-Rad iQ5 software and expression of endothelial genes was calculated relative to GAPDH levels by the comparative ΔCT method. 33 Primers used in the study are platelet endothelial cell adhesion molecule-1 (CD31) F 5′ TCTATGACCTCGCCCTCCACAAA 3′, R 5′ GAACGGTGTCTTCAGGTTGGTATTTCA 3′; VEGF receptor 2 (KDR) F 5′ ACTTTGGAAGACAGAACCAAATTATCTC 3′, R 5′ TGGGCACCATTCCACCA 3′; vWF F 5′ TGCTGACACCAGAAAAGTGC 3′, R 5′ AGTCCCCAATGGACTCACAG 3′; GAPDH F 5′ CGCTCTCTGCTCCTCCTGTT 3′, R 5′ CCATGGTGTCTGAGCGATGT 3′.
Microarray analysis
To analyze the gene expression profile of hMSCs, microarray analysis was used. RNA was hybridized to the Human Genome U133A 2.0 Array (Affymetrix) and scanned with a GeneChip G3000 scanner (Affymetrix). To normalize the measurements, we used a normalization method, which removes hybridization, amplification, and array location-based technical effects. Further data analysis and statistical testing were performed using R and Bioconductor statistical software (www.bioconductor.org/). A linear modeling approach with empirical Bayesian methods, as implemented in Limma package, 34 was used to determine differential gene expression. To analyze the donor variation in terms of endothelial differentiation ability, we scored the different donor-derived MSCs based on their CD31 expression on one hand and KDR expression on the other hand. Subsequently, a list of genes ranked on fold change between the best and poorest endothelial differentiating donors was generated.
Statistics
Each experiment was performed in triplicate. Data that required multiple comparison tests were analyzed in SPSS (PASW statistics) using one-way Anova followed by the Tukey's multiple comparison test (p<0.05). Error bars on graphs represent standard deviation.
Ethics statement
hMSCs were isolated from human bone marrow from donors with written informed consent. This study was carried out in strict accordance with the recommendations of the Medisch Ethische Toetsings Commissie Twente (Medical Ethical Research Committee Twente) and was approved by this Committee.
Results
hMSC characterization
hMSCs from our bank of 62 donors have been characterized previously (unpublished data) according to the set of standards proposed by the Mesenchymal and Tissue Stem Cell Committee of the International Society for Cellular Therapy. 35 In this manuscript, we have randomly selected 20 donors from the donor bank. 1 After defrosting and brief expansion, a subsection of cells of all donors was subjected to osteogenic and adipogenic differentiation. As shown in Supplementary Figure S1 (Supplementary Data are available online at www.liebertpub.com/tea), MSCs from all donors readily acquired properties of adipocytes or osteoblasts, respectively, demonstrating their multipotency.
Effect of culture expansion on multipotency of hMSCs
We previously showed 1 that efficient differentiation of hMSCs is limited after prolonged expansion in vitro. To confirm these results, osteogenic and adipogenic differentiation of hMSCs from one donor was evaluated over 10 passages. Similar to previous observations, changes in cell morphology occurred during prolonged in vitro expansion (Fig. 1A). hMSCs between passage 7 and 10 lost their fibroblast-like shape and acquired a more spread morphology. The cells in culture became larger, with irregular and heterogeneous contours. Moreover, hMSCs expanded in vitro lost their proliferation capacity over time, being unable to reach confluency even after prolonged culture (data not shown).
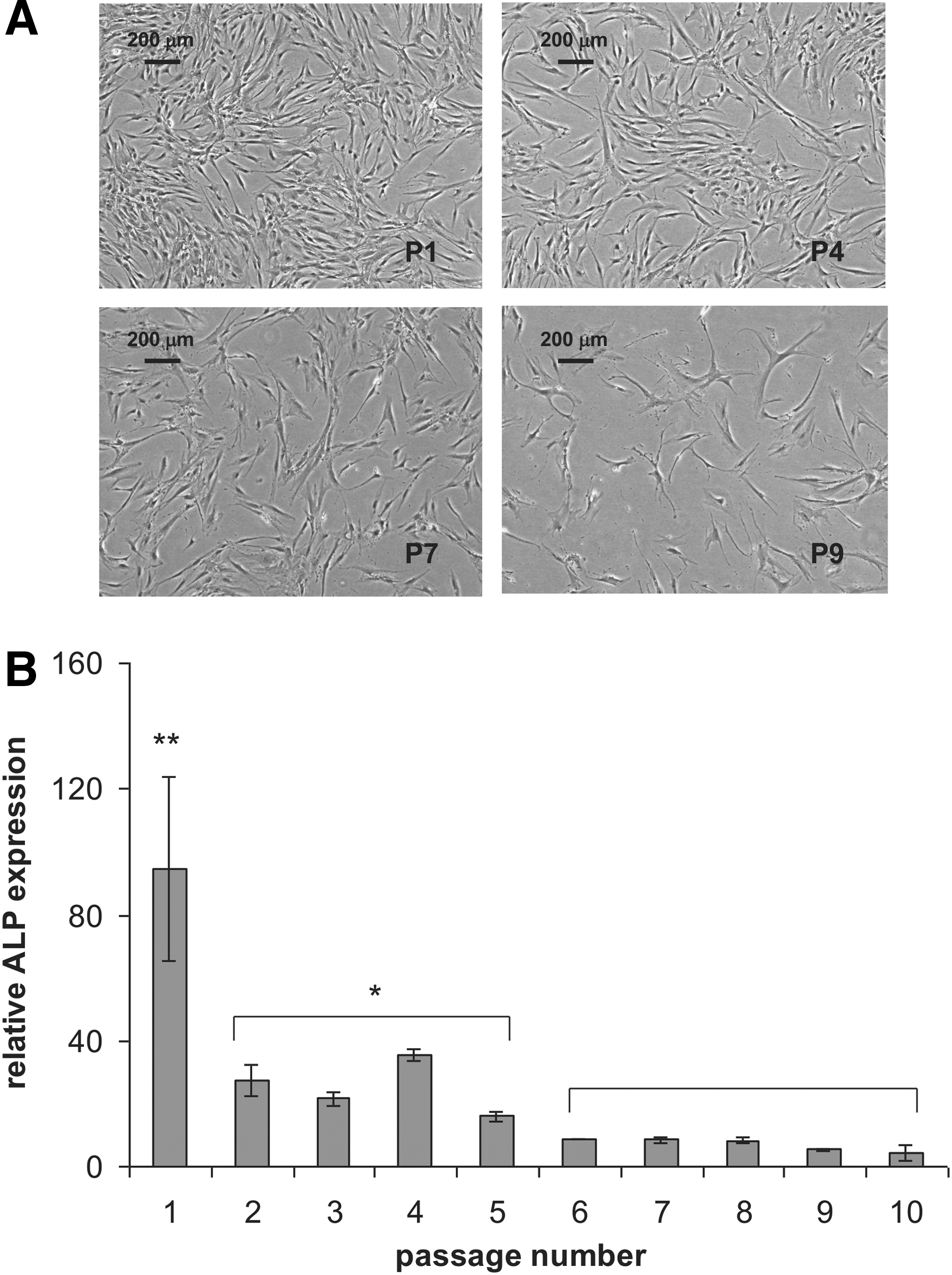
Expansion and osteogenic potential of serially passaged human mesenchymal stromal cells (hMSCs). Cell morphology
To document osteogenesis, we measured the dexamethasone-induced activity of the bone-maker ALP. 1 Cells from passage 1 showed a three times higher activity of ALP than cells from passage 2 to 5 (Fig. 1B) and the ALP activity declined again threefold in cells from passage 6 and higher (Fig. 1B). A similar trend was observed for adipogenic differentiation (Fig. 2A), where cells from passage 1 differentiated more efficiently than cells from passage 2 to 7. A gradual loss of adipogenic potential was observed (Fig. 2B). Next, we evaluated whether the endothelial differentiation capacity of hMSCs was lost as well upon culture expansion. In this case, we expanded cells from two donors (D42, D56) and first looked at their capacity to form tubes on Matrigel (Fig. 3A). Overall, MSCs did not lose their capacity to form tubes, up to passage 7. At higher passages, we were unable to retrieve sufficient number of cells to perform the assay. In the case of donor 42, prolonged expansion led to shorter CLS, although the number of branching points seemed to increase. With cells from donor 56, we observed the opposite, with longer capillary-like structures and fewer branching points at high passages. As a second readout of endothelial differentiation, we measured the expression of the endothelial marker CD31 in hMSCs exposed to the proliferation medium and endothelial differentiation medium of three donors at various passage numbers. At passage 3, we observed induction of CD31 in all three donors, but this was completely lost in donor 57. Surprisingly, the cells of the other two donors were still able to induce CD31 expression at passage 7, and even with a higher fold induction than at passage 3 (Fig. 3B). Our data show that endothelial differentiation is not as sensitive to culture-induced loss of differentiation capacity as adipogenic and osteogenic differentiation are.
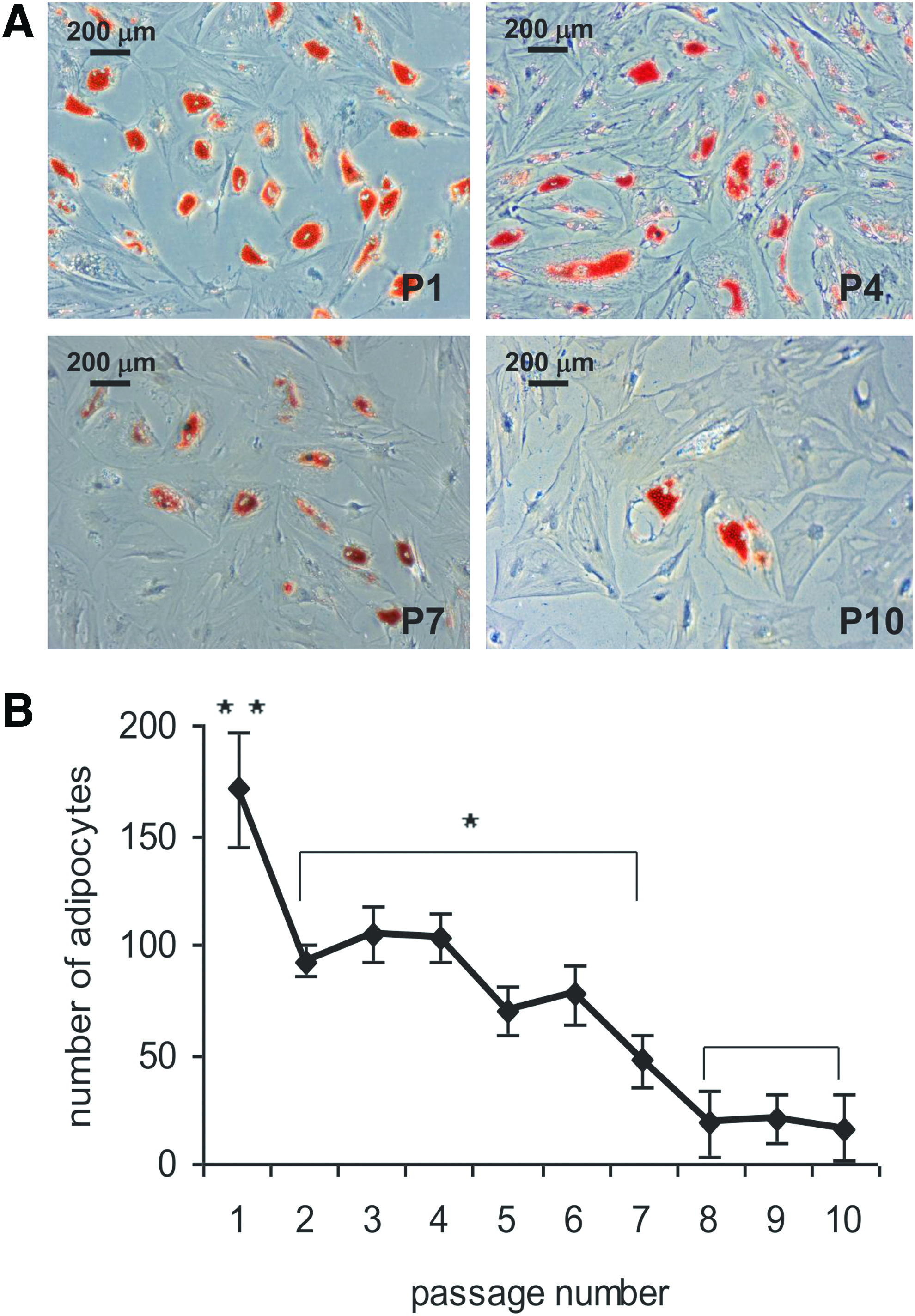
Adipogenic potential of serially passaged hMSCs. Adipogenic differentiation was visualized by staining with Oil Red O
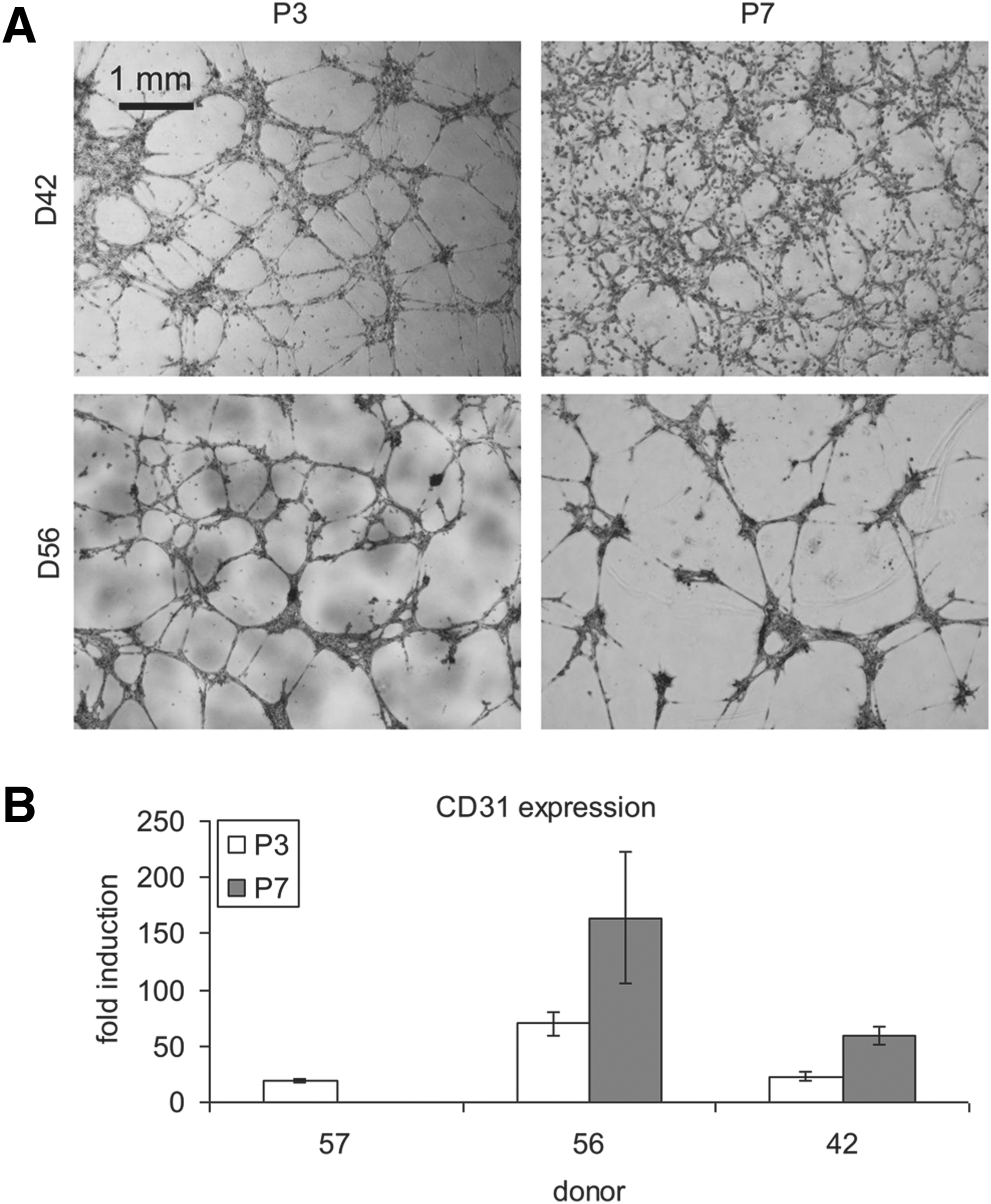
Endothelial potential of serially passaged hMSCs. Capillary-like structure formation was observed on Matrigel
Endothelial differentiation of hMSCs
To investigate whether the hMSC source (donor) influences endothelial differentiation, CLS formation was observed on Matrigel. Tube formation was recorded by phase-contrast microscopy and pictures were used for further study. As shown in Figure 4A, cells from different donors performed with very different outcome. The quantification of CLS formation was performed using in-house developed TubeCount software. HUVECs were used as a positive control in this assay and had a total tube length per analyzed area of 30 mm and more than 70 branching points. Measurement of these parameters with MSCs of 20 donors (Fig. 4B) revealed that cells from all donors were able to form tube-like structures although substantial interdonor variation was seen, ranging from equal efficiency as HUVECS to donors of whom the cells produced fivefold less tubes. It also occurred to us that cells from some donors started to form CLS at a later time point than most, but in the end reached the same level of CLS formation (data not shown). Overall, the total tube length and the number of branching points correlated to each other. Since all results were taken at an arbitrarily chosen time point (24 h) identical in case of all analyzed samples, this variability is present in the final quantification. Taken together, our data show that cells from all donors responded to the Matrigel stimulation, but with different efficiency. Yet, there was not a single donor from whom the cells did not respond at all.
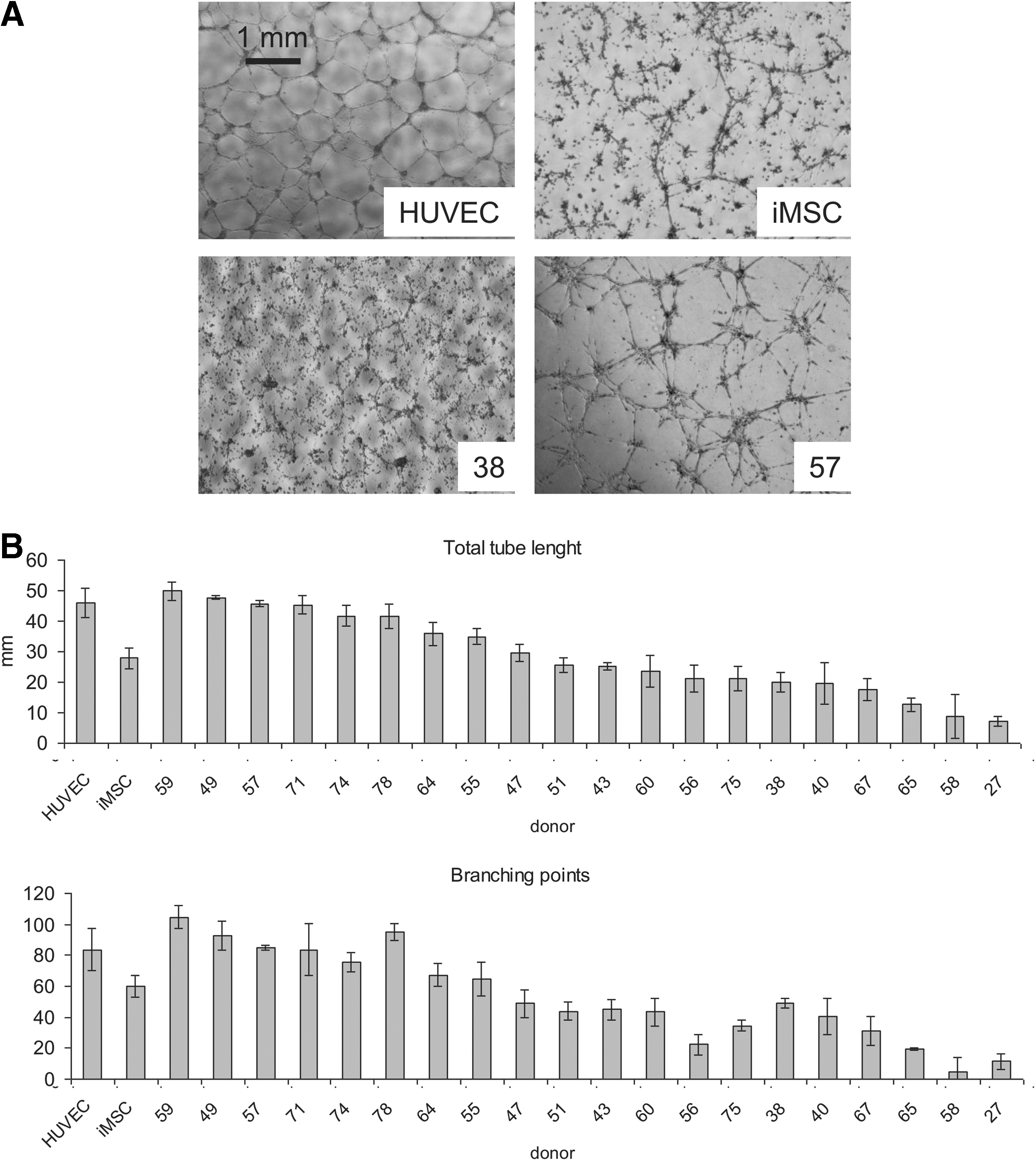
Tube assay on Matrigel. EL-hMSCs from different donors were cultured on Matrigel for 24 h in the EGM-2 medium
Endothelial gene expression profile in MSCs
For further classification of EL-MSCs from different donors, we analyzed endothelial-specific genes expression. As previously, cells were cultured for 10 days in EGM-2 on an orbital shaker and were then reseeded on Matrigel for another 24 h. This relative short time of culture was chosen because in some cases, the Matrigel coating was completely resorbed at later time points, indicating donor-specific matrix degrading activity. We performed qPCR on the platelet endothelial cell adhesion molecule (CD31), vascular endothelial growth factor receptor 2 (KDR), vWF, and vascular endothelial Cadherin (VE-Cadherin) and expressed it relative to GAPDH. We found a great variability of endothelial marker induction in cells from different donors. Cells from all donors responded to the differentiation protocol with increased CD31 expression (Fig. 5A), with a fold induction between 3 and 78. In fifteen donors out of twenty, we observed increased KDR expression (fold induction between 16 and 1000) and twelve of the 20 donors showed increased vWF expression (fold induction between 2 and 18) (Fig. 5B, C). Only six donors responded with significantly increased VE-Cadherin expression (fold induction between 32 and 88) (Fig. 5D). Not in all donors was VE-cadherin expression matched by other markers and we did not observe a robust correlation between total tube length or number of branching points and endothelial marker expression. The expression levels of all tested endothelial markers except KDR did not reach the level of its expression in HUVECs, however, we confirmed in our previous study with immunostainings 16 that these expression levels were relevant.

Donor variation in endothelial gene expression. Data are represented as fold induction compared to the gene expression level of undifferentiated MSCs from the same donor. Analyzed genes
For further analysis, we divided twenty donors into four groups, scoring their endothelial differentiation capability based on changes in CD31 and KDR expression: 25% of worst performing donors received grade 1, next 25% grade 2, third group grade 3, and the best performing 25% donors received grade 4 (Supplementary Table S1). These groups of samples allowed to discriminate between bone marrow-derived hMSCs able to differentiate efficiently into the endothelial lineage (grade 4) from hMSCs that do not have this differentiation capacity (grade 1) and were further used to find differences in gene expression of hMSCs at proliferating state.
Correlation between gene expression profile and ability to show endothelial gene expression
We investigated whether the differential expression of marker genes in endothelial-like cells is reflected in the gene expression repertoire of their undifferentiated ancestors, in other words, we tried to find genes predictive of the efficacy of induction of endothelial marker gene expression. To this end, we compared the expression profiles of MSCs between the four different groups defined above. The top 30 genes from the resulting list were ranked on p-value (Table 1). Interestingly, among the listed genes, we found three that are directly involved in the VEGF signaling pathway: CRYAB, EGR3, and IL7R; four that are typical for endothelial cells: GFPT2, EGR3, ILR7, and MEIS2; four involved in the proliferation and migration of cancer cells: SULF1, CRYZ, CA12, and PHLDA1; and six involved in cardiovascular system development and diseases: ELN, CRYAB, SLC5A3, CA12, PHLDA1, and FHL1 (Table 2).
(−) in the fold change value denotes downregulation when comparing donors with good and poor endothelial differentiation capabilities.
Donors were grouped based on their ability to differentiate into endothelial-like cells. Fold change between the best and poorest endothelial differentiating donors (based on CD31 and KDR expression) is specified in the table. Repeated gene names that appear in the table correspond to various probes for the same gene.
Donors were grouped based on their ability to differentiate into endothelial-like cells.
↑denotes upregulation;↓denotes downregulation when comparing donors with good and poor endothelial differentiation capabilities.
Discussion
hMSCs are multipotent cells with well-acknowledged capacity to differentiate into cell types from several lineages5–7,36,37 and have therefore been extensively tested in various tissue-engineering applications. One of the important questions that appear concerning the application of differentiated MSCs is the true nature of cells obtained upon various differentiation protocols. MSCs have been shown to differentiate toward various cell types based both on the expression of characteristic markers in vitro as well as on their ability to regenerate specific tissues in vivo. Still, the mechanism lying behind the ability of implanted MSCs to regenerate or improve regeneration of various tissues is not fully explored. 38 In our previous work, 16 we demonstrated that MSCs can differentiate toward endothelial-like cells. We based this conclusion on both the increased expression of endothelial markers in EL-MSCs, confirmed additionally by immunostainings, and on the effect on construct vascularization when EL-MSCs were implanted. Nonetheless, we did not claim to obtain endothelial cells from MSCs, but rather cells that are able to replace them in therapeutic applications. To prove the usefulness of the developed protocol, its robustness must be showed with respect to overpowering the variability among cells obtained from different donors.
In this study, we isolated and characterized hMSCs from 20 independent donors. First, we analyzed the cell performance in a Matrigel assay, where we found that the efficiency in tube formation was not the same between different donors. According to our observations at various time points (data not shown), some of the differences in results could be caused by the chosen observation time that was set for 24 h after seeding. Since cells from different donors started to form CLSs at different time points, the results were not fully reflecting the absolute potential of hMSCs to form CLSs. The differences between donors occurred rather as a result of differences in the speed of attachment and migration. CLS network formation on Matrigel occurs as an effect of cells attaching to the gel surface, spreading and getting in contact with each other and with the extracellular matrix (ECM). Since the cells used in this assay were freshly trypsinized, differences were likely to occur at the level of molecules responsible for the ECM binding on the cell surface. Therefore, it was to be expected that the speed of attachment may vary depending on the sample. Another reason for the differences in the speed of cell attachment and migration can be explained by the age of the donors, as Kasper et al. 39 demonstrated that those parameters are strongly correlated. Although donor variation in CLS formation was observed, CLS maturity was reached in all cases within 24 h. Endothelial cells of all origins are able to form tubules spontaneously, 40 but the exact time of this event can be different for various cells of this type. 41 Thus, since EL-MSCs from all investigated donors responded to Matrigel stimulation, the differences in time of response does not mean that those cells did not undergo endothelial differentiation.
Next, we analyzed the gene expression profile of differentiated hMSCs with respect to expression of endothelial markers: CD31, KDR, vWF, and VE-Cadherin. Here we found great variability of endothelial marker induction in cells from different donors. Upregulation in the expression of one selected gene was not necessarily followed by upregulation of the others. This phenomenon could be explained by the single time point that we have chosen for analysis. Cells from various donors were most likely at different levels of differentiation after 24 h of Matrigel culture. As demonstrated by Levenberg at al., 42 the expression of endothelial-specific genes in differentiating cells is not correlated with each other in time. Therefore, for microarray studies, we decided to focus on these two genes, namely, CD31 and KDR, which are commonly used as endothelial markers and for which, upregulation occurred in most of the donors.
Based on the performed microarray study, we selected 30 genes that were differentially expressed. Among those, we identified 17 genes whose expression is involved in VEGF signaling, expression of markers typical for endothelial cells, development and diseases of cardiovascular system as well as cell proliferation and migration in malignant cancers. hMSCs from various donors have, in general, a similar gene expression profile in their undifferentiated state. Therefore, many differentially expressed genes connected with endothelial cells might give the possibility to predict the potential of a given hMSC for endothelial differentiation. We consider that from genes involved in VEGF signaling, the upregulation of expression of EGR3 in proliferating hMSCs is of major importance. EGR3 is a transcription factor that plays a positive role in VEGFR1 transcription. 43 The abundance of EGR3 in hMSCs might make them more responsive to VEGF present in culture media and by that more prone to become EL-MSCs. Additionally EGR3 is a critical determinant of VEGF signaling in activated endothelial cells, where the EGR3 expression level influences VEGF-mediated proliferation, migration, and tube formation. 44 It is possible that EGR3 plays a similar role in differentiating hMSCs. The observed downregulation of two other genes involved in VEGF signaling that we found as differentially expressed in MSCs is probably connected to the fact that both CRYAB and IL7R are playing a role in VEGF signaling mostly in cases where cancer cells are involved.45–47
Among genes that are specific to endothelial cells and were differentially expressed in proliferating hMSCs, the expression of three was upregulated (GFPT2, EGR3, and MEIS2) while one, IL7R, typical for microvascular cells, was downregulated. Since GFPT2 48 and EGR3 are typical for all endothelial cells, MEIS2 is a marker of macrovascular endothelial cells and is not highly expressed in microvascular endothelial cells 49 and IL7R is present on microvascular endothelial cells; 50 we suggest that hMSCs might be more susceptible to differentiate toward endothelial cells that are different in their characteristics from microvascular endothelial cells.
Our results do not allow us to draw firm conclusions about hMSC potential to differentiate toward endothelial cells based on the differentially expressed genes involved in cell survival, proliferation, and migration or the one involved in cardiovascular system development and diseases. Further studies are necessary to conclude whether differences in these gene expressions are anyhow important for their application as therapeutic cells.
The applicability of hMSCs in regenerative medicine depends largely on their ability to expand in vitro without losing their potential to differentiate before reaching a clinically relevant cell number. Since EL-MSCs are required in rather large quantities to be used in TE applications, we performed a series of experiments to check whether extended proliferation will influence multipotency of hMSCs. To compare our results with the ones previously obtained,1,51 we evaluated osteogenic and adipogenic differentiation along with endothelial differentiation on cells from all 10 passages. Our results concerning the osteogenic and adipogenic potential of serially passaged hMSCs were consistent with those obtained earlier. Interestingly, the endothelial differentiation was not influenced by a prolonged expansion phase, as showed both in the Matrigel assay as well as in the gene expression studies. In the Matrigel assay, we observed that the parameters of structures formed after prolonged expansion are donor dependent, therefore, no conclusions about the influence of the passage number can be made. According to our unpublished results, we can conclude that in this assay, it is rather the number of seeded cells that is crucial for the final outcome, therefore, even cells from late passages that did not lose their ability to attach, spread, and migrate could efficiently form CLSs. Cells from one investigated donor stopped proliferating at passage 6, which excluded them from the endothelial senescence study, but in all other cases, no negative effect of in vitro expansion on endothelial potential was observed. To our knowledge, this is the first observation of such phenomenon in hMSCs. From this study, we conclude that hMSCs can be expanded in vitro at least up to passage 6, while keeping endothelial potential for effective use in tissue regeneration applications.
To summarize, our report provides several candidates for molecular markers that can be used for prediction of hMSC potential to differentiate into endothelial-like cells. We also showed that this potential is not affected by prolonged in vitro expansion as long as the cells keep proliferating. Altogether, our results indicate that hMSCs from various donors perform well enough to be considered in potential clinical applications.
Footnotes
Acknowledgments
This work was sponsored by a research grant from STW (De Stichting voor de Technische Wetenschappen) of J.deB. We acknowledge the financial support from the TeRM Smart Mix Program of the Netherlands Ministry of Economic Affairs and the Netherlands Ministry of Education, Culture, and Science.
Disclosure Statement
No competing financial interests exist.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.